Effective Targeting of Colon Cancer Cells with Piperine Natural Anticancer Prodrug Using Functionalized Clusters of Hydroxyapatite Nanoparticles

 ,
,  ,
,  , ,
, , 
Abstract
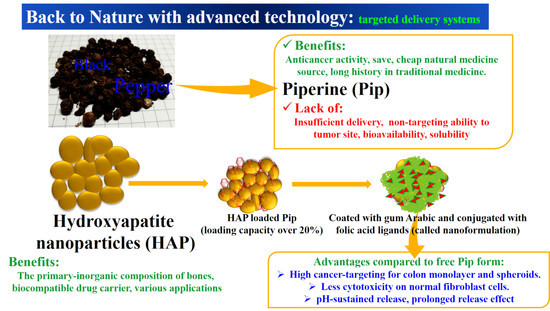
1. Introduction
2. Materials and Methods
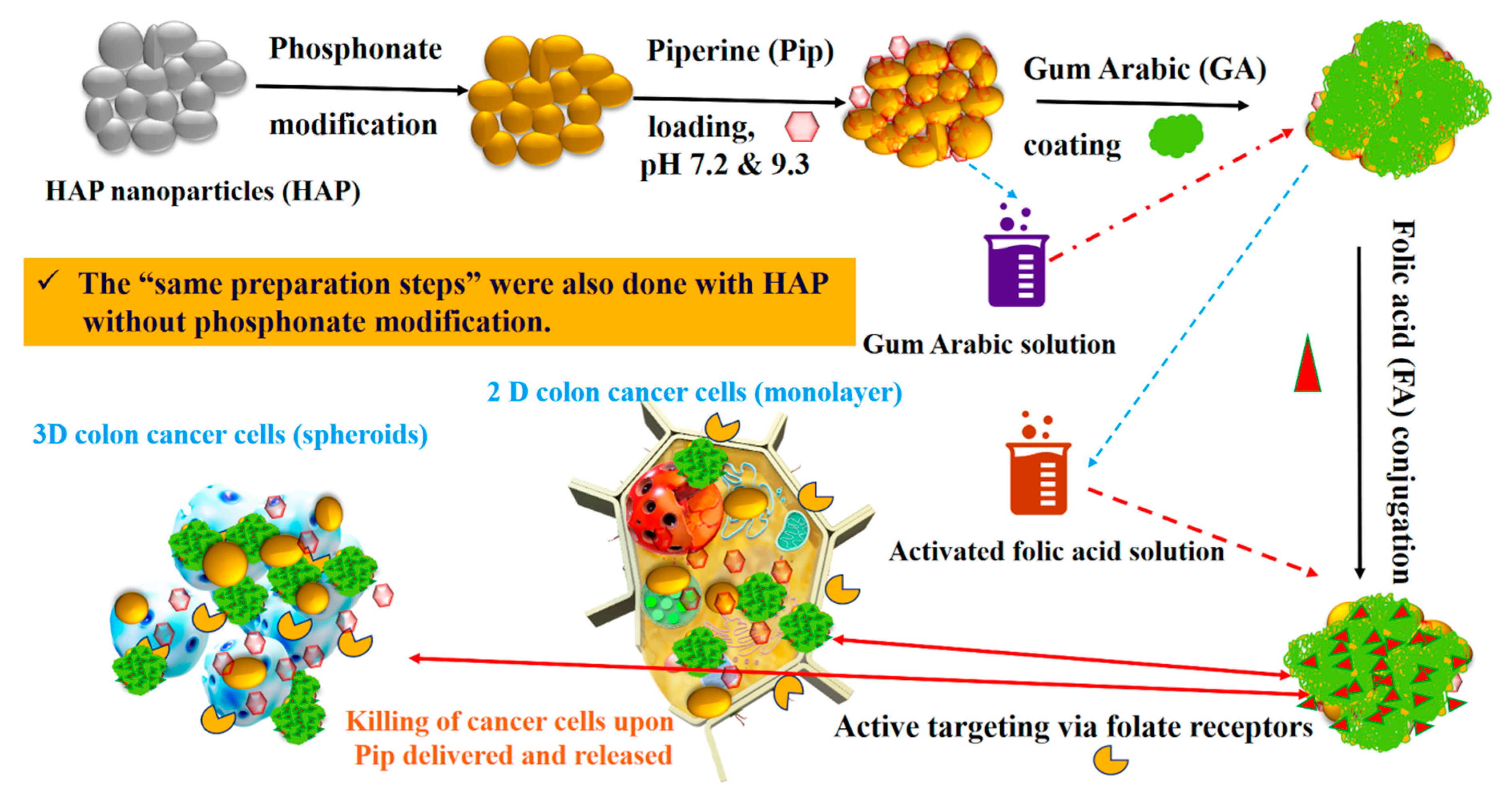
2.1. Synthesis and Surface Modification of Hydroxyapatite Nanoparticles
2.2. Piperine Loading
2.3. Gum Arabic Coating and Folic acid Conjugation
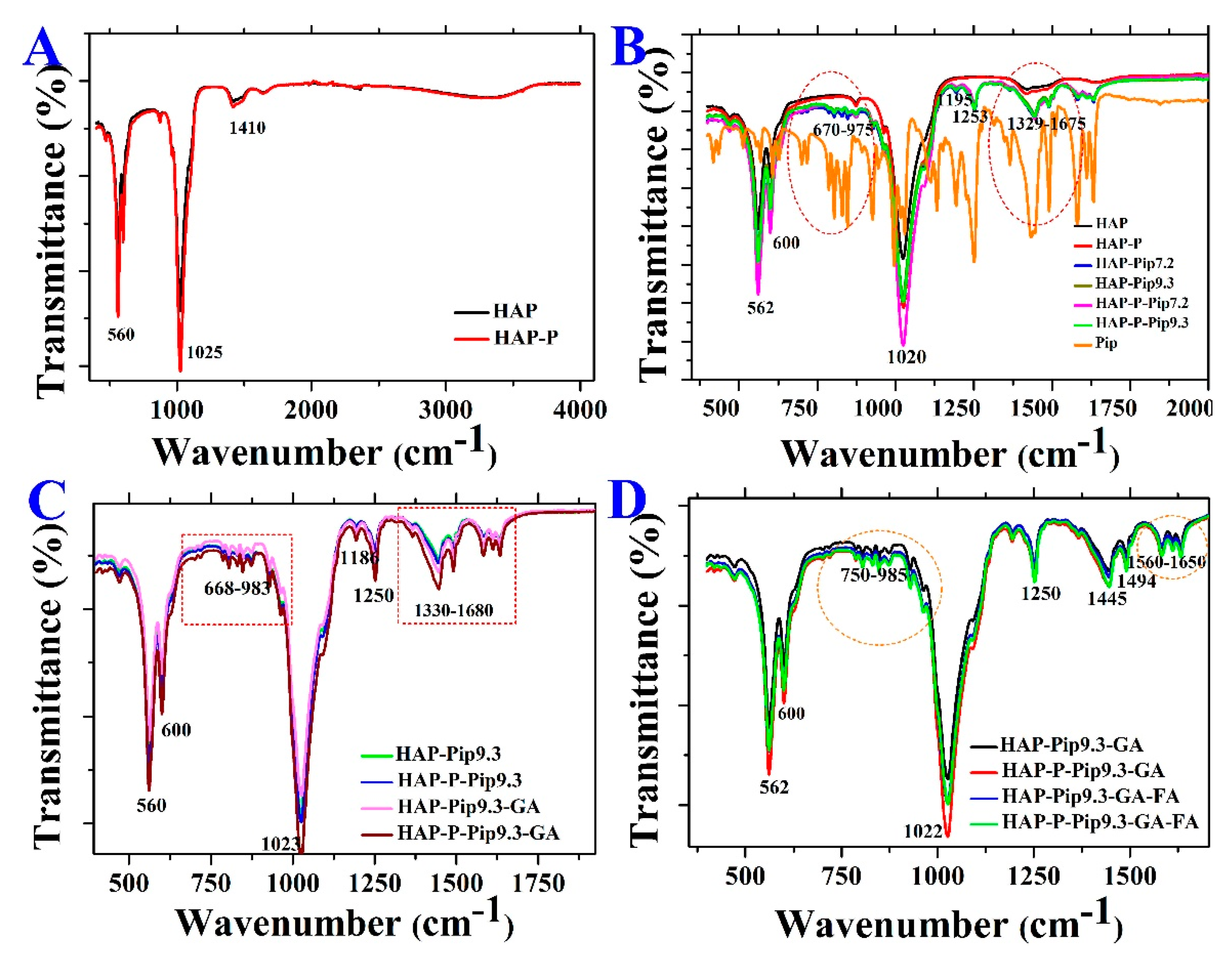
2.4. Characterization Techniques
2.5. In Vitro Drug Release
2.6. Cell Culture
2.7. In Vitro Cytotoxicity
2.8. Anticancer Effects
2.9. Statistical Analysis
3. Results
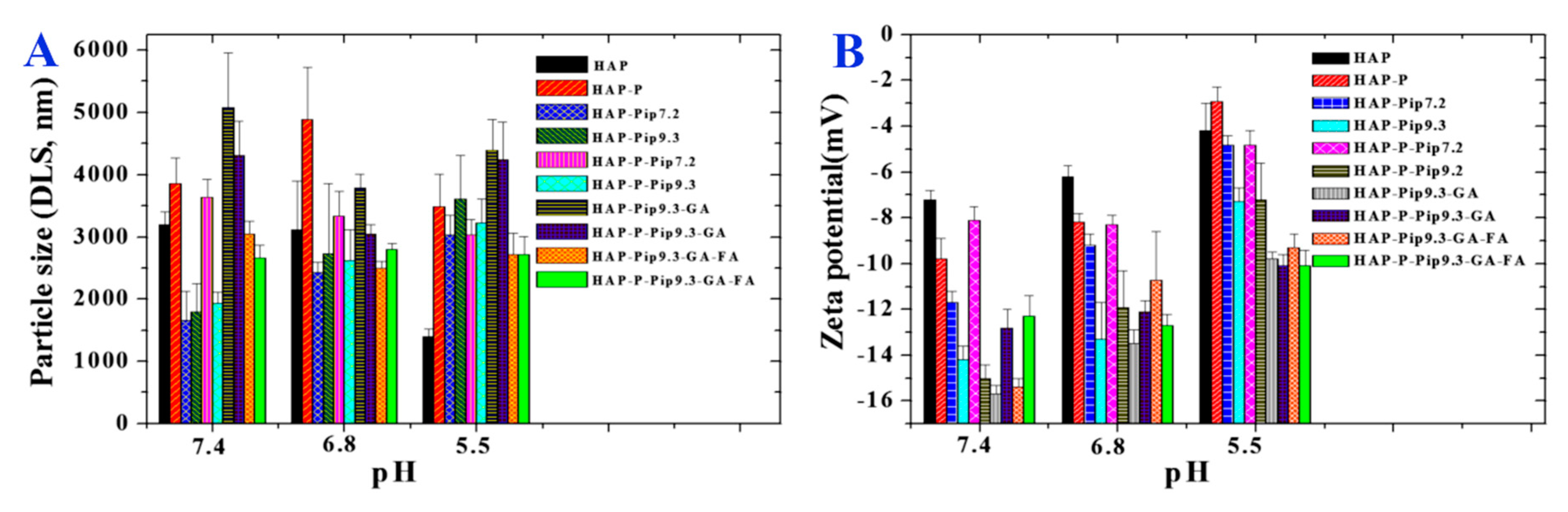
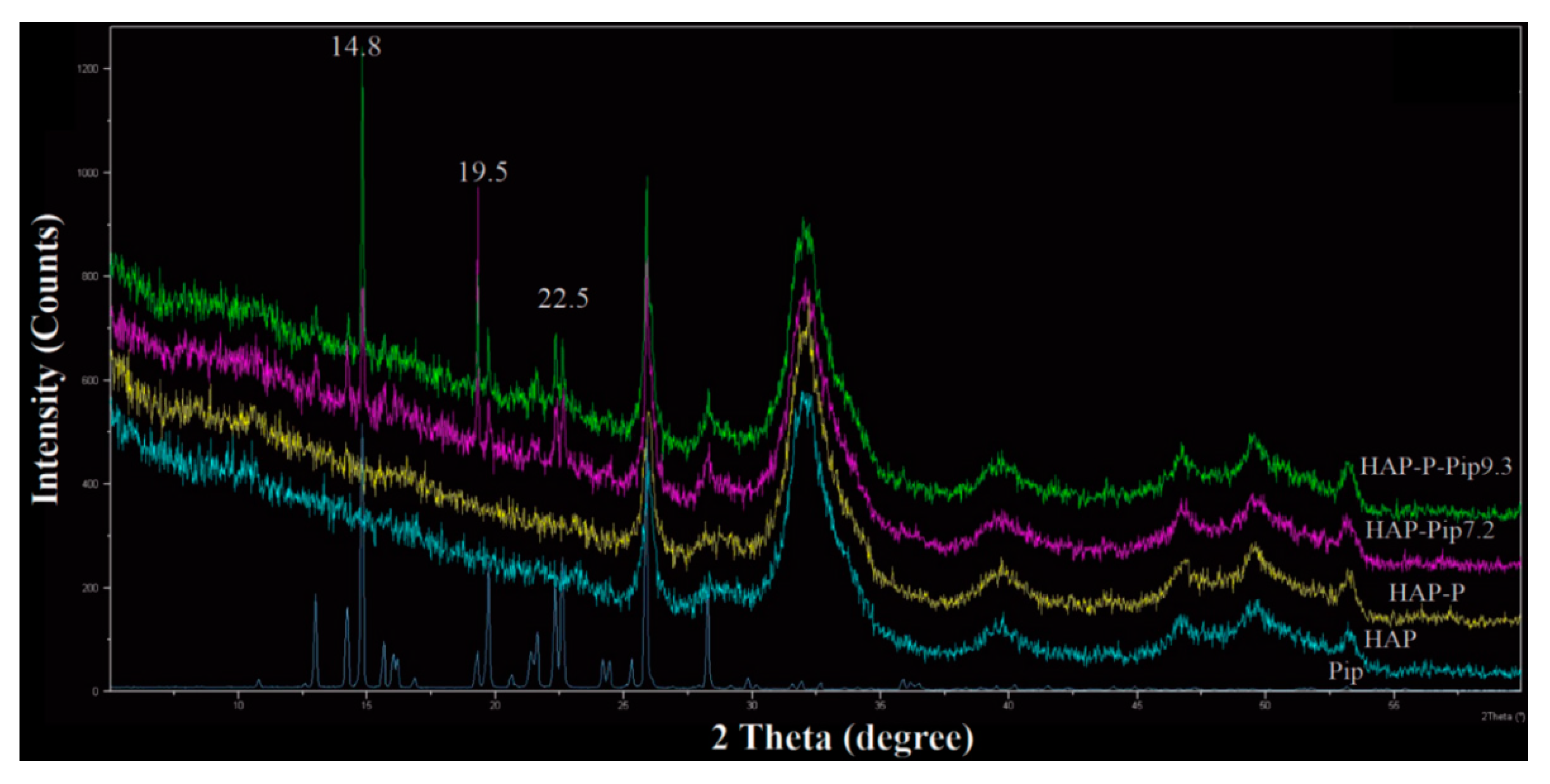
3.1. Morphology, Size, and Zeta Potential Measurements
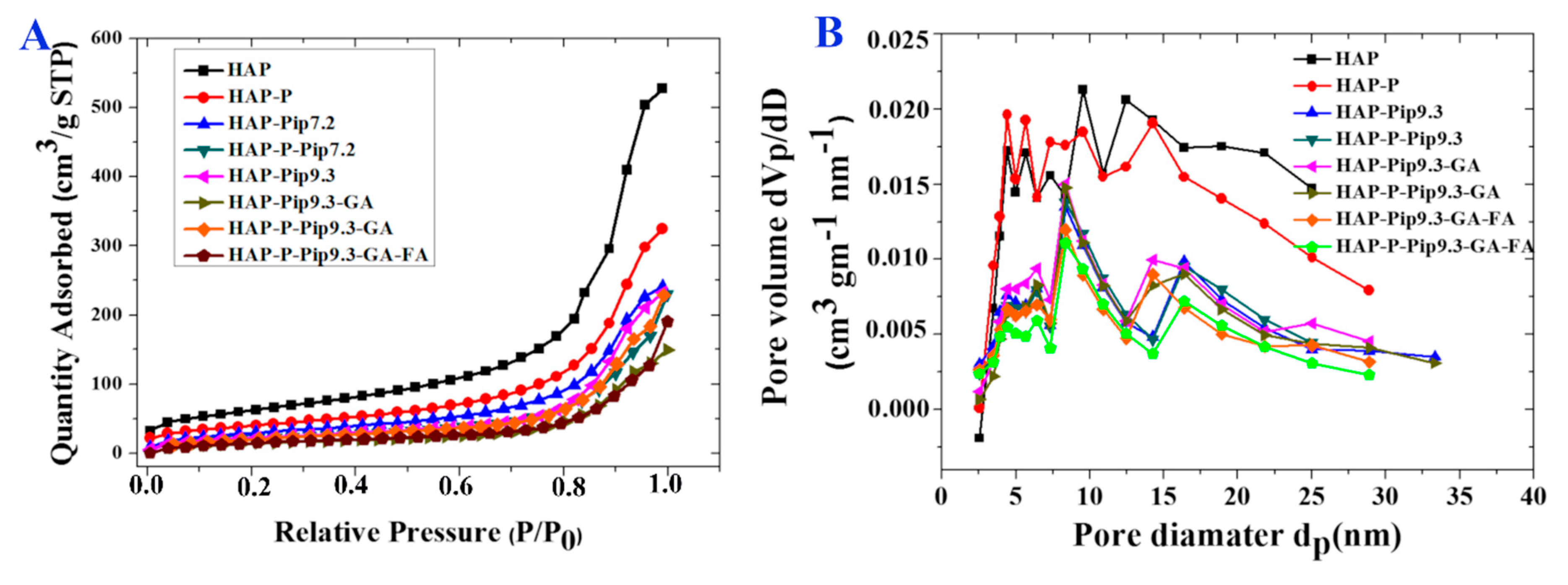
3.2. Specific Surface Area and Pore Size Distribution
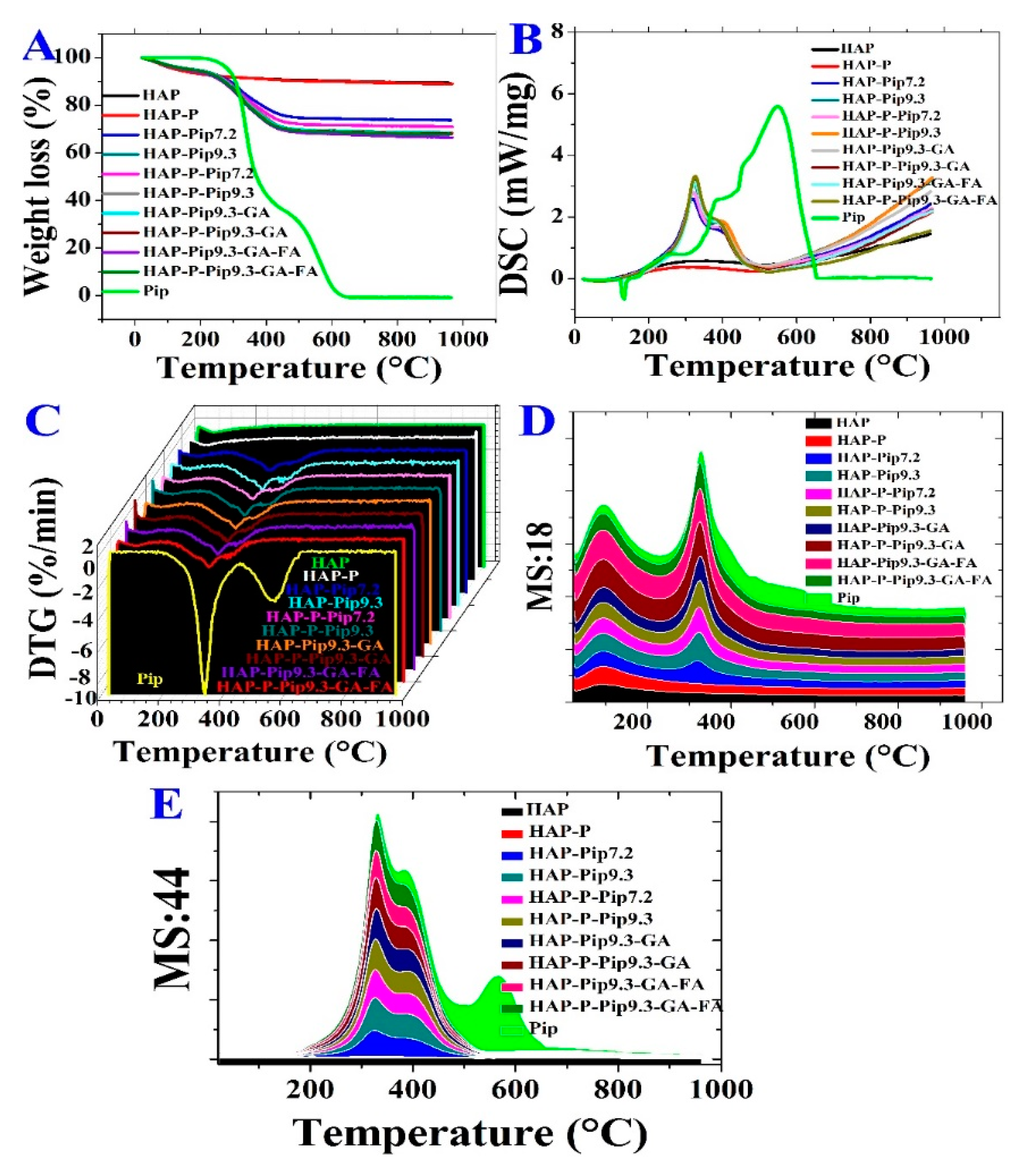
3.3. STA-DSC-MS Analysis
3.4. EDS Analysis
3.5. Pip Loading
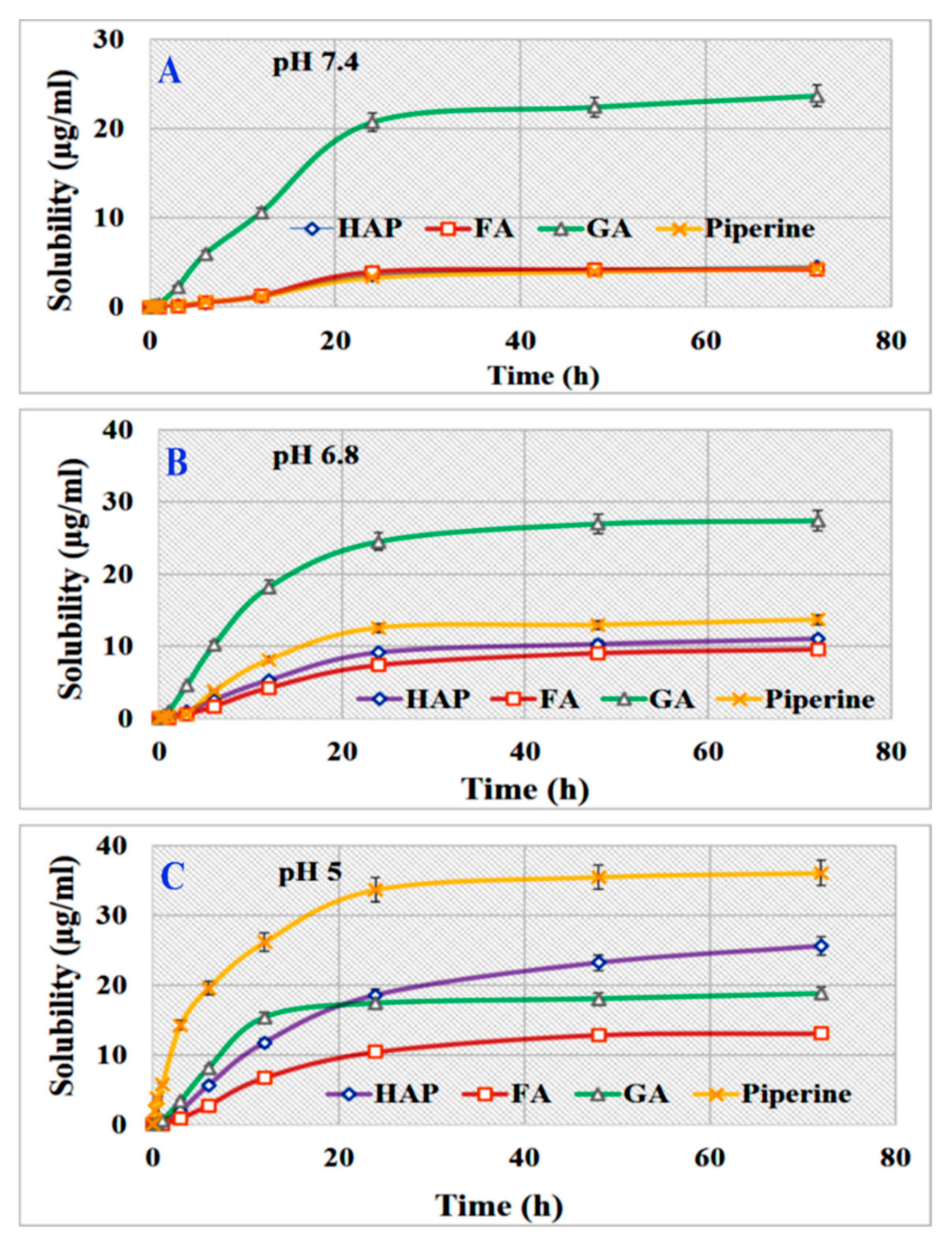
3.6. Solubility Measurements
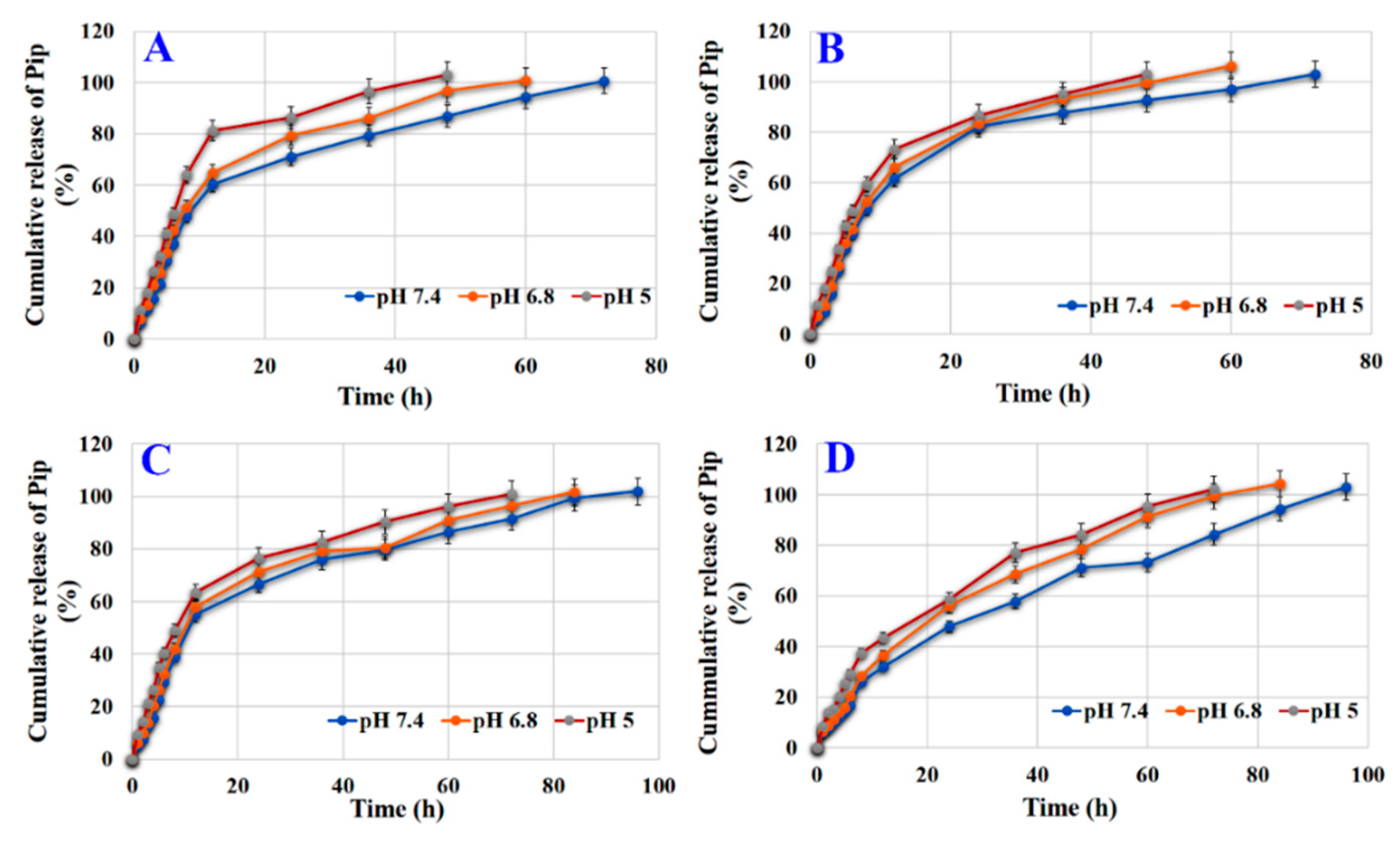
3.7. Effect of Loading Condition and Surface Modification on Release
3.8. Effect of Coating on Release
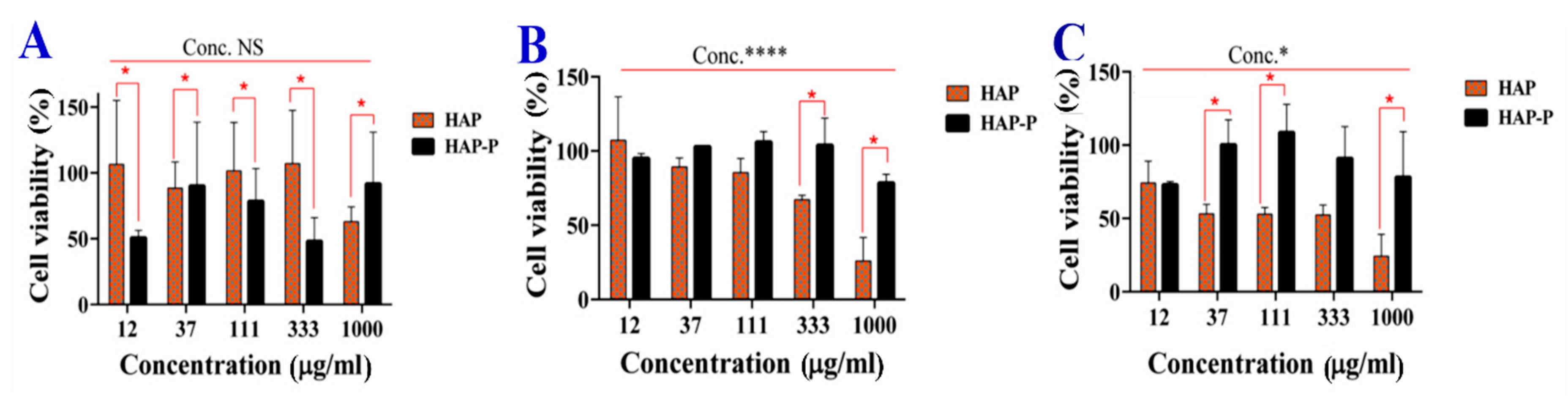
3.9. Cytotoxicity of HAPs
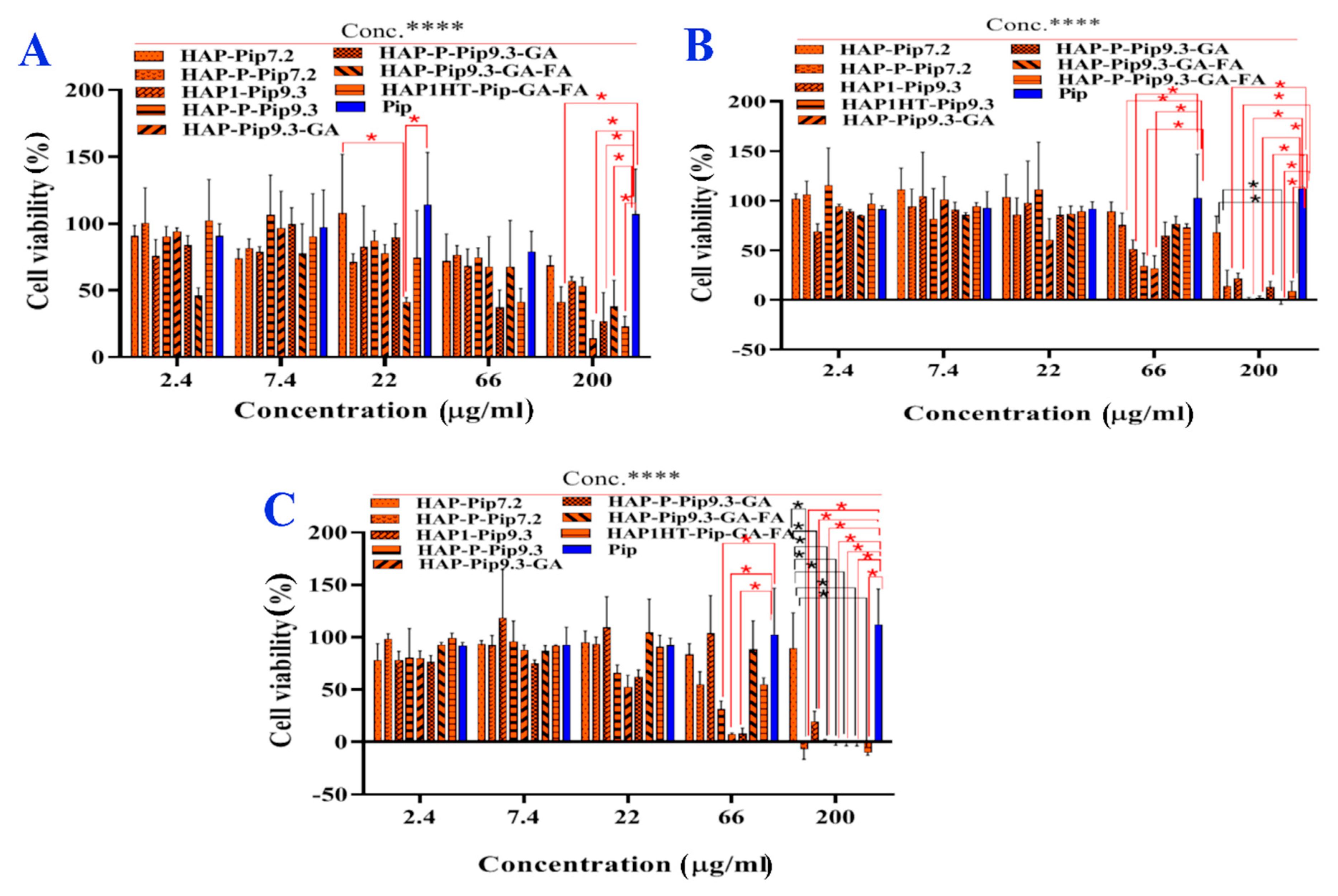
3.10. In Vitro Anticancer Effects
3.11. Microscopic Observations
3.12. Cellular Uptakes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Dyba, T.; Randi, G.; Bettio, M.; Gavin, A.; Visser, O.; Bray, F. Cancer incidence and mortality patterns in Europe: Estimates for 40 countries and 25 major cancers in 2018. Eur. J. Cancer 2018, 103, 356–387. [Google Scholar] [CrossRef] [PubMed]
- Wirsdorfer, F.; de Leve, S.; Jendrossek, V. Combining radiotherapy and immunotherapy in lung cancer: Can we expect limitations due to altered normal tissue toxicity? Int. J. Mol. Sci. 2018, 20, 24. [Google Scholar] [CrossRef] [PubMed]
- Herskovic, A.; Martz, K.; al-Sarraf, M.; Leichman, L.; Brindle, J.; Vaitkevicius, V.; Cooper, J.; Byhardt, R.; Davis, L.; Emami, B. Combined chemotherapy and radiotherapy compared with radiotherapy alone in patients with cancer of the esophagus. N. Engl. J. Med. 1992, 326, 1593–1598. [Google Scholar] [CrossRef] [PubMed]
- Cragg, G.M.; Newman, D.J. Plants as a source of anti-cancer agents. J. Ethnopharmacol. 2005, 100, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Ji, H.-F.; Li, X.-J.; Zhang, H.-Y. Natural products and drug discovery. Can thousands of years of ancient medical knowledge lead us to new and powerful drug combinations in the fight against cancer and dementia? EMBO Rep. 2009, 10, 194–200. [Google Scholar] [CrossRef] [PubMed]
- Atanasov, A.G.; Waltenberger, B.; Pferschy-Wenzig, E.M.; Linder, T.; Wawrosch, C.; Uhrin, P.; Temml, V.; Wang, L.; Schwaiger, S.; Heiss, E.H.; et al. Discovery and resupply of pharmacologically active plant-derived natural products: A review. Biotechnol. Adv. 2015, 33, 1582–1614. [Google Scholar] [CrossRef]
- Watkins, R.; Wu, L.; Zhang, C.; Davis, R.M.; Xu, B. Natural product-based nanomedicine: Recent advances and issues. Int. J. Nanomed. 2015, 10, 6055–6074. [Google Scholar] [CrossRef]
- Aljuffali, I.A.; Fang, C.L.; Chen, C.H.; Fang, J.Y. Nanomedicine as a strategy for natural compound delivery to prevent and treat cancers. Curr. Pharm. Des. 2016, 22, 4219–4231. [Google Scholar] [CrossRef]
- Patra, J.K.; Das, G.; Fraceto, L.F.; Campos, E.V.R.; Rodriguez-Torres, M.D.P.; Acosta-Torres, L.S.; Diaz-Torres, L.A.; Grillo, R.; Swamy, M.K.; Sharma, S.; et al. Nano based drug delivery systems: Recent developments and future prospects. J. Nanobiotechnol. 2018, 16, 71. [Google Scholar] [CrossRef]
