3D-Printed Solid Dispersion Drug Products



Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
2.2.1. Filament Preparation
2.2.2. Solid State Characterization of Filaments
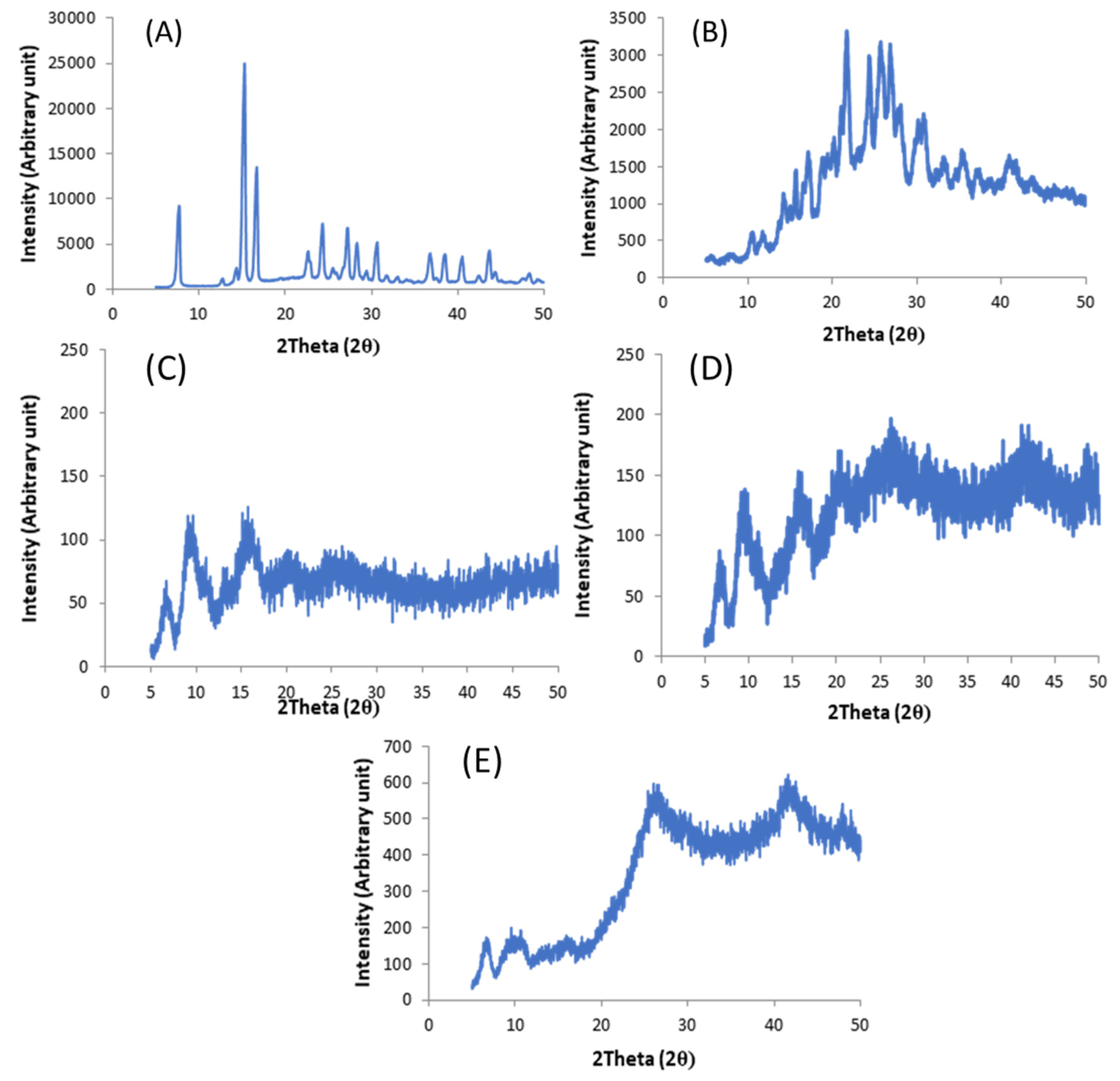
X-Ray Powder Diffration
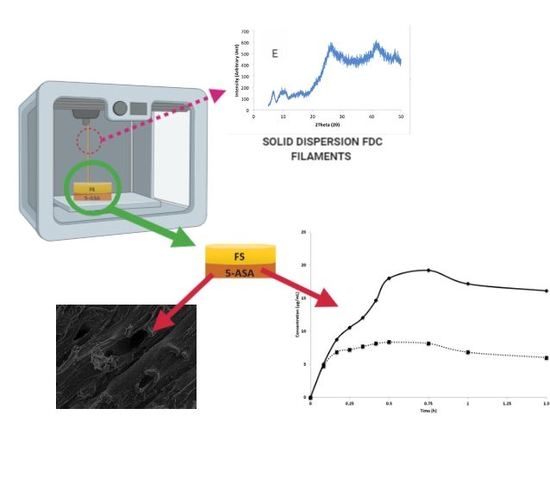
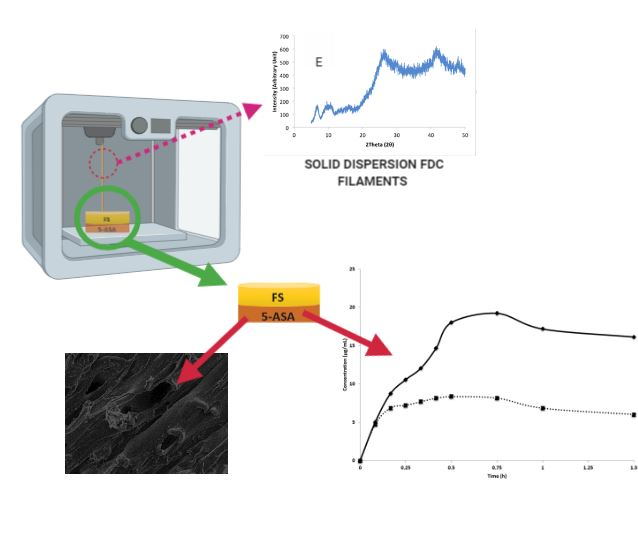
2.2.3. 3D-Printed Drug Product Design and Optimization
2.2.4. Morphology Studies
Scanning Electron Microscopy
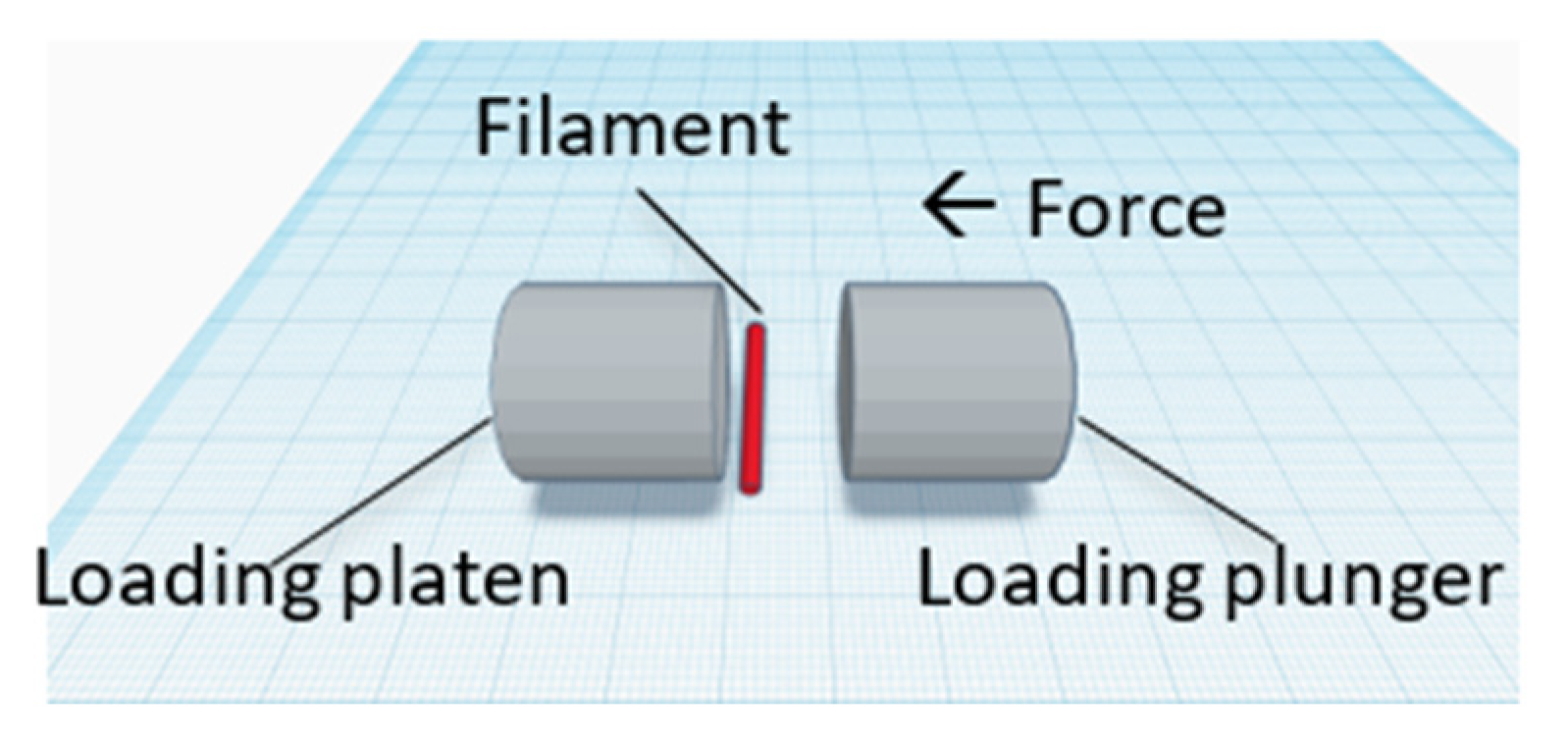
2.2.5. Crushing Strength
2.2.6. Solubility, Drug Content, and In Vitro Drug Dissolution Studies
Solubility Studies
