Nanoparticles Formulations of Artemisinin and Derivatives as Potential Therapeutics for the Treatment of Cancer, Leishmaniasis and Malaria



Abstract
1. Introduction
2. Anticancer Drugs
2.1. Classification of Anticancer Drugs
2.2. Multi-Drug Resistance of Anticancer Drugs
3. Antimalarial Drugs
3.1. Classification of Antimalarials Based on Their Mode of Action in the Malaria Life Cycle
3.2. Drug Resistance of Antimalarial Drugs
4. Antileishmanial Drugs
4.1. Classification of Antileishmanial Drugs
4.2. Drug Resistance of Antileishmanial Drugs
5. Nanoparticles
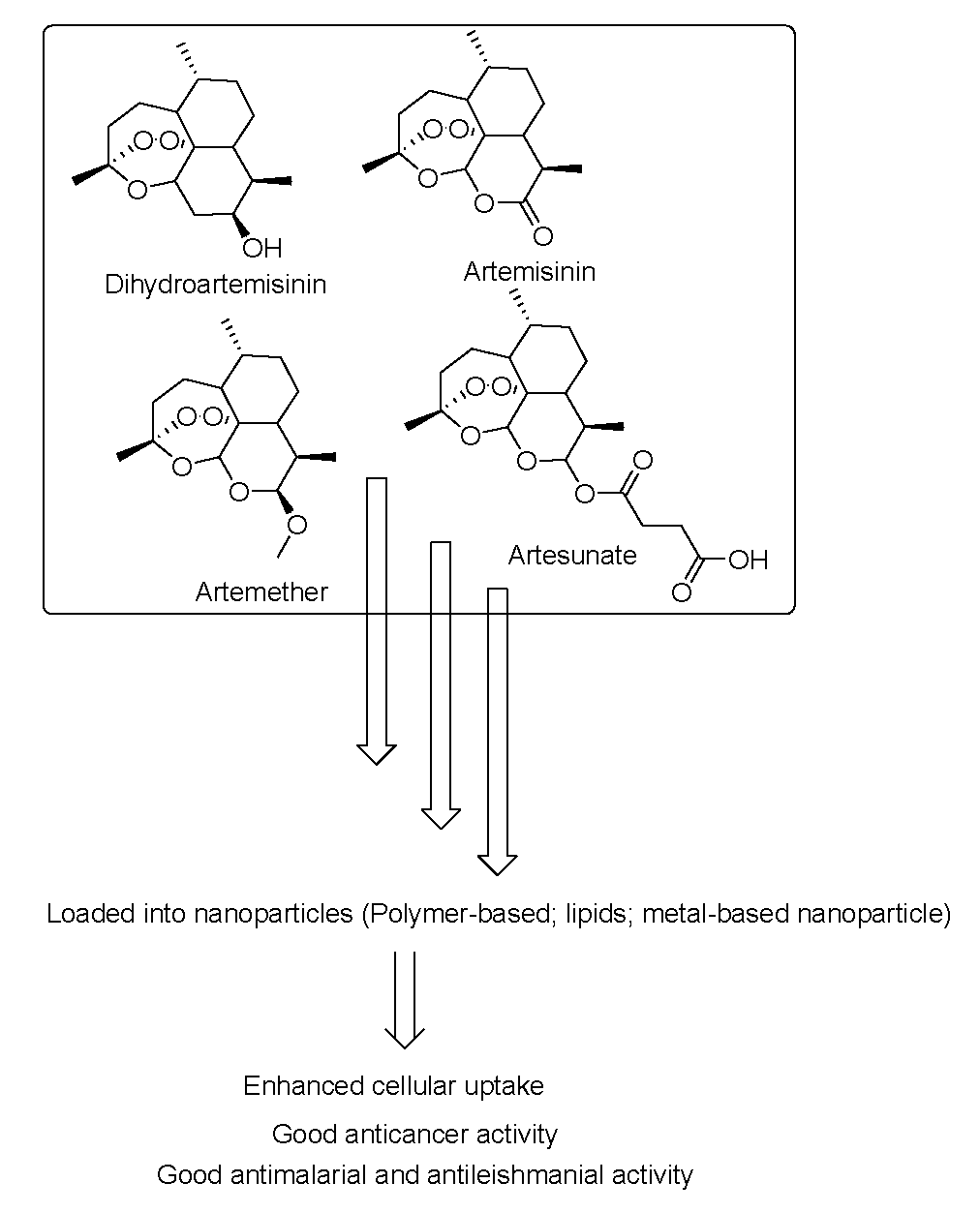
5.1. Nanoparticles Containing Artemisinins for Cancer Treatment
5.1.1. Polymer-Based Nanoparticles Loaded with Artemisinin and Derivatives with Anticancer Activity
5.1.2. Lipid-Based Nanoparticles Containing Artemisinin and Derivatives with Anticancer Activity
5.1.3. Metal-Based Nanoparticles Containing Artemisinin and Derivatives with Anticancer Activity
5.1.4. Carbon-Based Nanoparticles Loaded with Artemisinin and Derivatives as Anticancer Therapeutics
5.2. Nanoparticles Containing Artemisinins for Malaria Treatment
5.2.1. Polymer-Based Nanoparticles Loaded with Artemisinin and Derivatives with Antimalarial Activity
5.2.2. Lipid-Based Nanoparticles Loaded with Artemisinin and Derivatives with Antimalarial Activity
5.2.3. Metal-Based Nanoparticles Loaded with Artemisinin and Derivatives with Antimalarial Activity
5.3. Nanoparticles Containing Artemisinins for Leishmaniasis Treatment
5.3.1. Polymer-Based Nanoparticles Loaded with Artemisinin and Derivatives with Anti-Leishmanial Activity
5.3.2. Lipid-Based Nanoparticles Loaded with Artemisinin and Derivatives with Anti-Leishmanial Activity
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sharma, B.; Singh, S.; Kanwar, S.S. L-Methionase: A therapeutic enzyme to treat malignancies. BioMed Res. Int. 2014, 2014, 506287. [Google Scholar] [PubMed]
- Singh, S.; Sharma, B.; Kanwar, S.S.; Kumar, A. Lead phytochemicals for anticancer drug development. Front. Plant Sci. 2016, 7, 8973. [Google Scholar] [CrossRef]
- Ferlay, J.; Shin, H.-R.; Bray, F.; Forman, D.; Mathers, C.; Parkin, D.M. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int. J. Cancer 2008, 127, 2893–2917. [Google Scholar]
- Peter, S.; Aderibigbe, B.A. Ferrocene-based compounds with antimalaria/anticancer activity. Molecules 2019, 24, 3604. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Latest Global Cancer Data: Cancer Burden Rises to 18.1 Million New Cases and 9.6 Million Cancer Deaths in 2018; International Agency for Research on Cancer: Lyon, France, 2018; pp. 1–3. [Google Scholar]
- Shim, J.S.; Liu, J.O. Recent advances in drug repositioning for the discovery of new anticancer drugs. Int. J. Biol. Sci. 2014, 10, 654–663. [Google Scholar] [CrossRef]
- Alven, S.; Aderibigbe, B.A.; Balogun, M.O.; Matshe, W.M.R.; Ray, S.S. Polymer-drug conjugates containing antimalarial drugs and antibiotics. J. Drug Deliv. Sci. Technol. 2019, 53, 101171. [Google Scholar] [CrossRef]
- Estevanez, I.C.; Hernández-mora, M.G. Pulmonary complications of malaria: An update. Med. Clínica (English Ed.) 2016, 146, 354–358. [Google Scholar] [CrossRef]
- World Health Organization. Malaria. Available online: https://www.who.int/news-room/fact-sheets/detail/malaria (accessed on 27 October 2019).
- Nqoro, X.; Tobeka, N.; Aderibigbe, B.A. Quinoline-based hybrid compounds with antimalarial activity. Molecules 2017, 22, 2268. [Google Scholar]
- Kobets, T.; Grekov, I.; Lipoldova, M. Leishmaniasis: Prevention, parasite detection and treatment. Curr. Med. Chem. 2012, 19, 1443–1474. [Google Scholar]
- Mhlwatika, Z.; Aderibigbe, B.A. Application of dendrimers for the treatment of infectious diseases. Molecules 2018, 23, 2205. [Google Scholar]
- Alemayehu, B.; Alemayehu, M. Leishmaniasis: A review on parasite, vector and reservoir host. Heal. Sci. J. 2017, 11, 519. [Google Scholar] [CrossRef]
- Leishmaniasis. 2 March 2020. Available online: https://www.who.int/news-room/fact-sheets/detail/leishmaniasis (accessed on 15 April 2020).
- Croft, S.T.; Sndar, S.; Fairlamb, A.H. Is Technology Making Us Stupid (and Smarter)? Clin. Microbiol. Revews 2013, 19, 111–126. [Google Scholar] [CrossRef] [PubMed]
- Lamotte, S.; Späth, G.F.; Rachidi, N.; Prina, E. The enemy within: Targeting host–parasite interaction for antileishmanial drug discovery. PLoS Negl. Trop. Dis. 2017, 11, e0005480. [Google Scholar] [CrossRef] [PubMed]
- Neglected Tropical Diseases—The Present and the Future. Available online: https://tidsskriftet.no/en/2018/01/global-helse/neglected-tropical-diseases-present-and-future (accessed on 11 April 2020).