- Damanhouri, Z.; Ahmad, A. A review on therapeutic potential of Piper nigrum L. (black pepper): The king of spices. Med. Aromat. Plants 2014, 3, 161. [Google Scholar] [CrossRef]
- Gorgani, L.; Mohammadi, M.; Najafpour, G.D.; Nikzad, M. Piperine—The bioactive compound of black pepper: From isolation to medicinal formulations. Compr. Rev. Food Sci. Food Saf. 2017, 16, 124–140. [Google Scholar] [CrossRef]
- Koleva, I.I.; van Beek, T.A.; Soffers, A.E.; Dusemund, B.; Rietjens, I.M. Alkaloids in the human food chain—Natural occurrence and possible adverse effects. Mol. Nutr. Food Res. 2012, 56, 30–52. [Google Scholar] [CrossRef] [PubMed]
- Kasibhatta, R.; Naidu, M.U. Influence of piperine on the pharmacokinetics of nevirapine under fasting conditions: A randomised, crossover, placebo-controlled study. Drugs R D 2007, 8, 383–391. [Google Scholar] [CrossRef] [PubMed]
- Shoba, G.; Joy, D.; Joseph, T.; Majeed, M.; Rajendran, R.; Srinivas, P.S. Influence of piperine on the pharmacokinetics of curcumin in animals and human volunteers. Planta Med. 1998, 64, 353–356. [Google Scholar] [CrossRef]
- Bang, J.S.; Oh, D.H.; Choi, H.M.; Sur, B.-J.; Lim, S.-J.; Kim, J.Y.; Yang, H.-I.; Yoo, M.C.; Hahm, D.-H.; Kim, K.S. Anti-inflammatory and antiarthritic effects of piperine in human interleukin 1beta-stimulated fibroblast-like synoviocytes and in rat arthritis models. Arthritis Res. Ther. 2009, 11, R49. [Google Scholar] [CrossRef]
- Yang, W.; Chen, Y.H.; Liu, H.; Qu, H.D. Neuroprotective effects of piperine on the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced Parkinson’s disease mouse model. Int. J. Mol. Med. 2015, 36, 1369–1376. [Google Scholar] [CrossRef]
- Selvendiran, K.; Singh, J.P.; Krishnan, K.B.; Sakthisekaran, D. Cytoprotective effect of piperine against benzo[a]pyrene induced lung cancer with reference to lipid peroxidation and antioxidant system in Swiss albino mice. Fitoterapia 2003, 74, 109–115. [Google Scholar] [CrossRef]
- Yoo, E.S.; Choo, G.S.; Kim, S.H.; Woo, J.S.; Kim, H.J.; Park, Y.S.; Kim, B.S.; Kim, S.K.; Park, B.K.; Cho, S.D.; et al. Antitumor and apoptosis-inducing effects of piperine on human melanoma cells. Anticancer Res. 2019, 39, 1883–1892. [Google Scholar] [CrossRef]
- Si, L.; Yang, R.; Lin, R.; Yang, S. Piperine functions as a tumor suppressor for human ovarian tumor growth via activation of JNK/p38 MAPK-mediated intrinsic apoptotic pathway. Biosci. Rep. 2018, 38. [Google Scholar] [CrossRef]
- Yaffe, P.B.; Power Coombs, M.R.; Doucette, C.D.; Walsh, M.; Hoskin, D.W. Piperine, an alkaloid from black pepper, inhibits growth of human colon cancer cells via G1 arrest and apoptosis triggered by endoplasmic reticulum stress. Mol. Carcinog. 2015, 54, 1070–1085. [Google Scholar] [CrossRef]
- Selvendiran, K.; Banu, S.M.; Sakthisekaran, D. Protective effect of piperine on benzo(α)pyrene-induced lung carcinogenesis in Swiss albino mice. Clinica Chimica Acta 2004, 350, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Samykutty, A.; Shetty, A.V.; Dakshinamoorthy, G.; Bartik, M.M.; Johnson, G.L.; Webb, B.; Zheng, G.; Chen, A.; Kalyanasundaram, R.; Munirathinam, G. Piperine, a bioactive component of pepper spice exerts therapeutic effects on androgen dependent and androgen independent prostate cancer cells. PLoS ONE 2013, 8, e65889. [Google Scholar] [CrossRef] [PubMed]
- Do, M.T.; Kim, H.G.; Choi, J.H.; Khanal, T.; Park, B.H.; Tran, T.P.; Jeong, T.C.; Jeong, H.G. Antitumor efficacy of piperine in the treatment of human HER2-overexpressing breast cancer cells. Food Chem. 2013, 141, 2591–2599. [Google Scholar] [CrossRef] [PubMed]
- Gunasekaran, V.; Elangovan, K.; Niranjali Devaraj, S. Targeting hepatocellular carcinoma with piperine by radical-mediated mitochondrial pathway of apoptosis: An in vitro and in vivo study. Food Chem. Toxicol. 2017, 105, 106–118. [Google Scholar] [CrossRef]