Calculation of Drug Content in PVA Filaments
In Vitro Drug Dissolution Studies
2.2.7. Statistical Analysis
3. Results
3.1. Solvent Choice Optimization
3.2. Filament Characterization
3.2.1. Filament Hardness
3.2.2. Solid State Characterization of Filaments
3.3. Characterization of 3D-Printed Dosage Forms
Morphology Studies
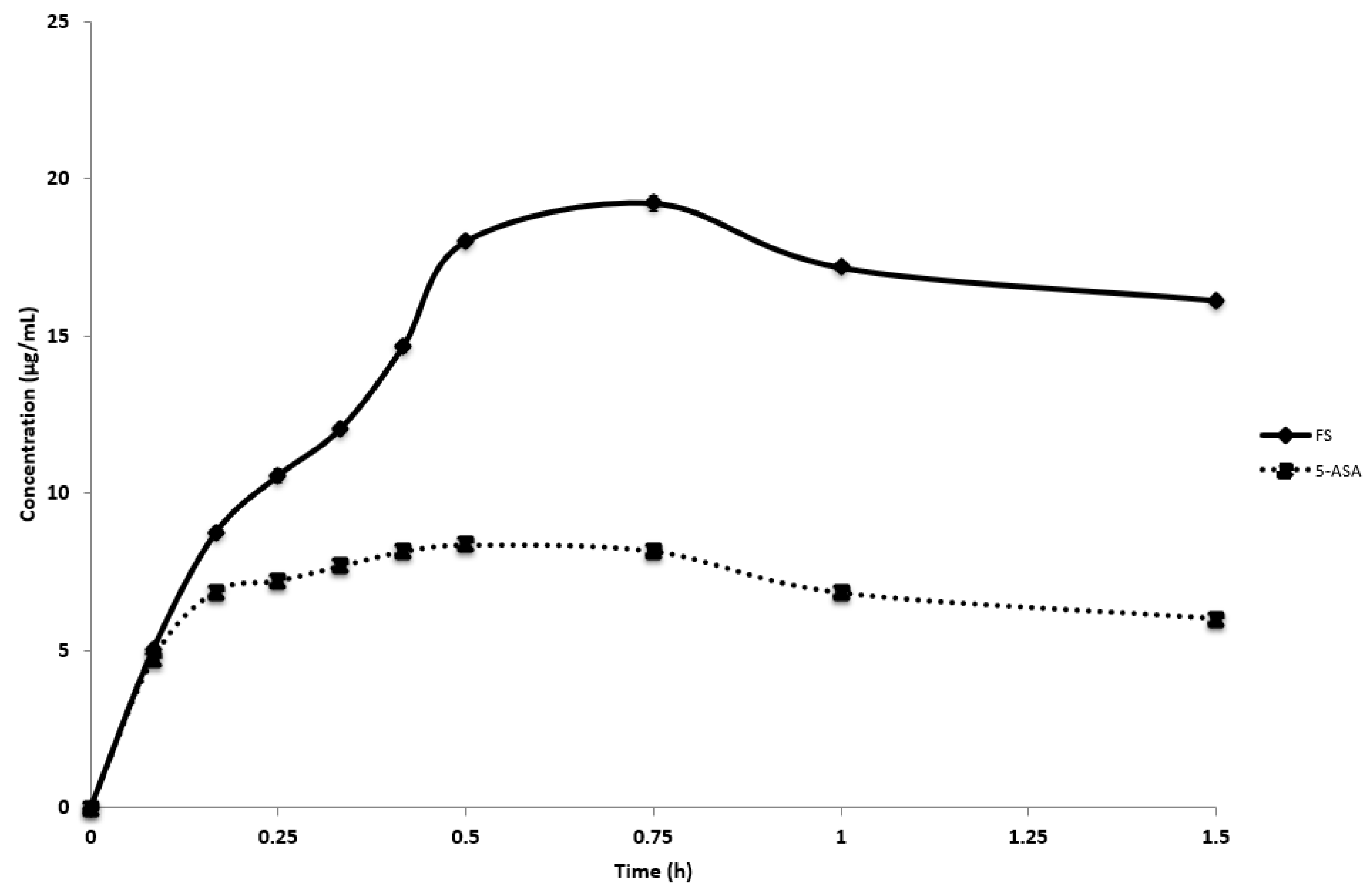
3.4. In Vitro Dissolution
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Desai, D.; Wang, J.; Wen, H.; Li, X. Timmins P. Formulation design, challenges, and development considerations for fixed dose combination (FDC) of oral solid dosage forms. Pharm. Dev. Technol. 2013, 18, 1265–1276. [Google Scholar] [CrossRef] [PubMed]