- WHO Neglected Tropical Disease. Available online: https://www.who.int/neglected_diseases/en/ (accessed on 11 April 2020).
- Cancer. Available online: https://www.who.int/news-room/fact-sheets/detail/cancer (accessed on 11 April 2020).
- Nasr, M.; Awadallah, A.; Al-karaki, R.; Madanat, F.; Alsunna, L.; Sami, K. Exploring the unique anticancer properties of curcumin nanoparticles. Clin. Oncol. Res. 2019, 2, 2–5. [Google Scholar]
- Agrawal, P.; Gupta, U.; Jain, N.K.A. Glycoconjugated peptide dendrimers-based nanoparticulate system for the delivery of chloroquine phosphate. Biomaterials. 2007, 28, 3349–3359. [Google Scholar] [PubMed]
- Ma, W.; Guo, Q.; Li, Y.; Wang, X.; Wang, J.; Tu, P. Co-assembly of doxorubicin and curcumin targeted micelles for synergistic delivery and improving anti-tumor efficacy. Eur. J. Pharm. Biopharm. 2017, 112, 209–223. [Google Scholar] [CrossRef]
- Dragojevic, S.; Ryu, J.S.; Raucher, D. Polymer-based prodrugs: Improving tumor targeting and the solubility of small molecule drugs in cancer therapy. Molecules 2015, 20, 21750–21769. [Google Scholar] [CrossRef]
- Hoare, T.R.; Kohane, D.S. Hydrogels in drug delivery: Progress and challenges. Polymer 2008, 49, 1993–2007. [Google Scholar]
- Chand, P.; Gnanarajan, G.; Kothiyal, P. In situ gel: A Review. Indian J. Pharm. Biol. Res. 2016, 4, 11–19. [Google Scholar]
- Chen, Y.; Lin, X.; Park, H.; Greever, R. Study of artemisinin nanocapsules as anticancer drug delivery systems. Nanomed. Nanotechnol. Biol. Med. 2009, 5, 316–322. [Google Scholar] [CrossRef]
- Marques, J.; Moles, E.; Urban, P.; Prohens, R.; Busquets, M.A.; Sevrin, C. Application of heparin as a dual agent with antimalarial and liposome targeting activities toward Plasmodium-infected red blood cells. Nanomedicine 2014, 10, 1719–1728. [Google Scholar] [CrossRef] [PubMed]
- Chamundeeswari, M.; Jeslin, J.; Verma, M.L. Nanocarriers for drug delivery applications. Environ. Chem. Lett. 2019, 17, 849–865. [Google Scholar] [CrossRef]
- Yan, G.; Li, A.; Zhang, A.; Sun, Y.; Liu, J. Polymer-based nanocarriers for co-delivery and combination of diverse therapies against cancers. Nanomaterials 2018, 8, 85. [Google Scholar] [CrossRef] [PubMed]
- Ndamase, A.S.; Aderibigbe, B.A.; Sadiku, E.R.; Labuschagne, P.; Lemmer, Y.; Ray, S.S.; Mwamadi, M. Synthesis, characterization and in vitro cytotoxicity evaluation of polyamidoamine conjugate containing pamidronate and platinum drug. J. Drug Deliv. Sci. Technol. 2018, 43, 267–273. [Google Scholar] [CrossRef]
- ITO, Y. Chemotherapy and hormone therapy for breast cancer: Current status and perspective. J. Jpn. Med. Assoc. 2002, 45, 424–433. [Google Scholar]
- Kim, J.H.; Choi, A.R.; Kim, Y.K.; Yoon, S. Co-treatment with the anti-malarial drugs mefloquine and primaquine highly sensitizes drug-resistant cancer cells by increasing P-gp inhibition. Biochem. Biophys. Res. Commun. 2013, 441, 655–660. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Ju, D.; Chang, C.; Reddy, P.M.; Velmurugan, B.K. A review on the effects of current chemotherapy drugs and natural agents in treating non-small cell lung cancer. BioMedicine 2017, 7, 12–23. [Google Scholar] [CrossRef] [PubMed]
- Efferth, T. Cancer combination therapies with artemisinin-type drugs. Biochem. Pharmacol. 2017, 139, 56–70. [Google Scholar] [CrossRef]
- Hevener, K.E.; Verstak, T.A.; Lutat, K.E.; Riggsbee, D.L.; Mooney, J.W. Recent developments in topoisomerase-targeted cancer chemotherapy. Acta Pharm. Sin. B 2018, 8, 844–861. [Google Scholar] [CrossRef]
- Goftar, M.K.; Rayeni, N.A.; Rasouli, S. Topoisomerase Inhibitors and Types of Them. Int. J. Adv. Biol. Biomed. Res. 2014, 2, 2431–2436. [Google Scholar]
- Vogus, D.R.; Evan, M.A.; Pusuluri, A.; Barajas, A.; Zhang, M.; Krishnan, V.; Nowak, M.; Menegatti, S.; Helgeson, M.E.; Squires, T.M.; et al. A hyaluronic acid conjugate engineered to synergistically and sequentially deliver gemcitabine and doxorubicin to treat triple negative breast cancer. J. Control. Release 2017, 267, 191–202. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Wan, J.; Zhang, Z.; Wang, F.; Guo, J.; Wang, C. Localized Fe(II)-induced cytotoxic reactive oxygen species generating nanosystem for enhanced anticancer therapy. ACS Appl. Mater. Interfaces 2018, 10, 4439–4449. [Google Scholar] [CrossRef] [PubMed]
- Budhraja, A.; Turnis, M.E.; Churchman, M.L.; Kothari, A.; Yang, X.; Xu, H.; Kaminska, E.; Panetta, J.C.; Finkelstein, D.; Mullighan, C.G.; et al. Modulation of navitoclax sensitivity by dihydroartemisinin-mediated MCL-1 repression in BCR-ABL+ B-Lineage acute lymphoblastic leukemia. Clin. Cancer Res. 2017, 23, 7558–7568. [Google Scholar] [PubMed]
- Lai, X.; Wang, H.; Cao, J.; Li, Y.; Dai, Y.; Xiang, Y.; Zhang, L. Circulating IL-27 is elevated in rheumatoid arthritis patients. Molecules 2016, 21, 1565. [Google Scholar]
- Xu, C.C.; Deng, T.; Fan, M.L.; Lv, W.B.; Liu, J.H.; Yu, B.Y. Synthesis and in vitro antitumor evaluation of dihydroartemisinin-cinnamic acid ester derivatives. Eur. J. Med. Chem. 2016, 107, 192–203. [Google Scholar]
- Lam, N.S.; Long, X.; Wong, J.W.; Griffin, R.C.; Doery, J.C. Artemisinin and its derivatives: A potential treatment for leukemia. Anti Cancer Drugs. 2019, 30, 1–18. [Google Scholar]
- Zhou, Y.; Wang, X.; Zhang, J.; He, A.; Wang, Y.L.; Han, K.; Su, Y.; Yin, J.; Lv, X.; Hu, H. Artesunate suppresses the viability and mobility of prostate cancer cells through UCA1, the sponge of miR-184. Oncotarget 2017, 8, 18260–18270. [Google Scholar]
- Nunes, J.J.; Pandey, S.K.; Yadav, A.; Goel, S.; Ateeq, B. Targeting NF-kappa B signaling by artesunate restores sensitivity of castrate-resistant prostate cancer cells to antiandrogens. Neoplasia 2017, 19, 333–345. [Google Scholar] [CrossRef]
- Kadioglu, O.; Chan, A.; Cong Ling Qiu, A.; Wong, V.K.W.; Colligs, V.; Wecklein, S.; Freund-Henni Rached, H.; Efferth, T.; Hsiao, W.L.W. Artemisinin derivatives target topoisomerase 1 and cause DNA damage in silico and in vitro. Front. Pharmacol. 2017, 8, 711. [Google Scholar]
- Ohtaka, M.; Itoh, M.; Tohda, S. BMI1 inhibitors down-regulate NOTCH signaling and suppress proliferation of acute leukemia cells. Anticancer Res. 2017, 37, 6047–6053. [Google Scholar]
- Lai, H.C.; Singh, N.P.; Sasaki, T. Development of artemisinin compounds for cancer treatment. Investig. New Drugs 2013, 31, 230–246. [Google Scholar]
- Gravett, A.M.; Liu, W.M.; Krishna, S.; Chan, W.C.; Haynes, R.K.; Wilson, N.L.; Dalgleish, A.G. In vitro study of the anti-cancer effects of artemisone alone or in combination with other chemotherapeutic agents. Cancer Chemother. Pharmacol. 2011, 67, 569–577. [Google Scholar] [PubMed]
- Azimi Mohamadabadi, M.; Mohammad Hassan, Z.; Hosseini, A.Z.; Gholamzad, M.; Noori, S.; Mahdavi, M.; Maroof, H. Arteether exerts antitumor activity and reduces CD4+ CD25+ FOXP3+ T-reg cells in vivo. Iran. J. Immunol. 2013, 10, 139–149. [Google Scholar] [PubMed]