- Anissian, D.; Ghasemi-Kasman, M.; Khalili-Fomeshi, M.; Akbari, A.; Hashemian, M.; Kazemi, S.; Moghadamnia, A.A. Piperine-loaded chitosan-STPP nanoparticles reduce neuronal loss and astrocytes activation in chemical kindling model of epilepsy. Int. J. Biol. Macromol. 2018, 107, 973–983. [Google Scholar] [CrossRef]
- Bhalekar, M.R.; Madgulkar, A.R.; Desale, P.S.; Marium, G. Formulation of piperine solid lipid nanoparticles (SLN) for treatment of rheumatoid arthritis. Drug Dev. Ind. Pharm. 2017, 43, 1003–1010. [Google Scholar] [CrossRef]
- Katiyar, S.S.; Muntimadugu, E.; Rafeeqi, T.A.; Domb, A.J.; Khan, W. Co-delivery of rapamycin- and piperine-loaded polymeric nanoparticles for breast cancer treatment. Drug Deliv. 2016, 23, 2608–2616. [Google Scholar] [CrossRef]
- Elnaggar, Y.S.R.; Etman, S.M.; Abdelmonsif, D.A.; Abdallah, O.Y. Intranasal piperine-loaded chitosan nanoparticles as brain-targeted therapy in Alzheimer’s disease: Optimization, biological efficacy, and potential toxicity. J. Pharm. Sci. 2015, 104, 3544–3556. [Google Scholar] [CrossRef]
- Shao, B.; Cui, C.; Ji, H.; Tang, J.; Wang, Z.; Liu, H.; Qin, M.; Li, X.; Wu, L. Enhanced oral bioavailability of piperine by self-emulsifying drug delivery systems: In vitro, in vivo and in situ intestinal permeability studies. Drug Deliv. 2015, 22, 740–747. [Google Scholar] [CrossRef]
- AbouAitah, K.; Swiderska-Sroda, A.; Farghali, A.A.; Wojnarowicz, J.; Stefanek, A.; Gierlotka, S.; Opalinska, A.; Allayeh, A.K.; Ciach, T.; Lojkowski, W. Folic acid-conjugated mesoporous silica particles as nanocarriers of natural prodrugs for cancer targeting and antioxidant action. Oncotarget 2018, 9, 26466–26490. [Google Scholar] [CrossRef]
- Shahein, S.A.; Aboul-Enein, A.M.; Higazy, I.M.; Abou-Elella, F.; Lojkowski, W.; Ahmed, E.R.; Mousa, S.A.; AbouAitah, K. Targeted anticancer potential against glioma cells of thymoquinone delivered by mesoporous silica core-shell nanoformulations with pH-dependent release. Int. J. Nanomed. 2019, 14, 5503–5526. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Carmona, M.; Gun’ko, Y.; Vallet-Regí, M. ZnO nanostructures for drug delivery and theranostic applications. Nanomaterials 2018, 8, 268. [Google Scholar] [CrossRef] [PubMed]
- Albinali, K.E.; Zagho, M.M.; Deng, Y.; Elzatahry, A.A. A perspective on magnetic core-shell carriers for responsive and targeted drug delivery systems. Int. J. Nanomed. 2019, 14, 1707–1723. [Google Scholar] [CrossRef] [PubMed]
- Palazzo, B.; Iafisco, M.; Laforgia, M.; Margiotta, N.; Natile, G.; Bianchi, C.L.; Walsh, D.; Mann, S.; Roveri, N. Biomimetic hydroxyapatite–drug nanocrystals as potential bone substitutes with antitumor drug delivery properties. Adv. Funct. Mater. 2007, 17, 2180–2188. [Google Scholar] [CrossRef]
- Zhu, S.H.; Huang, B.Y.; Zhou, K.C.; Huang, S.P.; Liu, F.; Li, Y.M.; Xue, Z.G.; Long, Z.G. Hydroxyapatite nanoparticles as a novel gene carrier. J. Nanoparticle Res. 2004, 6, 307–311. [Google Scholar] [CrossRef]
- Li, D.; Huang, X.; Wu, Y.; Li, J.; Cheng, W.; He, J.; Tian, H.; Huang, Y. Preparation of pH-responsive mesoporous hydroxyapatite nanoparticles for intracellular controlled release of an anticancer drug. Biomater. Sci. 2016, 4, 272–280. [Google Scholar] [CrossRef] [PubMed]
- Ignjatović, N.L.; Penov-Gaši, K.M.; Wu, V.M.; Ajduković, J.J.; Kojić, V.V.; Vasiljević-Radović, D.; Kuzmanović, M.; Uskoković, V.; Uskoković, D.P. Selective anticancer activity of hydroxyapatite/chitosan-poly(d,l)-lactide-co-glycolide particles loaded with an androstane-based cancer inhibitor. Colloids Surf. B Biointerfaces 2016, 148, 629–639. [Google Scholar] [CrossRef]