- EMA/CHMP/158268/2017, (CHMP) C for HMP. Guideline on Clinical Development of Fixed Combination Medicinal Products. 2017. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-clinical-development-fixed-combination-medicinal-products-revision-2_en.pdf (accessed on 2 October 2018).
- Kavanagh, O.N.; Albadarin, A.B.; Croker, D.M.; Healy, A.M.; Walker, G.M. Maximising success in multidrug formulation development: A review. J. Control. Release 2018, 283, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Sawicki-Wrzask, D.; Thomsen, M.; Bjerrum, O.J. An Analysis of the Fixed-Dose Combinations Authorized by the European Union, 2009–2014. Ther. Innov. Regul. Sci. 2015, 49, 553–559. [Google Scholar] [CrossRef] [PubMed]
- Bangalore, S.; Kamalakkannan, G.; Parkar, S.; Messerli, F.H. Fixed-Dose Combinations Improve Medication Compliance: A Meta-Analysis. Am. J. Med. 2007, 120, 713–719. [Google Scholar] [CrossRef] [PubMed]
- FDA. Fixed Dose Combinations, Co-Packaged Drug Products, and Single-Entity Versions of Previously Approved Antiretrovirals for the Treatment of HIV; Center for Drug Evaluation and Research (CDER): Rockville, MD, USA, 2006.