- Alcântara, D.D.; Ribeiro, H.F.; Cardoso, P.C.; Araújo, T.M.; Burbano, R.R.; Guimarães, A.C.; Khayat, A.S.; de Oliveira Bahia, M. In vitro evaluation of the cytotoxic and genotoxic effects of artemether, an antimalarial drug, in a gastric cancer cell line (PG100). J. Appl. Toxicol. 2013, 33, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, Y.; Kong, R.; Xue, D.; Pan, S.; Chen, H.; Sun, B. Dihydroartemisinin suppresses pancreatic cancer cells via a microRNA-mRNA regulatory network. Oncotarget 2016, 7, 62460–62473. [Google Scholar]
- Kong, R.; Jia, G.; Cheng, Z.X.; Wang, Y.W.; Mu, M.; Wang, S.J.; Pan, S.H.; Gao, Y.; Jiang, H.C.; Dong, D.L.; et al. Dihydroartemisinin enhances Apo2L/TRAIL-mediated apoptosis in pancreatic cancer cells via ROS-mediated up-regulation of death receptor 5. PLoS ONE 2012, 7, e37222. [Google Scholar] [CrossRef]
- Jirangkul, P.; Srisawat, P.; Punyaratabandhu, T.; Songpattanaslip, T.; Mungthin, M. Cytotoxic effect of artemisinin and its derivatives on human osteosarcoma cell lines. J. Med. Assoc. Thai. 2014, 97, S215–S221. [Google Scholar]
- Li, Z.; Ding, X.; Wu, H.; Liu, C. Artemisinin inhibits angiogenesis by regulating p38 MAPK/CREB/TSP-1 signaling pathway in osteosarcoma. J. Cell. Biochem. 2019, 120, 11462–11470. [Google Scholar] [CrossRef]
- Drenberg, C.D.; Buaboonnam, J.; Orwick, S.J.; Hu, S.; Li, L.; Fan, Y.; Shelat, A.A.; Guy, R.K.; Rubnitz, J.; Baker, S.D. Evaluation of artemisinins for the treatment of acute myeloid leukemia. Cancer Chemother. Pharmacol. 2016, 77, 1231–1243. [Google Scholar]
- Zhu, S.; Liu, W.; Ke, X.; Li, J.; Hu, R.; Cui, H.; Song, G. Artemisinin reduces cell proliferation and induces apoptosis in neuroblastoma. Oncol. Rep. 2014, 32, 1094–1100. [Google Scholar] [CrossRef]
- Qi, L.; Yang, Y.; Liu, Y.C.; Zhu, T.X.; Jin, S.; Zang, L.; Zhang, Y.Y.; Ren, K. The inhibitory effect of dihydroartemisinin on the growth of neuroblastoma cells. Asian Pac. J. Trop. Biomed. 2016, 6, 279–282. [Google Scholar] [CrossRef]
- Liao, K.; Li, J.; Wang, Z. Dihydroartemisinin inhibits cell proliferation via AKT/GSK3β/cyclinD1 pathway and induces apoptosis in A549 lung cancer cells. Int. J. Clin. Exp. Pathol. 2014, 7, 8684–8691. [Google Scholar] [PubMed]
- Zhou, H.J.; Zhang, J.L.; Li, A.; Wang, Z.; Lou, X.E. Dihydroartemisinin improves the efficiency of chemotherapeutics in lung carcinomas in vivo and inhibits murine Lewis lung carcinoma cell line growth in vitro. Cancer Chemother Pharmacol. 2010, 66, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Gao, S.; Zhu, J.; Zheng, Y.; Zhang, H.; Sun, H. Dihydroartemisinin induces apoptosis and inhibits proliferation, migration, and invasion in epithelial ovarian cancer via inhibition of the hedgehog signaling pathway. Cancer Med. 2018, 7, 5704–5715. [Google Scholar] [CrossRef] [PubMed]
- Greenshields, A.L.; Shepherd, T.G.; Hoskin, D.W. Contribution of reactive oxygen species to ovarian cancer cell growth arrest and killing by the anti-malarial drug artesunate. Mol. Carcinog. 2017, 56, 75–93. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Li, J.; Shi, X.; Li, S.; Tang, P.M.; Li, Z.; Li, H.; Wei, C. Antimalarial Dihydroartemisinin triggers autophagy within HeLa cells of human cervical cancer through Bcl-2 phosphorylation at Ser70. Phytomedicine 2019, 52, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Gong, X.; Zhou, D.; Cao, J.; Fan, S.; Zhu, W. Cytotoxic effect and radiation enhancement of artemisinin in uterine cervical carcinoma cell line HeLa. Suzhou Univ. J. Med. Sci. 2010, 30, 224–226. [Google Scholar]
- Paccez, J.D.; Duncan, K.; Sekar, D.; Correa, R.G.; Wang, Y.; Gu, X.; Bashin, M.; Chibale, K.; Libermann, T.A.; Zerbini, L.F. Dihydroartemisinin inhibits prostate cancer via JARID2/miR-7/miR-34a-dependent downregulation of Axl. Oncogenesis 2019, 8, 14. [Google Scholar] [CrossRef]
- Zheng, L.; Pan, J. The anti-malarial drug artesunate blocks Wnt/β-catenin pathway and inhibits growth, migration and invasion of uveal melanoma cells. Curr. Cancer Drug Targets 2018, 18, 988–998. [Google Scholar] [CrossRef]
- Cabello, C.M.; Lamore, S.D.; Bair, W.B.; Qiao, S.; Azimian, S.; Lesson, J.L.; Wondrak, G.T. The redox antimalarial dihydroartemisinin targets human metastatic melanoma cells but not primary melanocytes with induction of NOXA-dependent apoptosis. Investig. New Drugs 2012, 30, 1289–1301. [Google Scholar] [CrossRef]
- Gao, X.; Luo, Z.; Xiang, T.; Wang, K.; Li, J.; Wang, P. Dihydroartemisinin induces endoplasmic reticulum stress-mediated apoptosis in HepG2 human hepatoma cells. Tumori J. 2011, 97, 771–780. [Google Scholar] [CrossRef]
- Liang, L.I.; Liu, J.H.; Chen, Y.T.; Ju, Y.C.; Jing, W.A.; Zhou, R.M.; Huang, X.; Xiang-Ran, H.U.; Tian, Y.J. Inhibitory effect and its mechanism of artesunate on the growth of hepatoma cells. Med. J. Chin. Peoples Lib. Army 2018, 43, 594–599. [Google Scholar]
- Patel, G.M.; Patel, J.D. Single Core Osmotic Pump (SCOP): Development of Single Layer Osmotic controlled release tablet for poorly soluble drug. Int. J. Pharm. Sci. Drug Res. 2012, 4, 1. [Google Scholar] [CrossRef]
- Mansoori, B.; Mohammadi, A.; Davudian, S.; Shirjang, S.; Baradaran, B. The different mechanisms of cancer drug resistance: A brief review. Adv. Pharm. Bull. 2017, 7, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Alven, S.; Aderibigbe, B.A. Combination therapy strategies for the treatment. Molecules 2019, 24, 3601. [Google Scholar] [CrossRef]
- Foley, M.; Tilley, L. Quinoline Antimalarials: Mechanisms of action and resistance and prospects for new agents. Pharmacol. Ther. 1998, 79, 55–87. [Google Scholar] [CrossRef]
- Biamonte, M.A.; Wanner, J.; Roch, K.G.L. Rrecent advances in malaria drug discovery. Bioorganic Med. Chem. Lett. 2014, 23, 2829–2843. [Google Scholar] [CrossRef]
- Singh, S.; Agarwal, D.; Sharma, K.; Sharma, M.; Nielsen, M.A.; Aligrangis, M.; Singh, A.K.; Gupta, R.D.; Awasthi, S.K. 4-Aminoquinoline derivatives: Synthesis, in vitro and in vivo antiplasmodial activity against chloroquine-resistant parasites. Eur. J. Med. Chem. 2016, 122, 394–407. [Google Scholar] [CrossRef]
- Capela, R.; Cabal, G.G.; Rosenthal, P.J.; Gut, J.; Mota, M.M.; Moreira, R.; Lopes, F.; Prudencio, M. Design and evaluation of Primaquine-Artemisinin hybrids as a multistage antimalarial strategy. Antimicrob. Agents Chemother. 2011, 55, 4698–4706. [Google Scholar] [CrossRef]
- Malaria. Available online: https://www.malariafreefuture.org/malaria (accessed on 11 April 2020).
- Philip, J. Rosenthal, Antiprotozoal Drugs. Available online: https://basicmedicalkey.com/antiprotozoal-drugs/ (accessed on 11 April 2020).
- WHO. Guidelines for the Treatment of Malaria, 3rd ed.; WHO: Geneva, Switzerland, 2015; Available online: https://www.who.int/docs/default-source/documents/publications/gmp/guidelines-for-the-treatment-of-malaria-eng.pdf?sfvrsn=a0138b77_2 (accessed on 11 April 2020).