- Xiong, H.; Du, S.; Zhang, P.; Jiang, Z.; Zhou, J.; Yao, J. Primary tumor and pre-metastatic niches co-targeting “peptides-lego” hybrid hydroxyapatite nanoparticles for metastatic breast cancer treatment. Biomater. Sci. 2018, 6, 2591–2604. [Google Scholar] [CrossRef]
- Ignjatovic, N.L.; Sakac, M.; Kuzminac, I.; Kojic, V.; Markovic, S.; Vasiljevic-Radovic, D.; Wu, V.M.; Uskokovic, V.; Uskokovic, D.P. Chitosan oligosaccharide lactate coated hydroxyapatite nanoparticles as a vehicle for the delivery of steroid drugs and the targeting of breast cancer cells. J. Mater. Chem. B 2018, 6, 6957–6968. [Google Scholar] [CrossRef]
- Higuchi, J.; Fortunato, G.; Wozniak, B.; Chodara, A.; Domaschke, S.; Meczynska-Wielgosz, S.; Kruszewski, M.; Dommann, A.; Lojkowski, W. Polymer membranes sonocoated and electrosprayed with nano-hydroxyapatite for periodontal tissues regeneration. Nanomaterials 2019, 9, 1625. [Google Scholar] [CrossRef]
- Rogowska-Tylman, J.; Locs, J.; Salma, I.; Woźniak, B.; Pilmane, M.; Zalite, V.; Wojnarowicz, J.; Kędzierska-Sar, A.; Chudoba, T.; Szlązak, K.; et al. In vivo and in vitro study of a novel nanohydroxyapatite sonocoated scaffolds for enhanced bone regeneration. Mater. Sci. Eng. C 2019, 99, 669–684. [Google Scholar] [CrossRef] [PubMed]
- Kuśnieruk, S.; Wojnarowicz, J.; Chodara, A.; Chudoba, T.; Gierlotka, S.; Lojkowski, W. Influence of hydrothermal synthesis parameters on the properties of hydroxyapatite nanoparticles. Beilstein J. Nanotechnol. 2016, 7, 1586–1601. [Google Scholar] [CrossRef] [PubMed]
- Ammar, H.O.; Ghorab, M.M.; Mahmoud, A.A.; Higazy, I.M. Lamotrigine loaded poly-ε-(d,l-lactide-co-caprolactone) nanoparticles as brain delivery system. Eur. J. Pharm. Sci. Off. J. Eur. Fed. Pharm. Sci. 2018, 115, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Assaad, H.I.; Zhou, L.; Carroll, R.J.; Wu, G. Rapid publication-ready MS-Word tables for one-way ANOVA. SpringerPlus 2014, 3, 474. [Google Scholar] [CrossRef]
- Padmanabhan, V.P.; Kulandaivelu, R.; Santhana Panneer, D.; Vivekananthan, S.; Sagadevan, S.; Anita Lett, J. Microwave synthesis of hydroxyapatite encumbered with ascorbic acid intended for drug leaching studies. Mater. Res. Innov. 2019, 1–8. [Google Scholar] [CrossRef]
- Shingate, P.; Dongre, P.; Kannur, D. New method development for extraction and isolation of piperine from black pepper. Int. J. Pharm. Sci. Res. 2013, 4, 3165–3170. [Google Scholar]
- Dahiya, S.; Rani, R.; Dhingra, D.; Kumar, S.; Dilbaghi, N. Conjugation of epigallocatechin gallate and piperine into a zein nanocarrier: Implication on antioxidant and anticancer potential. Adv. Nat. Sci. Nanosci. Nanotechnol. 2018, 9, 035011. [Google Scholar] [CrossRef]
- Ibekwe, C.A.; Oyatogun, G.M.; Esan, T.A.; Oluwasegun, K.M. Synthesis and characterization of chitosan/gum Arabic nanoparticles for bone regeneration. Am. J. Mater. Sci. Eng. 2017, 5, 28–36. [Google Scholar] [CrossRef]
- Pal, K.; Roy, S.; Parida, P.K.; Dutta, A.; Bardhan, S.; Das, S.; Jana, K.; Karmakar, P. Folic acid conjugated curcumin loaded biopolymeric gum acacia microsphere for triple negative breast cancer therapy in invitro and in vivo model. Mater. Sci. Eng. C Mater. Biol. Appl. 2019, 95, 204–216. [Google Scholar] [CrossRef]
- Tiwari, A.; Singh, A.; Debnath, A.; Kaul, A.; Garg, N.; Mathur, R.; Singh, A.; Randhawa, J.K. Multifunctional magneto-fluorescent nanocarriers for dual mode imaging and targeted drug delivery. ACS Appl. Nano Mater. 2019, 2, 3060–3072. [Google Scholar] [CrossRef]
- Li, J.; Shen, S.; Kong, F.; Jiang, T.; Tang, C.; Yin, C. Effects of pore size on in vitro and in vivo anticancer efficacies of mesoporous silica nanoparticles. RSC Adv. 2018, 8, 24633–24640. [Google Scholar] [CrossRef]