- TheKing’sFund. Polypharmacy and Medicines Optimisation: Making it Safe and Sound. Published 2013. Available online: https://www.kingsfund.org.uk/publications/polypharmacy-and-medicines-optimisation (accessed on 6 December 2018).
- Rea, F.; Corrao, G.; Merlino, L.; Mancia, G. Early cardiovascular protection by initial two-drug fixed-dose combination treatment vs. monotherapy in hypertension. Eur. Heart J. 2018, 39, 3654–3661. [Google Scholar] [CrossRef] [PubMed]
- Pourkavoos, N. Unique Risks, Benefits, and Challenges of Developing Drug-Drug Combination Products in a Pharmaceutical Industrial Setting. Comb. Prod. Ther. 2012, 2, 2. [Google Scholar] [CrossRef]
- Jamróz, W.; Szafraniec, J.; Kurek, M.; Jachowicz, R. 3D Printing in Pharmaceutical and Medical Applications—Recent Achievements and Challenges. Pharm. Res. 2018, 35, 176. [Google Scholar] [CrossRef]
- Richey, R.H.; Hughes, C.; Craig, J.V.; Shah, U.U.; Ford, J.L.; Barker, C.E.; Peak, M.; Nunn, A.J.; Turner, M.A. A systematic review of the use of dosage form manipulation to obtain required doses to inform use of manipulation in paediatric practice. Int. J. Pharm. 2017, 518, 155–166. [Google Scholar] [CrossRef]
- Shastry, B.S. Pharmacogenetics and the concept of individualized medicine. Pharmacogenom. J. 2006, 6, 16–21. [Google Scholar] [CrossRef]
- Acosta-Vélez, G.F. 3D Pharming: Direct Printing of Personalized Pharmaceutical Tablets. Polym. Sci. 2016, 2. [Google Scholar] [CrossRef]
- Konta, A.; García-Piña, M.; Serrano, D. Personalised 3D Printed Medicines: Which Techniques and Polymers Are More Successful? Bioengineering 2017, 4, 79. [Google Scholar] [CrossRef] [PubMed]
- Norman, J.; Madurawe, R.D.; Moore, C.M.V.; Khan, M.A.; Khairuzzaman, A. A new chapter in pharmaceutical manufacturing: 3D-printed drug products. Adv. Drug Deliv. Rev. 2017, 108, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Khaled, S.A.; Burley, J.C.; Alexander, M.R.; Yang, J.; Roberts, C.J. 3D printing of five-in-one dose combination polypill with defined immediate and sustained release profiles. J. Control. Release 2015, 217, 308–314. [Google Scholar] [CrossRef] [PubMed]
- Goyanes, A.; Robles Martinez, P.; Buanz, A.; Basit, A.W.; Gaisford, S. Effect of geometry on drug release from 3D printed tablets. Int. J. Pharm. 2015, 494, 657–663. [Google Scholar] [CrossRef] [PubMed]
- Kadry, H.; Al-Hilal, T.A.; Keshavarz, A.; Alam, F.; Xu, C.; Joy, A.; Ahsan, F. Multi-purposable filaments of HPMC for 3D printing of medications with tailored drug release and timed-absorption. Int. J. Pharm. 2018, 544, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Sadia, M.; Isreb, A.; Abbadi, I.; Isreb, M.; Aziz, D.; Selo, A.; Timmins, P.; Alhnan, M.A. From ‘fixed dose combinations’ to ‘a dynamic dose combiner’: 3D printed bi-layer antihypertensive tablets. Eur. J. Pharm. Sci. 2018, 123, 484–494. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Goyanes, A.; Gaisford, S.; Basit, A.W. Stereolithographic (SLA) 3D printing of oral modified-release dosage forms. Int. J. Pharm. 2016, 503, 207–212. [Google Scholar] [CrossRef]