- Saifi, M.A.; Beg, T.; Harrath, A.H.; Suleman, F.; Altayalan, H.; Al Quraishy, S. Antimalarial drugs: Mode of action and status of resistance. Afr. J. Pharm. Pharmacol. 2013, 7, 148–156. [Google Scholar] [CrossRef]
- Bray, P.; Janneh, O.; Ward, S. Chloroquine uptake and activity is determined by binding to ferriprotoporphyrin IX in Plasmodium falciparum. Novartis Fundam. Symp. 1999, 226, 252–264. [Google Scholar]
- Grewal, R.S. Pharmacology of 8-aminoquinolines. Bull. World Health Organ. 1981, 59, 397–406. [Google Scholar] [PubMed]
- Sa, J.M.; Twu, O.; Hayton, K.; Reyes, S.; Fay, M.P.; Ringwald, P.; Wellems, T.E. Geographic patterns of Plasmodium falciparum drug resistance distinguished by differential responses to amodiaquine and chloroquine. Proc. Natl. Acad. Sci. USA 2009, 106, 18883–18889. [Google Scholar] [CrossRef] [PubMed]
- Petersen, I.; Eastman, R.; Lanzer, M. Drug-resistant malaria: Molecular mechanisms and implications for public health. FEBS Lett. 2011, 585, 1551–1562. [Google Scholar] [PubMed]
- Muangphrom, P.; Seki, H.; Fukushima, E.O.; Muranaka, T. Artemisinin-based antimalarial research: Application of biotechnology to the production of artemisinin, its mode of action, and the mechanism of resistance of Plasmodium parasites. J. Nat. Med. 2016, 70, 318–334. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Pandey, S.C.; Samant, M. Slow pace of antileishmanial drug development. Parasitol Open. 2018, 4, 1–11. [Google Scholar] [CrossRef]
- Ghorbani, M.; Farhodi, R. Leishmaniasis in humans: Drug or vaccine therapy? Drug Des. Devel. Ther. 2018, 12, 25–40. [Google Scholar] [CrossRef]
- Parasites-Leishmaniasis. Available online: https://www.cdc.gov/parasites/leishmaniasis/health_professionals/index.html#tx (accessed on 11 April 2020).
- Rosenthal, E.; Delaunay, P.; Jeandel, P.Y.; Haas, H.; Pomares-Estran, C.; Marty, P. Liposomal amphotericin B as treatment for visceral leishmaniasis in Europe, 2009. Med. Mal. Infect. 2009, 39, 741–744. [Google Scholar] [CrossRef]
- Nagle, A.S.; Khare, S.; Kumar, A.B.; Supek, F.; Buchynskyy, A.; Mathison, C.J.; Chennamaneni, N.K.; Pendem, N.; Buckner, F.S.; Gelb, M.H.; et al. Recent developments in drug discovery for leishmaniasis and human African trypanosomiasis. Chem. Rev. 2014, 114, 11305–11347. [Google Scholar] [CrossRef]
- Sen, R.; Ganguly, S.; Saha, P.; Chatterjee, M. Efficacy of artemisinin in experimental visceral leishmaniasis. Int. J. Antimicrob. Agents. 2010, 36, 43–49. [Google Scholar] [CrossRef]
- Sarkar, A.; Saha, P.; Mandal, G.; Mukhopadhyay, D.; Roy, S.; Singh, S.K.; Das, S.; Goswami, R.P.; Saha, B.; Kumar, D.; et al. Monitoring of intracellular nitric oxide in leishmaniasis: Its applicability in patients with visceral leishmaniasis. Cytom. A. 2011, 79, 35–45. [Google Scholar]
- Ghaffarifar, F.; Heydari, F.E.; Dalimi, A.; Hassan, Z.M.; Delavari, M.; Mikaeiloo, H. Evaluation of apoptotic and antileishmanial activities of Artemisinin on promastigotes and BALB/C mice infected with Leishmania major. Iran. J. Parasitol. 2015, 10, 258–267. [Google Scholar] [PubMed]
- Ebrahimisadr, P.; Ghaffarifar, F.; Hassan, Z.M. In-vitro evaluation of antileishmanial activity and toxicity of artemether with focus on its apoptotic effect. Iran J. Pharm. Res. 2013, 12, 903–904. [Google Scholar] [PubMed]
- Hendrickx, S.; Guerin, P.J.; Caljon, G.; Croft, S.L.; Maes, L. Evaluating drug resistance in visceral leishmaniasis: The challenges. Parasitology 2018, 145, 453–463. [Google Scholar] [CrossRef] [PubMed]
- Maltezou, H.C. Drug resistance in visceral leishmaniasis. J. Biomed. Biotechnol. 2009, 2010, 617521. [Google Scholar] [CrossRef] [PubMed]
- Ponte-Sucre, A.; Gamarro, F.; Dujardin, J.C.; Barrett, M.P.; Lopez-Velez, R.; Garcia-Hernandez, R.; Pountain, A.W.; Mwenechanya, R.; Papadopoulou, B. Drug resistance and treatment failure in leishmaniasis: A 21st century challenge. PLoS Negl. Trop. Dis. 2017, 11, 24. [Google Scholar]
- Basselin, M.; Denise, H.; Coombs, G.H.; Barrett, M.P. Resistance to Pentamidine in Leishmania mexicana Involves Exclusion of the Drug from the Mitochondrion. Antimicrob. Agents Chemother. 2002, 46, 3731–3738. [Google Scholar] [CrossRef]
- Garcia-salcedo, J.A.; Unciti-broceta, J.D.; Valverde-pozo, J.; Tica, A.A. New approaches to overcome transport related drug resistance in Trypanosomatid Parasitese. Front. Pharmacol. 2016, 7, 351. [Google Scholar] [CrossRef]
- Capela, R.; Moreira, R.; Lopes, F. An overview of drug resistance in Protozoal diseases. Intern. J. Mol. Sci. 2019, 20, 5478. [Google Scholar] [CrossRef]
- Dennis, E.; People, V.A.; Johnson, F.; Topps, D.; Bopta-Waffo, A.; Coast, M.T. Utilizing nanotechnology to combat malaria. Infect. Dis. Ther. 2015, 3, 1000229. [Google Scholar]
- Pan, J.; Rostamizadeh, K.; Filipczak, N.; Torchilin, V.P. Polymeric Co-delivery systems in cancer treatment: An overview on component drugs’ dosage ratio effect. Molecules 2019, 24, 1035. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Dilbaghi, N.; Saharan, R.; Bhanjana, G. Nanotechnology as emerging tool for enhancing solubility of poorly water-soluble drugs. Bionanoscience 2012, 2, 227–250. [Google Scholar]
- Rao, J.P.; Geckeler, K.E. Mint: Polymer nanoparticles: Preparation techniques and size-control parameters. Prog. Polym Sci. 2011, 36, 887–913. [Google Scholar] [CrossRef]
- Vijay, R.; Angayarkanny, S.; Reddy, B.S.R.; Mandal, A.B.; Baskar, G. Mint: Adsorption and emulsification properties of amphiphilic poly(styrene-co-octadecyl maleamic acid salt) with comb-like architecture. J. Colloid Interface Sci. 2010, 346, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Torchilin, V. Tumor delivery of macromolecular drugs based on the EPR effect. Adv. Drug Deliv. Rev. 2011, 63, 131–135. [Google Scholar] [CrossRef] [PubMed]
- Volpedo, G.; Costa, L.; Ryan, N.; Halsey, G.; Satoskar, A.; Oghumu, S. Nanoparticulate drug delivery systems for the treatment of neglected tropical protozoan diseases. J. Venom. Anim. Toxins Incl. Trop. Dis. 2019, 25, e144118. [Google Scholar]
- Dreaden, E.C.; Alkilany, A.M.; Huang, X.; Murphy, C.J.; El-Sayed, M.A. The golden age: Gold nanoparticles for biomedicine. Chem. Soc. Rev. 2012, 41, 2740–2779. [Google Scholar]
- Bozzuto, G.; Molinari, A. Liposomes as nanomedical devices. Int. J. Nanomed. 2015, 10, 975–999. [Google Scholar] [CrossRef]
- Nisini, R.; Poerio, N.; Mariotti, S.; De Santis, F.; Fraziano, M. The multirole of liposomes in therapy and prevention of infectious diseases. Front. Immunol. 2019, 9, 155. [Google Scholar] [CrossRef]
- Jaiswal, M.; Dudhe, R.; Sharma, P.K. Nanoemulsion: An advanced mode of drug delivery system. 3 Biotech. 2015, 5, 123–127. [Google Scholar]
- Chime, S.A.; Kenechukwu, F.C.; Attama, A.A. Nanoemulsions—Advances in Formulation, Characterization and Applications in Drug Delivery, Application of Nanotechnology in Drug Delivery; Demir, S.A., Ed.; IntechOpen: London, UK, 2014; Available online: https://www.intechopen.com/books/application-of-nanotechnology-in-drug-delivery/nanoemulsions-advances-in-formulation-characterization-and-applications-in-drug-delivery (accessed on 11 April 2020). [CrossRef]
- Ghasemiyeh, P.; Mohammadi-Samani, S. Solid lipid nanoparticles and nanostructured lipid carriers as novel drug delivery systems: Applications, advantages and disadvantages. Res. Pharm. Sci. 2018, 13, 288–303. [Google Scholar] [PubMed]
- Yoon, G.; Park, J.W.; Yoon, I.S. Solid lipid nanoparticles (SLNs) and nanostructured lipid carriers (NLCs): Recent advances in drug delivery. J. Pharm. Investig. 2013, 43, 353–362. [Google Scholar] [CrossRef]
- Weber, S.; Zimmer, A.; Pardeike, J. Solid Lipid Nanoparticles (SLN) and Nanostructured Lipid Carriers (NLC) for pulmonary application: A review of the state of the art. Eur. J. Pharm. Biopharm. 2014, 86, 7–22. [Google Scholar] [CrossRef] [PubMed]
- Garcês, A.; Amaral, M.H.; Sousa Lobo, J.M.; Silva, A.C. Formulations based on solid lipid nanoparticles (SLN) and nanostructured lipid carriers (NLC) for cutaneous use: A review. Eur. J. Pharm. Sci. 2018, 112, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Gupta, Y.K.; Dinda, A.K. Nano-pharmaceuticals in medicine. Proc. Indian Natl. Sci. Acad. 2018, 84, 255–266. [Google Scholar] [CrossRef]
- Sykes, E.A.; Dai, Q. Tailoring nanoparticle designs to target cancer based on tumor pathophysiology. Proc. Natl. Acad. Sci. USA 2016, 113, E1142–E1151. [Google Scholar] [CrossRef]
- Jain, R.K. Barriers to drug delivery in solid tumors. Sci. Am. 1994, 271, 58–65. [Google Scholar] [CrossRef]
- De Menezes, D.E.L.; Pilarski, L.M.; Allen, T.M. In vitro and in vivo targeting of immunoliposomal doxorubicin to human B-cell lymphoma. Cancer Res. 1998, 58, 3320–3330. [Google Scholar]
- Benefits of Nanotechnology for Cancer. Available online: https://www.cancer.gov/nano/cancer-nanotechnology/benefits (accessed on 13 April 2020).