- Bavnhøj, C.G.; Knopp, M.M.; Madsen, C.M.; Löbmann, K. The role interplay between mesoporous silica pore volume and surface area and their effect on drug loading capacity. Int. J. Pharm. X 2019, 1, 100008. [Google Scholar] [CrossRef]
- Manayi, A.; Nabavi, S.M.; Setzer, W.N.; Jafari, S. Piperine as a potential anti-cancer agent: A review on preclinical studies. Curr. Med. Chem. 2018, 25, 4918–4928. [Google Scholar] [CrossRef]
- Lai, L.H.; Fu, Q.H.; Liu, Y.; Jiang, K.; Guo, Q.M.; Chen, Q.Y.; Yan, B.; Wang, Q.Q.; Shen, J.G. Piperine suppresses tumor growth and metastasis in vitro and in vivo in a 4T1 murine breast cancer model. Acta Pharmacologica Sinica 2012, 33, 523–530. [Google Scholar] [CrossRef]
- Rather, R.A.; Bhagat, M. Cancer chemoprevention and piperine: Molecular mechanisms and therapeutic opportunities. Front. Cell Dev. Biol. 2018, 6, 10. [Google Scholar] [CrossRef]
- Syed, S.B.; Arya, H.; Fu, I.H.; Yeh, T.K.; Periyasamy, L.; Hsieh, H.P.; Coumar, M.S. Targeting P-glycoprotein: Investigation of piperine analogs for overcoming drug resistance in cancer. Sci. Rep. 2017, 7, 7972. [Google Scholar] [CrossRef]
- Wu, F.; Lin, D.D.W.; Chang, J.H.; Fischbach, C.; Estroff, L.A.; Gourdon, D. Effect of the materials properties of hydroxyapatite nanoparticles on fibronectin deposition and conformation. Cryst. Growth Des. 2015, 15, 2452–2460. [Google Scholar] [CrossRef] [PubMed]
- Ślósarczyk, A.; Szymura-Oleksiak, J.; Mycek, B. The kinetics of pentoxifylline release from drug-loaded hydroxyapatite implants. Biomaterials 2000, 21, 1215–1221. [Google Scholar] [CrossRef]
- Oberbek, P.; Bolek, T.; Chlanda, A.; Hirano, S.; Kusnieruk, S.; Rogowska-Tylman, J.; Nechyporenko, G.; Zinchenko, V.; Swieszkowski, W.; Puzyn, T. Characterization and influence of hydroxyapatite nanopowders on living cells. Beilstein J. Nanotechnol. 2018, 9, 3079–3094. [Google Scholar] [CrossRef] [PubMed]
- Jauregui-Osoro, M.; Williamson, P.A.; Glaria, A.; Sunassee, K.; Charoenphun, P.; Green, M.A.; Mullen, G.E.D.; Blower, P.J. Biocompatible inorganic nanoparticles for [18F]-fluoride binding with applications in PET imaging. Dalton Trans. 2011, 40, 6226–6237. [Google Scholar] [CrossRef]
- Lobaz, V.; Konefał, R.; Pánek, J.; Vlk, M.; Kozempel, J.; Petřík, M.; Novy, Z.; Gurská, S.; Znojek, P.; Štěpánek, P.; et al. In situ in vivo radiolabeling of polymer-coated hydroxyapatite nanoparticles to track their biodistribution in mice. Colloids Surf. B Biointerfaces 2019, 179, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Sayed, E.; Karavasili, C.; Ruparelia, K.; Haj-Ahmad, R.; Charalambopoulou, G.; Steriotis, T.; Giasafaki, D.; Cox, P.; Singh, N.; Giassafaki, L.-P.N.; et al. Electrosprayed mesoporous particles for improved aqueous solubility of a poorly water soluble anticancer agent: In vitro and ex vivo evaluation. J. Control. Release 2018, 278, 142–155. [Google Scholar] [CrossRef] [PubMed]
- Han, C.; Huang, H.; Dong, Y.; Sui, X.; Jian, B.; Zhu, W. A comparative study of the use of mesoporous carbon and mesoporous silica as drug carriers for oral delivery of the water-insoluble drug carvedilol. Molecules 2019, 24, 1770. [Google Scholar] [CrossRef] [PubMed]
- Le, T.T.; Elzhry Elyafi, A.K.; Mohammed, A.R.; Al-Khattawi, A. Delivery of poorly soluble drugs via mesoporous silica: Impact of drug overloading on release and thermal profiles. Pharmaceutics 2019, 11, 269. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.; Wu, Y.; Liu, Y.; Wu, D. High drug-loading nanomedicines: Progress, current status, and prospects. Int. J. Nanomed. 2017, 12, 4085–4109. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Wang, C.; Wang, Y.; Xu, Y.; Zhao, J.; Gao, M.; Ding, Y.; Peng, J.; Li, L. Development and evaluation of a novel drug delivery: Soluplus®/TPGS mixed micelles loaded with piperine in vitro and in vivo. Drug Dev. Ind. Pharm. 2018, 44, 1409–1416. [Google Scholar] [CrossRef]