- Feuerbach, T.; Kock, S.; Thommes, M. Characterisation of fused deposition modeling 3D printers for pharmaceutical and medical applications. Pharm. Dev. Technol. 2018, 23, 1136–1145. [Google Scholar] [CrossRef]
- Tran, T.Q.; Chinnappan, A.; Lee, J.K.Y.; Loc, N.H.; Tran, L.T.; Wang, T.; Vijay Kumar, V.; Jayathilaka, W.A.D.M.; Ji, D.; Doddamani, M.; et al. 3D Printing of Highly Pure Copper. Metals 2019, 9, 756. [Google Scholar] [CrossRef]
- Goyanes, A.; Wang, J.; Buanz, A.; Martínez-Pacheco, R.; Telford, R.; Gaisford, S.; Basit, A.W. 3D Printing of Medicines: Engineering Novel Oral Devices with Unique Design and Drug Release Characteristics. Mol. Pharm. 2015, 12, 4077–4084. [Google Scholar] [CrossRef]
- Skowyra, J.; Pietrzak, K.; Alhnan, M.A. Fabrication of extended-release patient-tailored prednisolone tablets via fused deposition modelling (FDM) 3D printing. Eur. J. Pharm. Sci. 2015, 68, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Rahman, Z.; Barakh Ali, S.F.; Ozkan, T.; Charoo, N.A.; Reddy, I.K.; Khan, M.A. Additive Manufacturing with 3D Printing: Progress from Bench to Bedside. AAPS J. 2018, 20, 101. [Google Scholar] [CrossRef] [PubMed]
- Solanki, N.G.; Tahsin, M.; Shah, A.V.; Serajuddin, A.T.M. Formulation of 3D Printed Tablet for Rapid Drug Release by Fused Deposition Modeling: Screening Polymers for Drug Release, Drug-Polymer Miscibility and Printability. J. Pharm. Sci. 2018, 107, 390–401. [Google Scholar] [CrossRef] [PubMed]
- Alhnan, M.A.; Okwuosa, T.C.; Sadia, M.; Wan, K.-W.; Ahmed, W.; Arafat, B. Emergence of 3D Printed Dosage Forms: Opportunities and Challenges. Pharm. Res. 2016, 33, 1817–1832. [Google Scholar] [CrossRef] [PubMed]
- Modica de Mohac, L.; Keating, A.; de Fátima Pina, M.; Raimi-Abraham, B. Engineering of Nanofibrous Amorphous and Crystalline Solid Dispersions for Oral Drug Delivery. Pharmaceutics 2018, 11, 7. [Google Scholar] [CrossRef] [PubMed]
- Goyanes, A.; Buanz, A.B.M.; Hatton, G.B.; Gaisford, S.; Basit, A.W. 3D printing of modified-release aminosalicylate (4-ASA and 5-ASA) tablets. Eur. J. Pharm. Biopharm. 2015, 89, 157–162. [Google Scholar] [CrossRef]
- Goyanes, A.; Buanz, A.B.M.; Basit, A.W.; Gaisford, S. Fused-filament 3D printing (3DP) for fabrication of tablets. Int. J. Pharm. 2014, 476, 88–92. [Google Scholar] [CrossRef]
- Tagami, T.; Fukushige, K.; Ogawa, E.; Hayashi, N.; Ozeki, T. 3D Printing Factors Important for the Fabrication of Polyvinylalcohol Filament-Based Tablets. Biol. Pharm. Bull. 2017, 40, 357–364. [Google Scholar] [CrossRef]
- Hayes, A.W.; Kruger, C.L. Hayes’ Principles and Methods of Toxicology; CRC Press: Boca Raton, FL, USA, 2014. [Google Scholar]
- Hassan, S.; Adam, F.; Abu Bakar, M.R.; Abdul Mudalip, S.K. Evaluation of solvents’ effect on solubility, intermolecular interaction energies and habit of ascorbic acid crystals. J. Saudi Chem. Soc. 2019, 23, 239–248. [Google Scholar] [CrossRef]