- Torchilin, V.P. Passive and active drug targeting: Drug delivery to tumors as an example. In Drug Delivery; Springer: Berlin/Heidelberg, Germany, 2010; pp. 3–53. [Google Scholar]
- Natesan, S.; Ponnusamy, C.; Sugumaran, A.; Chelladurai, S.; Palaniappan, S.S.; Palanichamy, R. Artemisinin loaded chitosan magnetic nanoparticles for the efficient targeting to the breast cancer. Int. J. Biol. Macromol. 2017, 104, 1853–1859. [Google Scholar] [CrossRef]
- Chen, Z.; Kang, X.; Wu, Y.; Xiao, H.; Cai, X.; Sheng, S.; Wang, X.; Chen, S. A mitochondria targeting artesunate prodrug-loaded nanoparticle exerting anticancer activity via iron-mediated generation of the reactive oxygen species. Chem. Commun. 2019, 55, 4781–4784. [Google Scholar] [CrossRef]
- Liu, R.; Yu, X.; Su, C.; Shi, Y.; Zhao, L. Nanoparticle delivery of artesunate enhances the anti-tumor efficiency by activating mitochondria-mediated cell apoptosis. Nanoscale Res. Lett. 2017, 12, 403. [Google Scholar] [CrossRef] [PubMed]
- Modica-Napolitano, J.S.; Weissig, V. Treatment strategies that enhance the efficacy and selectivity of mitochondria-targeted anticancer agents. Int. J. Mol. Sci. 2015, 16, 17394–17421. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Dai, L.; Li, C.; Liu, J.; Wang, L.; Lei, J. Self-assembled targeted nanoparticles based on transferrin-modified eight-arm-polyethylene glycol–dihydroartemisinin conjugate. Sci. Rep. 2016, 6, 29461. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.T.; Tran, T.H.; Kim, J.O.; Yong, C.S.; Nguyen, C.N. Enhancing the in vitro anti-cancer efficacy of artesunate by loading into poly-d,l-lactide-co-glycolide (PLGA) nanoparticles. Arch. Pharm. Res. 2015, 38, 716–724. [Google Scholar]
- Li, Z.; Zhu, J.; Wang, Y.; Zhou, M.; Li, D.; Zheng, S.; Luo, C.; Zhang, H.; Zhong, L.; Li, W.; et al. In situ apolipoprotein E-enriched corona guides dihydroartemisinin-decorating, nanoparticles towards LDLr-mediated tumor-homing chemotherapy. Asian J. Pharm. Sci. 2019. [Google Scholar] [CrossRef]
- Ma, W.; Xu, A.; Ying, J.; Li, B.; Jin, Y. Biodegradable core-shell copolymer-phospholipid nanoparticles for combination chemotherapy: An in vitro study. J. Biomed. Nanotechnol. 2015, 11, 1193–1200. [Google Scholar]
- Wang, Z.; Duan, X.; Lv, Y.; Zhao, Y. Low density lipoprotein receptor (LDLR)-targeted lipid nanoparticles for the delivery of sorafenib and Dihydroartemisinin in liver cancers. Life Sci. 2019, 239, 117013. [Google Scholar]
- Zhang, Y.J.; Zhan, X.; Wang, L.; Ho, R.J.; Sasaki, T. pH-responsive artemisinin dimer in lipid nanoparticles are effective against human breast cancer in a xenograft model. J. Pharm. Sci. 2015, 104, 1815–1824. [Google Scholar]
- Zhang, Y.J.; Gallis, B.; Taya, M.; Wang, S.; Ho, R.J.; Sasaki, T. pH-responsive artemisinin derivatives and lipid nanoparticle formulations inhibit growth of breast cancer cells in vitro and induce down-regulation of HER family members. PLoS ONE 2013, 8, e59086. [Google Scholar] [CrossRef]
- Li, X.Y.; Zhao, Y.; Sun, M.G.; Shi, J.F.; Ju, R.J.; Zhang, C.X.; Li, X.T.; Zhao, W.Y.; Mu, L.M.; Zeng, F.; et al. Multifunctional liposomes loaded with paclitaxel and artemether for treatment of invasive brain glioma. Biomaterials 2014, 35, 5591–5604. [Google Scholar] [PubMed]
- Righeschi, C.; Coronnello, M.M.; Leto, I.; Bergonzi, M.C.; Bilia, A.R. Transferrin-targeted stealth liposomes loaded with artemisinin: The Trojan horse to enhance its selectivity and anticancer activity. Planta Med. 2014, 80, SL27. [Google Scholar] [CrossRef]
- Leto, I.; Coronnello, M.; Righeschi, C.; Bergonzi, M.C.; Mini, E.; Bilia, A.R. Enhanced efficacy of artemisinin loaded in transferrin-conjugated liposomes versus stealth liposomes against HCT-8 colon cancer cells. ChemMedChem 2016, 11, 1745–1751. [Google Scholar] [CrossRef] [PubMed]
- Kang, X.J.; Wang, H.Y.; Peng, H.G.; Chen, B.F.; Zhang, W.Y.; Wu, A.H.; Xu, Q.; Huang, Y.Z. Codelivery of dihydroartemisinin and doxorubicin in mannosylated liposomes for drug-resistant colon cancer therapy. Acta Pharmacol. Sin. 2017, 38, 885–896. [Google Scholar] [PubMed]
- Righeschi, C.; Coronnello, M.; Mastrantoni, A.; Isacchi, B.; Bergonzi, M.C.; Mini, E.; Bilia, A.R. Strategy to provide a useful solution to effective delivery of dihydroartemisinin: Development, characterization and in vitro studies of liposomal formulations. Colloids Surf. B Biointerfaces 2014, 116, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.J.; Huang, X.R.; Zhou, X.B.; Zheng, B.Y.; Huang, J.D. Potential sonodynamic anticancer activities of artemether and liposome-encapsulated artemether. Chem. Commun. 2015, 51, 4681–4684. [Google Scholar] [CrossRef]
- Tian, L.; Liu, J.; Jia, Q.; Ying, Y.; Yang, Z.; Huang, G. Preparation and evaluation of artemether liposomes for enhanced anti-tumor therapy. AAPS PharmSciTech 2018, 19, 512–521. [Google Scholar] [CrossRef]
- Wang, S.; Wang, H.; Liang, W.; Huang, Y. An injectable hybrid nanoparticle-in-oil-in-water submicron emulsion for improved delivery of poorly soluble drugs. Nanoscale Res. Lett. 2012, 7, 219. [Google Scholar] [CrossRef]
- Wang, D.; Zhou, J.; Chen, R.; Shi, R.; Xia, G.; Zhou, S.; Liu, Z.; Zhang, N.; Wang, H.; Guo, Z.; et al. Magnetically guided delivery of DHA and Fe ions for enhanced cancer therapy based on pH-responsive degradation of DHA-loaded Fe3O4@C@MIL-100(Fe) nanoparticles. Biomaterials 2016, 107, 88–101. [Google Scholar] [CrossRef]
- Hou, L.; Shan, X.; Hao, L.; Feng, Q.; Zhang, Z. Copper sulfide nanoparticle-based localized drug delivery system as an effective cancer synergistic treatment and theranostic platform. Acta Biomater. 2017, 54, 307–320. [Google Scholar] [CrossRef]
- Feng, Q.; Zhang, Y.; Zhang, W.; Shan, X.; Yuan, Y.; Zhang, H.; Hou, L.; Zhang, Z. Tumor-targeted and multi-stimuli responsive drug delivery system for near-infrared light induced chemo-phototherapy and photoacoustic tomography. Acta Biomater. 2016, 38, 129–142. [Google Scholar] [CrossRef]
- Wang, S.; Riedinger, A.; Li, H.; Fu, C.; Liu, H.; Li, L.; Liu, T.; Tan, L.; Barthel, M.J.; Pugliese, G.; et al. Plasmonic copper sulfide nanocrystals exhibiting near-infrared photothermal and photodynamic therapeutic effects. ACS Nano 2015, 9, 1788–1800. [Google Scholar] [PubMed]
- Chen, J.; Guo, Z.; Wang, H.B.; Zhou, J.J.; Zhang, W.J.; Chen, Q.W. Multifunctional mesoporous nanoparticles as pH-responsive Fe2+ reservoirs and artemisinin vehicles for synergistic inhibition of tumor growth. Biomaterials 2014, 35, 6498–6507. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Han, Y.; Yang, Y.; Yang, J.; Guo, X.; Zhang, J.; Pan, L.; Xia, G. Effect of interaction of magnetic nanoparticles of Fe3O4 and artesunate on apoptosis of K562 cells. Int. J. Nanomed. 2011, 6, 1185–1192. [Google Scholar]
- Guo, S.; Yao, X.; Jiang, Q.; Wang, K.; Zhang, Y.; Peng, H.; Tang, J.; Yang, W. Dihydroartemisinin-loaded magnetic nanoparticles for enhanced Chemodynamic therapy. Front. Pharmacol. 2020, 11, 226. [Google Scholar]
- Liu, L.; Wei, Y.; Zhai, S.; Chen, Q.; Xing, D. Dihydroartemisinin and transferrin dual-dressed nano-graphene oxide for a pH-triggered chemotherapy. Biomaterials 2015, 62, 35–46. [Google Scholar] [CrossRef]
- Zhang, H.; Ji, Y.; Chen, Q.; Jiao, X.; Hou, L.; Zhu, X.; Zhang, Z. Enhancement of cytotoxicity of artemisinin toward cancer cells by transferrin-mediated carbon nanotubes nanoparticles. J. Drug Target. 2015, 23, 552–567. [Google Scholar] [CrossRef]