- Huang, X.S.; Lin, X.Z.; Guo, M.T.; Zou, Y. Characteristics of piperine solubility in multiple solvent. Adv. Mat. Res. 2011, 236–238, 2495–2498. [Google Scholar] [CrossRef]
- Barahuie, F.; Dorniani, D.; Saifullah, B.; Gothai, S.; Hussein, M.Z.; Pandurangan, A.K.; Arulselvan, P.; Norhaizan, M.E. Sustained release of anticancer agent phytic acid from its chitosan-coated magnetic nanoparticles for drug-delivery system. Int. J. Nanomed. 2017, 12, 2361–2372. [Google Scholar] [CrossRef]
- Laha, A.; Yadav, S.; Majumdar, S.; Sharma, C.S. In-vitro release study of hydrophobic drug using electrospun cross-linked gelatin nanofibers. Biochem. Eng. J. 2016, 105, 481–488. [Google Scholar] [CrossRef]
- Selvakumar, M.; Kumar, P.S.; Das, B.; Dhara, S.; Chattopadhyay, S. Structurally tuned antimicrobial mesoporous hydroxyapatite nanorods by cyclic oligosaccharides regulation to release a drug for osteomyelitis. Cryst. Growth Des. 2017, 17, 433–445. [Google Scholar] [CrossRef]
- Poletto, F.S.; Jäger, E.; Ré, M.I.; Guterres, S.S.; Pohlmann, A.R. Rate-modulating PHBHV/PCL microparticles containing weak acid model drugs. Int. J. Pharm. 2007, 345, 70–80. [Google Scholar] [CrossRef] [PubMed]
- Peppas, N.A.; Narasimhan, B. Mathematical models in drug delivery: How modeling has shaped the way we design new drug delivery systems. J. Control. Release Off. J. Control. Release Soc. 2014, 190, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Yu, F.; Cole, A.J.; Chertok, B.; David, A.E.; Wang, J.; Yang, V.C. Gum arabic-coated magnetic nanoparticles for potential application in simultaneous magnetic targeting and tumor imaging. AAPS J. 2009, 11, 693–699. [Google Scholar] [CrossRef] [PubMed]
- Khan, K.A. The concept of dissolution efficiency. J. Pharm. Pharmacol. 1975, 27, 48–49. [Google Scholar] [CrossRef] [PubMed]
- Rinaki, E.; Dokoumetzidis, A.; Macheras, P. The mean dissolution time depends on the dose/solubility ratio. Pharm. Res. 2003, 20, 406–408. [Google Scholar] [CrossRef] [PubMed]
- Hornung, A.; Poettler, M.; Friedrich, R.P.; Zaloga, J.; Unterweger, H.; Lyer, S.; Nowak, J.; Odenbach, S.; Alexiou, C.; Janko, C. Treatment efficiency of free and nanoparticle-loaded mitoxantrone for magnetic drug targeting in multicellular tumor spheroids. Molecules 2015, 20, 18016–18030. [Google Scholar] [CrossRef]
- Pereira, P.M.R.; Berisha, N.; Bhupathiraju, N.V.S.D.K.; Fernandes, R.; Tomé, J.P.C.; Drain, C.M. Cancer cell spheroids are a better screen for the photodynamic efficiency of glycosylated photosensitizers. PLoS ONE 2017, 12, e0177737. [Google Scholar] [CrossRef]
- Nunes, A.S.; Barros, A.S.; Costa, E.C.; Moreira, A.F.; Correia, I.J. 3D tumor spheroids as in vitro models to mimic in vivo human solid tumors resistance to therapeutic drugs. Biotechnol. Bioeng. 2019, 116, 206–226. [Google Scholar] [CrossRef]
- Yang, S.-J.; Lin, F.-H.; Tsai, K.-C.; Wei, M.-F.; Tsai, H.-M.; Wong, J.-M.; Shieh, M.-J. Folic acid-conjugated chitosan nanoparticles enhanced protoporphyrin IX accumulation in colorectal cancer cells. Bioconjugate Chem. 2010, 21, 679–689. [Google Scholar] [CrossRef]
- Soe, Z.C.; Poudel, B.K.; Nguyen, H.T.; Thapa, R.K.; Ou, W.; Gautam, M.; Poudel, K.; Jin, S.G.; Jeong, J.-H.; Ku, S.K.; et al. Folate-targeted nanostructured chitosan/chondroitin sulfate complex carriers for enhanced delivery of bortezomib to colorectal cancer cells. Asian J. Pharm. Sci. 2019, 14, 40–51. [Google Scholar] [CrossRef]