- Babagowda Kadadevara Math, R.S.; Goutham, R.; Srinivas Prasad, K. Study of Effects on Mechanical Properties of PLA Filament which is blended with Recycled PLA Materials. IOP Conf. Ser. Mater. Sci. Eng. 2018, 310, 012103. [Google Scholar] [CrossRef]
- Fell, J.T.; Newton, J.M. Determination of Tablet Strength by the Diametral-Compression Test. J. Pharm. Sci. 1970, 59, 688–691. [Google Scholar] [CrossRef] [PubMed]
- Banić-Tomišić, Z.; Kojić-Prodić, B.; Širola, I. Hydrogen bonds in the crystal packings of mesalazine and mesalazine hydrochloride. J. Mol. Struct. 1997, 416, 209–220. [Google Scholar] [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2016, 72, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Michael ARepka Nigel Langley, J.D. Melt Extrusion: Materials, Technology and Drug Product Design (AAPS Advances in the Pharmaceutical Sciences Series Book; Springer Science & Business Media: Berlin, Germany, 2013. [Google Scholar] [CrossRef]
- Wlodarski, K.; Zhang, F.; Liu, T.; Sawicki, W.; Kipping, T. Synergistic Effect of Polyvinyl Alcohol and Copovidone in Itraconazole Amorphous Solid Dispersions. Pharm. Res. 2018, 35, 16. [Google Scholar] [CrossRef]
- Konno, H.; Handa, T.; Alonzo, D.E.; Taylor, L.S. Effect of polymer type on the dissolution profile of amorphous solid dispersions containing felodipine. Eur. J. Pharm. Biopharm. 2008, 70, 493–499. [Google Scholar] [CrossRef]
- Konno, H.; Taylor, L.S. Influence of Different Polymers on the Crystallization Tendency of Molecularly Dispersed Amorphous Felodipine. J. Pharm. Sci. 2006, 95, 2692–2705. [Google Scholar] [CrossRef]
- Gupta, D.; Jassal, M.; Agrawal, A.K. The electrospinning behavior of poly(vinyl alcohol) in DMSO–water binary solvent mixtures. RSC Adv. 2016, 6, 102947–102955. [Google Scholar] [CrossRef]
- Paruta, A.N.; Sciarrone, B.J.; Lordi, N.G. Correlation between solubility parameters and dielectric constants. J. Pharm. Sci. 1962, 51, 704–705. [Google Scholar] [CrossRef]
- ThermoFisher. Kaolin Asp® 400p Safety Data Sheet; Emirates U.A.: Fair Lawn, NJ, USA, 2012; pp. 8–10. [Google Scholar]
- Åkerlöf, G. Dielectric constants of some organic solvent-water mixtures at various temperatures. J. Am. Chem. Soc. 1932, 54, 4125–4139. [Google Scholar] [CrossRef]
- Chernyak, Y. Dielectric constant, dipole moment, and solubility parameters of some cyclic acid esters. J. Chem. Eng. Data 2006, 51, 416–418. [Google Scholar] [CrossRef]
- Mohsin, M.; Hossin, A.; Haik, Y. Thermomechanical properties of poly(vinyl alcohol) plasticized with varying ratios of sorbitol. Mater. Sci. Eng. A 2011, 528, 925–930. [Google Scholar] [CrossRef]
- Goyanes, A.; Chang, H.; Sedough, D.; Hatton, G.B.; Wang, J.; Buanz, A.; Gaisford, S.; Basit, A.W. Fabrication of controlled-release budesonide tablets via desktop (FDM) 3D printing. Int. J. Pharm. 2015, 496, 414–420. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Jang, B.N.; Park, B.J.; Joung, Y.K.; Han, D.K. Effect of Solvent on Drug Release and a Spray-Coated Matrix of a Sirolimus-Eluting Stent Coated with Poly(lactic-co-glycolic acid). Langmuir 2014, 30, 10098–10106. [Google Scholar] [CrossRef] [PubMed]