- Zhang, H.; Hou, L.; Jiao, X.; Ji, Y.; Zhu, X.; Zhang, Z. Transferrin-mediated fullerenes nanoparticles as Fe2+-dependent drug vehicles for synergistic anti-tumor efficacy. Biomaterials 2015, 37, 353–366. [Google Scholar] [CrossRef]
- Devalapally, H.; Chakilam, A.; Amiji, M.M. Role of nanotechnology in pharmaceutical product development. J. Pharm. Sci. 2007, 96, 2547–2565. [Google Scholar] [CrossRef]
- Mosqueira, V.C.F.; Loiseau, P.M.; Bories, C.; Legrand, P.; Devissaguet, J.P.; Barratt, G. Efficacy and pharmacokinetics of intravenous nanocapsule formulations of halofantrine in Plasmodium berghei-infected mice. Antimicrob. Agents Chemother. 2004, 48, 1222–1228. [Google Scholar] [CrossRef]
- Barratt, G. Colloidal drug carriers: Achievements and perspectives. Cell. Mol. Life Sci. 2003, 60, 21–37. [Google Scholar] [CrossRef]
- Santos-Magalhães, N.S.; Mosqueira, V.C.F. Nanotechnology applied to the treatment of malaria. Adv. Drug Deliv. Rev. 2010, 62, 560–575. [Google Scholar]
- Scherphof, G.L.; Velinova, M.; Kamps, J.; Donga, J.; Van der Want, H.; Kuipers, F.; Havekes, L.; Daemen, T. Modulation of pharmacokinetic behavior of liposomes. Adv. Drug Deliv. Rev. 1997, 24, 179–191. [Google Scholar]
- Torchilin, V.P. Multifunctional nanocarriers. Adv. Drug Deliv. Rev. 2006, 58, 1532–1555. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.; Sukla, S.; Panday, A. Kinetic modeling and release behavior of PLGA-loaded nanoparticle of anti-malarial drug using dialysis membrane. Nanomed. Nanotechnol. J. 2019, 2, 123. [Google Scholar]
- Oyeyemi, O.; Morenkeji, O.; Afolayan, F.; Dauda, K.; Busari, Z.; Meene, J.; Panda, A. Curcumin-Artesunate based polymeric nanoparticle; Antiplasmodial and toxicological evaluation in murine model. Front. Pharmacol. 2018, 9, 562. [Google Scholar] [PubMed]
- Yaméogo, J.B.G.; Annabelle, G.; Choisnard, L.; Putaux, J.-L.; Gansane, A.; Sirima, S.B.; Semde, R.; Wouessidjewe, D. Self-assembled biotransesterified cyclodextrins as Artemisinin nanocarriers—I: Formulation, lyoavailability and in vitro antimalarial activity assessment. Eur. J. Pharm. Biopharm. 2012, 80, 508–517. [Google Scholar] [CrossRef]
- Yaméogo, J.B.G.; Mazet, R.; Wouessidjewe, D.; Choisnard, L.; Godin-Ribuot, D.; Putaux, J.-L.; Semde, R.; Geze, A. Pharmacokinetic study of intravenously administered artemisinin-loaded surface-decorated amphiphilic γ-cyclodextrin nanoparticles. Mater. Sci. Eng. C 2020, 106, 110281. [Google Scholar] [CrossRef]
- Chadha, R.; Gupta, S.; Pathak, N. Artesunate-loaded chitosan/lecithin nanoparticles: Preparation, characterization, and in vivo studies. Drug Dev. Ind. Pharm. 2012, 38, 1538–1546. [Google Scholar] [CrossRef]
- Masiiwa, W.L.; Gadaga, L.L. Intestinal Permeability of Artesunate-Loaded solid lipid nanoparticles using the everted gut method. J. Drug Deliv. 2018, 2018, 3021738. [Google Scholar] [CrossRef]
- Omwoyo, W.N.; Melariri, P.; Gathirwa, J.W.; Oloo, F.; Mahanga, G.M.; Kalombo, L.; Ogutu, B.; Swai, H.S. Development, characterization and antimalarial efficacy of dihydroartemisinin loaded solid lipid nanoparticles. Nanomed. Nanotechnol. Biol. Med. 2016, 12, 801–809. [Google Scholar] [CrossRef]
- Aditya, N.P.; Patankar, S.; Madhusudhan, B.; Murthy, R.S.R.; Souto, E.B. Arthemeter-loaded lipid nanoparticles produced by modified thin-film hydration: Pharmacokinetics, toxicological and in vivo anti-malarial activity. Eur. J. Pharm. Sci. 2010, 40, 448–455. [Google Scholar] [CrossRef] [PubMed]
- Boateng-Marfo, Y.; Dong, Y.; Loh, Z.H.; Lin, H.; Ng, W.K. Intravenous human serum albumin (HSA)-bound artemether nanoparticles for treatment of severe malaria. Colloids Surf. A Physicochem. Eng. Asp. 2018, 536, 20–29. [Google Scholar] [CrossRef]
- Attama, A.A.; Kenechukwu, F.C.; Onuigbo, E.B.; Nnamani, P.O.; Obitte, N.; Finke, J.H.; Pretor, S.; Müller-Goymann, C.C. Solid lipid nanoparticles encapsulating a fluorescent marker (coumarin 6) and antimalarials–artemether and lumefantrine: Evaluation of cellular uptake and antimalarial activity. Eur. J. Nanomed. 2016, 8, 129–138. [Google Scholar]
- Isacchi, B.; Arrigucci, S.; Marca, G.L.; Bergonzi, M.C.; Vannucchi, M.G.; Novelli, A.; Bilia, A.R. Conventional and long-circulating liposomes of artemisinin: Preparation, characterization, and pharmacokinetic profile in mice. J. Liposome Res. 2011, 21, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Ismail, M.; Ling, L.; Du, Y.; Yao, C.; Li, X. Liposomes of dimeric artesunate phospholipid: A combination of dimerization and self-assembly to combat malaria. Biomaterials 2018, 163, 76–87. [Google Scholar] [CrossRef] [PubMed]
- Waknine-Grinberg, J.H.; Even-Chen, S.; Avichzer, J.; Turjeman, K.; Bentura-Marciano, A.; Haynes, R.K.; Weiss, L.; Allon, N.; Ovadia, H.; Golenser, J.; et al. Glucocorticosteroids in nano-sterically stabilized liposomes are efficacious for elimination of the acute symptoms of experimental cerebral malaria. PLoS ONE 2013, 8, e72722. [Google Scholar]
- Aditya, N.P.; Chimote, G.; Gunalan, K.; Banerjee, R.; Patankar, S.; Madhusudhan, B. Curcuminoids-loaded liposomes in combination with arteether protects against Plasmodium berghei infection in mice. Exp. Parasitol. 2012, 131, 292–299. [Google Scholar] [CrossRef]
- Kakran, M.; Sahoo, N.G.; Li, L.; Judeh, Z.; Wang, Y.; Chong, K.; Loh, L. Fabrication of drug nanoparticles by evaporative precipitation of nanosuspension. Int. J. Pharm. 2010, 383, 285–292. [Google Scholar] [CrossRef]
- Ma, Y.; Lu, T.; Zhao, W.; Wang, Y.; Chen, T.; Mei, Q.; Chen, T. Enhanced antimalarial activity by a novel artemether-lumefantrine lipid emulsion for parenteral administration. Antimicrob. Agents Chemother. 2014, 58, 5658–5665. [Google Scholar] [CrossRef][Green Version]
- Ibrahim, N.; Ibrahim, H.; Sabater, A.M.; Mazier, D.; Valentin, A.; Nepveu, F. Artemisinin nanoformulation suitable for intravenous injection: Preparation, characterization and antimalarial activities. Int. J. Pharm. 2015, 495, 671–679. [Google Scholar] [CrossRef]
- Yang, Y.; Gao, H.; Zhou, S.; Kuang, X.; Wang, Z.; Liu, H.; Sun, J. Optimization and evaluation of lipid emulsions for intravenous co-delivery of artemether and lumefantrine in severe malaria treatment. Drug Deliv. Transl. Res. 2018, 8, 1171–1179. [Google Scholar] [PubMed]
- Memvanga, P.B.; Coco, R.; Préat, V. An oral malaria therapy: Curcumin-loaded lipid-based drug delivery systems combined with β-arteether. J. Control. Release 2013, 172, 904–913. [Google Scholar] [CrossRef] [PubMed]
- Dwivedi, P.; Khatik, R.; Chaturvedi, P.; Khandelwal, K.; Taneja, I.; Raju, K.S.; Dwivedi, H.; Kumar Singh, S.; Gupta, P.K.; Shukla, P.; et al. Arteether nanoemulsion for enhanced efficacy against Plasmodium yoelii nigeriensis malaria: An approach by enhanced bioavailability. Colloids Surf. B Biointerfaces 2015, 126, 467–475. [Google Scholar] [CrossRef] [PubMed]
- Memvanga, P.B.; Préat, V. Formulation design and in vivo antimalarial evaluation of lipid-based drug delivery systems for oral delivery of β-arteether. Eur. J. Pharm. Biopharm. 2012, 82, 112–119. [Google Scholar] [CrossRef]
- Parashar, D.; NP, A.; Murthy, R.S.R. Development of artemether and lumefantrine co-loaded nanostructured lipid carriers: Physicochemical characterization and in vivo antimalarial activity. Drug Deliv. 2016, 23, 123–129. [Google Scholar]
- Kannan, D.; Yadav, N.; Ahmad, S.; Namdev, P.; Bhattacharjee, S.; Lochab, B.; Singh, S. Pre-clinical study of iron oxide nanoparticles fortified artesunate for efficient targeting of malarial parasite. EBioMedicine 2019, 45, 261–277. [Google Scholar] [CrossRef]