- Virgone-Carlotta, A.; Lemasson, M.; Mertani, H.C.; Diaz, J.-J.; Monnier, S.; Dehoux, T.; Delanoë-Ayari, H.; Rivière, C.; Rieu, J.-P. In-depth phenotypic characterization of multicellular tumor spheroids: Effects of 5-Fluorouracil. PLoS ONE 2017, 12, e0188100. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Chen, J.; Gao, L.; Jiang, Z.; Zhang, Y.; Li, M.; Xiao, Q.; Lee, S.S.; Chen, X. Engineered nanomedicines with enhanced tumor penetration. Nano Today 2019, 29, 100800. [Google Scholar] [CrossRef]
- Ding, J.; Feng, X.; Jiang, Z.; Xu, W.; Guo, H.; Zhuang, X.; Chen, X. Polymer-mediated penetration-independent cancer therapy. Biomacromolecules 2019, 20, 4258–4271. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Cai, L.; Li, D.; Lao, Y.-H.; Liu, D.; Li, M.; Ding, J.; Chen, X. Tumor microenvironment-responsive hyaluronate-calcium carbonate hybrid nanoparticle enables effective chemotherapy for primary and advanced osteosarcomas. Nano Res. 2018, 11, 4806–4822. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | SBET (m2/g) a | Total Pore Volume b (cm3/g) | Mean Pore Size Diamter c (nm) | Weight Loss % wt. d | Weight Content % wt. e |
|---|---|---|---|---|---|
| HAP | 222 ± 3 | 0.815 | 12.8 | 10.69 ± 0.65 | - |
| HAP-P | 143 ± 3 | 0.501 | 11.6 | 11.46 ± 0.40 | 0.77 as P |
| HAP-Pip7.2 | 108 ± 3 | 0.372 | - | 28.78 ± 2.32 | - |
| HAP-P-Pip7.2 | 78 ± 3 | 0.356 | - | 31.76 ± 1.01 | - |
| HAP-Pip-9.3 | 83 ± 3 | 0.358 | 10.8 | 29.94 ± 0.89 | - |
| HAP-P-Pip 9.3 | 81 ± 3 | 0.358 | 10.4 | 31.30 ± 0.31 | - |
| HAP-Pip9.3-GA | 56 ± 3 | 0.231 | 11.2 | 31.83 ± 0.61 | 1.89 as GA |
| HAP-P-Pip9.3-GA | 79 ± 3 | 0.354 | 11.7 | 31.72 ± 0.62 | 0.42 as GA |
| HAP-Pip9.3-GA-FA | 70 ± 3 | 0.165 | 9.4 | 32.01 ± 1.45 | 0.18 as FA |
| HAP-P-Pip9.3-GA-FA | 61 ± 3 | 0.294 | 9.3 | 31.89 ± 1.16 | 0.17 as FA |
| Nanoformulation | TLC (%) | EE (%) | ||
|---|---|---|---|---|
| UV Method | TG Method | UV Method | TG Method | |
| HAP-Pip7.2 | 16.82 ± 3.52 a | 18.08 ± 2.60 cd | 77.42 ± 3.2 ce | 72.33 ± 10.41 cd |
| HAP-P-Pip7.2 | 20.14 ± 4.87 a | 18.71 ± 0.90 bd | 74.98 ± 2.60 e | 74.83 ± 3.61 bd |
| HAP-Pip9.3 | 22.49 ± 2.97 a | 20.31 ± 1.00 a | 79.97 ± 0.85 bc | 81.23 ± 3.98 a |
| HAP-P-Pip9.3 | 21.26 ± 3.17 a | 19.84 ± 0.59 a | 80.75 ± 3.33 e | 79.36 ± 2.36 a |
| HAP-Pip9.3-GA | 17.32 ± 5.49 a | 19.55 ± 0.59 ab | 77.90 ± 1.26 be | 78.20 ± 2.36 ab |
| HAP-P-Pip9.3-GA | 18.75 ± 1.33 a | 19.58 ± 0.59 ab | 76.58 ± 1.0 de | 78.32 ± 2.36 ab |
| HAP-Pip9.3-GA-FA | 17.27 ± 2.39 a | 18.15 ± 0.59 d | 85.05 ± 1.79 a | 72.60 ± 2.36 d |
| HAP-P-Pip9.3-GA-FA | 19.47 ± 1.20 a | 19.44 ± 0.59 abc | 78.61 ± 2.2 bcd | 77.76 ± 2.36 abc |
| Concentration (µg/mL) | HAP | HAP-P |
|---|---|---|
| 48 h | ||
| 12 ** | 90.16 ±6.9 | 110.7 ±5.0 |
| 37 ** | 96.09 ±14.8 | 117.05 ±1.3 |
| 111 ** | 78.29 ±9.6 | 107.56 ± 12.0 |
| 333 ** | 88.18 ±6.4 | 91.74 ±7.0 |
| 1000 ** | 78.69 ±6.9 | 88.58 ±5.0 |
| 72 h | ||
| 12 | 129.2 ±12.7 | 129.2 ±9.7 |
| 37 | 122.1 ±17.2 | 122.1 ±19.0 |
| 111 | 130.8 ±15.6 | 130.8 ±14.0 |
| 333 | 125.7 ±11.4 | 125.7 ±11.6 |
| 1000 | 120.7 ±6.7 | 120.7 ±4.3 |
| Concentration (µg/mL) | Treatment | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| HAP-Pip7.2 | HAP-P-Pip7.2 | HAP-Pip9.3 | HAP-P-Pip9.3 | HAP-Pip9.3-GA | HAP-P-Pip9.3-GA | HAP-Pip9.3-GA-FA | HAP-P-Pip9.3-GA-FA | Pip | |
| 48 h | |||||||||
| 2.4 | 118.7 ± 14 | 170.4 ± 9 | 93.2 ± 10 | 116.5 ± 8 | 105.2 ± 8 | 106.6 ± 2 | 95.9 ± 5 | 78.4 ± 26 | 110.9 ± 8 |
| 7.4 | 110.9 ± 32 | 135.4 ± 17 | 85.4 ± 10 | 81.7 ± 9 | 112.2 ± 6 | 90.7 ± 19 | 104.8 ± 14 | 88.8 ± 33 | 113.0 ± 6 |
| 22 | 117.7 ± 25 | 153.3 ± 48 | 97.6 ± 16 | 103.2 ± 11 | 107.5 ± 9 | 95.2 ± 2 | 136.2 ± 14 | 79.1 ± 12 | 102.6 ± 11 |
| 66 | 112.0 ± 19 | 142.5 ± 19 * | 104.8 ± 21 | 106.3 ± 14 | 114.5 ± 18 | 75.0 ± 4 | 155.2 ± 26 * | 71.8 ± 14 | 102.8 ± 19 * |
| 200 | 93.2 ± 17 | 141.7 ± 16 * | 85.6 ± 11 | 86.0 ± 8 | 109.2 ± 10 | 105.2 ± 13 | 170.2 ± 19 * | 107.5 ± 18 * | 90.5 ± 13 * |
| 72 h | |||||||||
| 2.4# | 94.2 ± 8 | 87.6 ± 7 | 108.0 ± 15 | 121.0 ± 25 | 111.4 ± 17 | 108.4 ± 7 | 105.4 ± 6 | 103.9 ± 7 | 125.9 ± 11 |
| 7.4# | 85.8 ± 4 | 86.9 ± 11 | 103.9 ± 4 | 104.9 ± 8 | 105.1 ± 53 | 106.7 ± 11 | 85.9 ± 7 | 105.2 ± 4 | 133.4 ± 9 |
| 22# | 90.0 ± 12 | 88.1 ± 3 | 99.8 ± 12 | 99.3 ± 12 | 93.2 ± 4 | 106.7 ± 7 | 86.9 ± 12 | 100.7 ± 15 | 132.5 ± 12 |
| 66# | 79.6 ± 30 * | 83.0 ± 9 * | 104.2 ± 23 * | 98.8 ± 2 * | 78.4 ± 7 * | 106.0 ± 9 * | 58.1 ± 15 * | 89.4 ± 6 * | 125.5 ± 19 * |
| 200# | 91.9 ± 6 | 67.4 ± 4 * | 74.8 ± 13 * | 70.3 ± 7 * | 56.2 ± 6 * | 78.1 ± 11 * | 53.8 ± 8 * | 54.2 ± 9 * | 125.2 ± 12 * |
| Piperine Delivery Method | Structure | Solubility and Release Kinetics | Targeting Specifications |
|---|---|---|---|
| Pip (free form) |  | Higher solubility in PBS at acidic pH 5 than 6.8, 7.4, and 9. | Moderate cytotoxicity towards normal WI-38 cells. Weak activity against HCT116 colon cancer cells (monolayer and spheroids). Not effective. Non-targeting for colon. |
| HAP-Pip or HAP-P-Pip—Pip-loaded HAP not modified or modified with phosphonate. Loading at pH 7.2 and 9.3 |  | Short-term release. Release 100% of Pip capacity ~36 h (pH 6.8) and 24 h (pH 5). | Moderate cytotoxicity towards normal cells. High anticancer effect on monolayer colon cancer cells but not spheroids. Not enough cancer targeting. No change in cell morphology in monolayer or spheroids. |
| HAP-P-Pip9.3-GA-FA—Pip-loaded HAP modified with phosphonate, coated with gum Arabic, and conjugated to folic acid |  | Long-term release. Release 100% of Pip capacity ~84 h (pH 6.8) and 72 h (pH 5). | Less cytotoxicity. High anticancer effect with full inhibition of monolayer HCT116 cells and ~60% inhibition for spheroids. High cancer-targeting of monolayer and spheroid HCT116 cells. Cancer targeting through folate receptors. Cell morphology changes: smaller size, shrinkage, and defragmentation of spheroids. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
AbouAitah, K.; Stefanek, A.; Higazy, I.M.; Janczewska, M.; Swiderska-Sroda, A.; Chodara, A.; Wojnarowicz, J.; Szałaj, U.; Shahein, S.A.; Aboul-Enein, A.M.; et al. Effective Targeting of Colon Cancer Cells with Piperine Natural Anticancer Prodrug Using Functionalized Clusters of Hydroxyapatite Nanoparticles. Pharmaceutics 2020, 12, 70. https://doi.org/10.3390/pharmaceutics12010070
AbouAitah K, Stefanek A, Higazy IM, Janczewska M, Swiderska-Sroda A, Chodara A, Wojnarowicz J, Szałaj U, Shahein SA, Aboul-Enein AM, et al. Effective Targeting of Colon Cancer Cells with Piperine Natural Anticancer Prodrug Using Functionalized Clusters of Hydroxyapatite Nanoparticles. Pharmaceutics. 2020; 12(1):70. https://doi.org/10.3390/pharmaceutics12010070
Chicago/Turabian StyleAbouAitah, Khaled, Agata Stefanek, Iman M. Higazy, Magdalena Janczewska, Anna Swiderska-Sroda, Agnieszka Chodara, Jacek Wojnarowicz, Urszula Szałaj, Samar A. Shahein, Ahmed M. Aboul-Enein, and et al. 2020. "Effective Targeting of Colon Cancer Cells with Piperine Natural Anticancer Prodrug Using Functionalized Clusters of Hydroxyapatite Nanoparticles" Pharmaceutics 12, no. 1: 70. https://doi.org/10.3390/pharmaceutics12010070
APA StyleAbouAitah, K., Stefanek, A., Higazy, I. M., Janczewska, M., Swiderska-Sroda, A., Chodara, A., Wojnarowicz, J., Szałaj, U., Shahein, S. A., Aboul-Enein, A. M., Abou-Elella, F., Gierlotka, S., Ciach, T., & Lojkowski, W. (2020). Effective Targeting of Colon Cancer Cells with Piperine Natural Anticancer Prodrug Using Functionalized Clusters of Hydroxyapatite Nanoparticles. Pharmaceutics, 12(1), 70. https://doi.org/10.3390/pharmaceutics12010070