- Kumara, R.; Sahoo, G.C.; Pandeya, K.; Dasa, V.; Yousuf, M.; Ansaria, S.R.; Dasa, P. PLGA-PEG Encapsulated sitamaquine nanoparticles drug delivery system against Leishmania donovani. J. Sci. Innov. Res. 2014, 3, 85–90. [Google Scholar]
- Akbari, M.; Oryan, A.; Hatam, G. Application of nanotechnology in treatment of leishmaniasis: A review. Acta Tropica. 2017, 172, 86–90. [Google Scholar]
- Want, M.Y.; Islamuddin, M.; Chouhan, G.; Ozbak, H.A.; Hemeg, H.A.; Dasgupta, A.K.; Chattopadhyay, A.P.; Afrim, F. Therapeutic efficacy of artemisinin-loaded nanoparticles in experimental visceral leishmaniasis. Colloids Surf. B Biointerfaces 2015, 130, 215–221. [Google Scholar]
- Want, M.Y.; Islamuddin, M.; Chouhan, G.; Dasgupta, A.K.; Chattopadhyay, A.P.; Afrin, F. A new approach for the delivery of artemisinin: Formulation, characterization, and ex-vivo antileishmanial studies. J. Colloid Interface Sci. 2014, 432, 258–269. [Google Scholar] [CrossRef]
- Want, M.Y.; Islammudin, M.; Chouhan, G.; Ozbak, H.A.; Hemeg, H.A.; Chattopadhyay, A.P.; Afrin, F. Nanoliposomal artemisinin for the treatment of murine visceral leishmaniasis. Int. J. Nanomed. 2017, 12, 2189. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Artemisinin and Derivatives | Cancer | Mode of Action | Ref. |
|---|---|---|---|
| Artemisone and Atremisinin | Breast (MCF-7) and colon (HCT116 and SW480) | Growth arrest, induction of apoptosis and blockage of the cell cycle. Decrease in the levels of its regulatory proteins CDK4 and cyclin D1 | [48] |
| Arteether | 4T1 cell line | Reduction of the cell growth of 4T1 cell line in a dose-dependent manner | [49] |
| Artemether | Gastric cancer cell lines (PG100) | Induced genotoxic and cytotoxic effects in the gastric cancer cell line. | [50] |
| Dihydroartemisinin | Pancreas cancer cell lines (PANC-1 and BxPC-3) | Up-regulation of intracellular perforin, granzyme B expression and IFN-γ production | [51] |
| The production of reactive oxygen species, the modulation of apoptosis-related proteins and the induction of death receptor 5 | [52] | ||
| Artesunate, dihydroartemisinin | Osteosarcoma (MG63 and 148B) | Inhibition of the growth of human osteosarcoma cells in vitro | [53] |
| Artemisinin | Inhibition of angiogenesis by regulating the p38 MAPK/CREB/TSP-1 signaling pathway in vivo | [54] | |
| Artemisinin, artesunate, and dihydroartemisinin | Leukemia (MV4-11, MOLM-13, or ML-2) | Induced reactive oxygen species (ROS)-mediated apoptosis. | [55] |
| Artemisinin | Neuroblastoma (SK-N-AS, SK-N-DZ and SHEP1) | Inhibition of cell growth and proliferation, cell cycle arrest in the G1 phase in neuroblastoma cell lines in vitro | [56] |
| Dihydroartemisinin | SH-SY5Y | Induced apoptosis by decreasing the expression of cyclin D1 protein and increasing the expression of caspase-3 protein | [57] |
| Dihydroartemisinin | Lung (A549) | Induced apoptosis by increasing the ratio of Bax/Bcl-2 and active caspase-3 and cytochrome-c | [58] |
| Inhibition of angiogenesis | [59] | ||
| Dihydroartemisinin | Human ovarian cancer (SKOV3, SKOV3-IP, HO8910, and HO8910-PM) and human ovarian surface epithelial cells)) | Inhibited proliferation, migration, and invasion of ovarian cancer cells, and induced apoptosis in vitro | [60] |
| Artesunate | Induction of reactive oxygen species (ROS) cell cycle arrest in the G2/M phase | [61] | |
| Dihydroartemisinin | Cervix carcinoma (HeLa) | Promoted autophagic cell death in vitro | [62] |
| Artemisinin | Artemisinin inhibited the G2/M phase | [63] | |
| Dihydroartemisinin | Prostate (Human PCa cell lines C4, C4-2, and C4-2B) | Inhibits Axl expression in PCa via regulation of microRNAs and proteins of the polycomb repressive complex 2 | [64] |
| Artesunate | DU145 and LNCaP | Inhibited the viability and mobility of the cell lines triggered by UCA1 down-regulation | [43] |
| Artesunate | Melanoma [Primary (92.1, Mel270) and metastatic (Omm1 and Omm2.3)] | Suppression of the phosphorylation of GSK3β at S9, and lowered protein level of β-catenin and its downstream targets (c-Myc, cyclin D1). Inhibition of cell viability and colony formation ability. Induced apoptosis with reduced migration and invasion of uveal melanoma cells. Induced upregulation of oxidative and genotoxic stress response genes | [65] |
| Dihydroartemisinin | A375, G361, LOX | Induced apoptosis with upregulation of cellular oxidative stress, phosphatidylserine externalization, and activational cleavage of procaspase 3. | [66] |
| Dihydroartemisinin | Hepatoma (HepG2 cell) | Induced apoptosis in HepG2 cell lines and increased the intracellular production of ROS | [67] |
| Artesunate | SMMC-7721 | Induction of apoptosis and cell cycle arrest | [68] |
| Type of Nanoparticle | Carrier | Artemisinin Derivative | Application | Therapeutic Outcome | Ref. |
|---|---|---|---|---|---|
| Polymeric nanoparticles | Chitosan | Artemisinin | Anticancer | High drug loading capacity. Enhanced accumulation of the nanoparticles in the 4T1 breast tumor tissues of BALB/c mice model in vivo. | [122] |
| Polymeric nanoparticles | N,N′-bis(dodecyl)-l-glutamic diamide | Artesunate | Anticancer | The formulation mediated ROS generation and targeted the mitochondria, a target for inducing cancer cell death. | [123] |
| Polymeric nanoparticles | Bovine serum albumin | Artesunate | Anticancer | High cytotoxic effect and significant apoptotic effect. | [124] |
| Polymeric nanoparticles | Polyethylene glycol | Dihydroartemisinin | A significant growth inhibition effect with prolonged circulation time. | [126] | |
| Polymeric nanoparticles | PLGA | Artesunate | Anticancer | Higher cytotoxicity against cancer cell lines in vitro. | [127] |
| Polymeric nanoparticles | PLGA | Dihydroartemisinin | anticancer | Sustained drug release kinetics and enhanced anticancer activity in vitro and in vivo. | [128] |
| Polymeric nanoparticles | PLGA | dihydroartemisinin | anticancer | High cell accumulation with enhanced cytotoxicity. | [129] |
| Lipid nanoparticles | cholesteryl oleatea and triolein | Dihydroartemisinin | Anticancer | A synergistic anticancer activity and high cell accumulation. | [130] |
| Lipid nanoparticles | egg phosphatidylcholine | Artemisinin | Anticancer | Effective against human breast cancer and non-toxic on non-tumorigenic cells. | [131] |
| Lipid nanoparticles | l-α-Phosphatidylcholine | Artemisinin | Anticancer | A down regulated of the anti-apoptotic protein, survivin, and cyclin D1 was observed in the breast cancer cell lines at low concentration of the formulation. A down regulated oncogenic protein HER2 and HER3 was observed in a HER2+ cell line with a reduction in the wild type epidermal growth factor receptor (EGFR or HER1) in a triple negative breast cancer cell line. | [132] |
| Lipid nanoparticles | Mannose-vitamin E derivative conjugate and a dequalinium-lipid derivative conjugate. | Artemether | Anticancer | Prolonged circulation time with a significant inhibitory effect and apoptosis-inducing effect against the brain cancer cells. | [133] |
| Lipid nanoparticles | Cholesterol | Artemisinin | Anticancer | Significant anticancer activity. | [134] |
| Lipid nanoparticles | Cholesterol | Artemisinin | Anticancer | The cell uptake and cytotoxicity studies of the formulation in HCT-8 cell line confirmed an enhanced uptake of the formulation due to the presence of iron ions. | [135] |
| Lipid nanoparticles | Soybean phosphatidylcholine, cholesterol, | Dihydroartemisinin | Anticancer | Downregulation of Bcl-xl, increased cancer cell apoptosis, and the induction of autophagy. | [136] |
| Lipid nanoparticles | Cholesterol, PEG | Dihydroartemisinin | Anticancer | High cellular uptake and the absence of toxicity | [137] |
| Lipid nanoparticles | Cholesterol | Artemether | Anticancer | High intracellular uptake and high generation of ROS in HepG2 cells. | [138] |
| Lipid nanoparticles | Cholesterol | Artemether | Anticancer | The in vitro drug release of artemether from the formulation was sustained. The growth inhibition rate was 1.54 times higher than the free drug solution. | [139] |
| Lipid nanoparticles | Lecithin, soy beans oil, poloxamer | Dihydroartemisinin | Anticancer | High tumor growth inhibition of 51.8% and extended half-life of the drugs. | [140] |
| Metal-based nanoparticle | Iron oxide | Dihydroartemisinin | Anticancer | Non-toxic and high anticancer efficacy in vitro. | [141] |
| Metal-based nanoparticle | CuS nanoparticles | Artesunate | Anticancer | A significant inhibition rate of 74.8% with a good tumor targeting ability and retention effect. | [142] |
| Metal-based nanoparticles | Iron oxide/silver | Artemisinin | Anticancer | Synergistic anticancer activity and good cellular uptake. | [145] |
| Metal-based nanoparticles | Iron oxide | Artesunate | Anticancer | Reduced cell viability. | [146] |
| Metal-based nanoparticles | Iron oxide | Dihydroartemisinin | Anticancer | Increased the amount of reactive oxygen species and significant killing effect on breast cancer cells, MCF-7 cells. | [147] |
| Polymeric nanoparticles | Graphene oxide | Dihydroartemisinin | Anticancer | A synergistic cytotoxicity activity with complete tumor cure. | [148] |
| Carbon-based nanoparticles | Carbon nanotubes | Artemisinin | Anticancer | A synergistic antitumor effect when compared to the free drug in vitro in MCF-7 cells and in vivo in tumor-bearing murine model. Increased intracellular drug uptake was significant with high inhibition effect. | [149] |
| Carbon-based nanoparticles | Fullerene | Artesunate | Anticancer | High drug loading efficacy of 162.4% and antitumor efficacy. The tumor inhibition rate. | [150] |
| Polymeric nanoparticles | PLGA | Artemisinin | Antimalarial | High drug encapsulation efficiency and controlled drug release mechanism. | [157] |
| Polymeric nanoparticles | PLGA | Artesunate | Antimalarial | Improved antimalarial activity in vivo with sustained and controlled drug release. | [158] |
| Polymeric nanoparticle | Cyclodextrin | Artemisinin | Antimalarial | Controlled drug release and parasite growth inhibition. | [159] |
| Polymeric nanoparticles | γ-cyclodextrin | Artemisinin | Antimalarial | Significant improved pharmacokinetic parameters. | [160] |
| Polymeric nanoparticles | Chitosan/lecithin | Artesunate and artemisinin | Antimalarial | Less mean percent parasitemia in vivo. | [161] |
| Lipid nanoparticles | Glyceryl monostearate | Artesunate | Antimalarial | Sustained drug release and enhanced drug intestinal permeability. | [162] |
| Lipid nanoparticles | steric acid | Dihydroartemisinin | Antimalarial | Good parasite chemosuppression in vivo. | [163] |
| Lipid nanoparticles | soybean oil (liquid lipid) and glyceryl trimyristate (as solid lipid) | Artemether | Antimalarial | Reduced hemolytic toxicity and good antiplasmodial efficacy in vivo. | [164] |
| Lipid nanoparticles | Human serum albumin | Artemether | Antimalarial | The nanoparticles displayed significantly enhanced solubility when compared to the free drug. | [165] |
| Lipid nanoparticles | Phospholipon®, theobroma oil and beeswax | Artemether | Antimalarial | High clearance of parasitemia with minimal side effects. | [166] |
| Lipid nanoparticles | PEG | Artemisinin | Antimalarial | Extended blood-circulation time and improved half-life of artemisinin by more than 5-fold. | [167] |
| Lipid nanoparticles | Glycerophosphorylcholine | Artesunate | Antimalarial | Longer retention half-life in the bloodstream. Enhanced parasites killing in P. berghei-infected mice in vivo with delayed recrudescence and improved survival when compared to free drug. | [168] |
| Lipid nanoparticles | Glucocorticoid prodrug | Artemisone | Antimalarial | Administration of artemisone after treatment with the liposome formulation resulted in a complete cure. The combination resulted in a reduced level of cerebral inflammation, hemorrhage and edema. | [169] |
| Lipid nanoparticles | Phosphatidylcholine | α/β Arteether | Antimalarial | High cure rate with the absence of recrudescence. | [170] |
| Lipid nanoparticles | - | Artemisinin | Antimalarial | The dissolution of drug-loaded nanoparticles was enhanced when compared to the free artesunate | [171] |
| Lipid nanoparticles | soybean oil, sodium oleate, glycerol, and egg lecithin, poloxamer | Artemether | Antimalarial | Decrease in the parasitemia levels after 3 days, and with parasitemia inhibition rate of 90%. | [172] |
| Lipid nanoparticles | Human serum albumin | Artesunate | Antimalarial | A 96% parasitemia inhibition at 10 mg/kg/day. Prolonged mean survival time with no recrudescence. | [173] |
| Lipid nanoparticles | soybean oil, oleic acid, egg lecithin | Artemether | Antimalarial | High Cmax of artemether and lumfantrine were 452.86 and 2844.15 ng/mL, respectively, in the lipid emulsion group when compared to the drug solution group which were 37.92 and 918.94 ng/mL. | [174] |
| Lipid nanoparticles | l-α-Phosphatidylcholine, Labrasol | β-arteether | Antimalarial | Increased survival rate and a significant delayed recrudescence. | [175] |
| Lipid nanoparticles | Tween 80, PEG 400 | Arteether | Antimalarial | Improved drug bioavailability. | [176] |
| Lipid nanoparticles | groundnut oil, Tween 80 | β-arteether | Antimalarial | A 100% cure for more than 45 days. | [177] |
| Lipid nanoparticles | Soybean oil | Artemether | Antimalarial | Excellent antimalarial activity with regards to parasitemia progression and survivability period. | [178] |
| Metal-based nanoparticles | Iron oxide | Artesunate | Antimalarial | Good intracellular drug uptake with the improved antimalarial activity. | [179] |
| Polymeric nanoparticles | PLGA | Artemisinin | Antileishmanial | Sustained in vitro drug release and ex vivo antileishmanial activity | [182] |
| Polymeric nanoparticles | PLGA | Artemisinin | Antileishmanial | Significant parasite burden reduction | [183] |
| Lipid nanoparticles | Artemisinin | Antileishmanial | Significantly reduced intracellular infection of Leishmania donovani amastigotes. | [184] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alven, S.; Aderibigbe, B.A. Nanoparticles Formulations of Artemisinin and Derivatives as Potential Therapeutics for the Treatment of Cancer, Leishmaniasis and Malaria. Pharmaceutics 2020, 12, 748. https://doi.org/10.3390/pharmaceutics12080748
Alven S, Aderibigbe BA. Nanoparticles Formulations of Artemisinin and Derivatives as Potential Therapeutics for the Treatment of Cancer, Leishmaniasis and Malaria. Pharmaceutics. 2020; 12(8):748. https://doi.org/10.3390/pharmaceutics12080748
Chicago/Turabian StyleAlven, Sibusiso, and Blessing Atim Aderibigbe. 2020. "Nanoparticles Formulations of Artemisinin and Derivatives as Potential Therapeutics for the Treatment of Cancer, Leishmaniasis and Malaria" Pharmaceutics 12, no. 8: 748. https://doi.org/10.3390/pharmaceutics12080748
APA StyleAlven, S., & Aderibigbe, B. A. (2020). Nanoparticles Formulations of Artemisinin and Derivatives as Potential Therapeutics for the Treatment of Cancer, Leishmaniasis and Malaria. Pharmaceutics, 12(8), 748. https://doi.org/10.3390/pharmaceutics12080748