- Raimi-Abraham, B.T.; Mahalingam, S.; Edirisinghe, M.; Craig, D.Q.M. Generation of poly(N-vinylpyrrolidone) nanofibres using pressurised gyration. Mater. Sci. Eng. C 2014, 39, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Modica de Mohac, L.; de Fátima Pina, M.; Raimi-Abraham, B.T. Solid microcrystalline dispersion films as a new strategy to improve the dissolution rate of poorly water soluble drugs: A case study using olanzapine. Int. J. Pharm. 2016, 508, 42–50. [Google Scholar] [CrossRef]
- Sun, D.D.; Lee, P.I. Probing the mechanisms of drug release from amorphous solid dispersions in medium-soluble and medium-insoluble carriers. J. Control. Release 2015, 211, 85–93. [Google Scholar] [CrossRef]
- Baghel, S.; Cathcart, H.; O’Reilly, N.J. Polymeric Amorphous Solid Dispersions: A Review of Amorphization, Crystallization, Stabilization, Solid-State Characterization, and Aqueous Solubilization of Biopharmaceutical Classification System Class II Drugs. J. Pharm. Sci. 2016, 105, 2527–2544. [Google Scholar] [CrossRef]
- Sun, D.D.; Lee, P.I. Evolution of Supersaturation of Amorphous Pharmaceuticals: The Effect of Rate of Supersaturation Generation. Mol. Pharm. 2013, 10, 4330–4346. [Google Scholar] [CrossRef]
- Craig, D.Q.M. The mechanisms of drug release from solid dispersions in water-soluble polymers. Int. J. Pharm. 2002, 231, 131–144. [Google Scholar] [CrossRef]
- Ilevbare, G.A.; Liu, H.; Edgar, K.J.; Taylor, L.S. Inhibition of solution crystal growth of ritonavir by cellulose polymers—Factors influencing polymer effectiveness. CrystEngComm 2012, 14, 6503. [Google Scholar] [CrossRef]
- Megrab, N.A.; Williams, A.C.; Barry, B.W. Oestradiol permeation through human skin and silastic membrane: Effects of propylene glycol and supersaturation. J. Control. Release 1995, 36, 277–294. [Google Scholar] [CrossRef]
- Curatolo, W.; Nightingale, J.A.; Herbig, S.M. Utility of Hydroxypropylmethylcellulose Acetate Succinate (HPMCAS) for Initiation and Maintenance of Drug Supersaturation in the GI Milieu. Pharm. Res. 2009, 26, 1419–1431. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Active Pharmaceutical Ingredient (API) | Ethanol (EtOH) | Methanol (MeOH) | Dimethyl Sulfoxide (DMSO) |
|---|---|---|---|
| PVA | |||
| Fluorescein sodium (FS) | 2.0% w/v (FS-EtOH) | 2.5% w/v (2.5%FS-MeOH) | 2.5% w/v (FS-DMSO) |
| 5-aminosalicyclic acid (5-ASA) | 1.0% w/v (5-ASA-EtOH) | 1.25% w/v (5-ASA-MeOH) | 1.25% w/v (5-ASA-DMSO) |
| FS and 5-ASA | 2.5% w/v FS and 1.25% w/v 5-ASA (FDC-MeOH) | ||
| Solvents | Drug-Loaded Filaments | Drug Loading (% w/w) |
|---|---|---|
| Ethanol | FS-EtOH | 1.19 ± 0.161 |
| 5-ASA-EtOH | 0.10 ± 0.001 | |
| Methanol | FS-MeOH | 4.89 ± 0.449 |
| 5-ASA-MeOH | 0.17 ± 0.007 | |
| FDC-MeOH | FS: 6.16 ± 0.197 5-ASA: 2.97 ± 0.362 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chew, S.L.; Modica de Mohac, L.; Tolulope Raimi-Abraham, B. 3D-Printed Solid Dispersion Drug Products. Pharmaceutics 2019, 11, 672. https://doi.org/10.3390/pharmaceutics11120672
Chew SL, Modica de Mohac L, Tolulope Raimi-Abraham B. 3D-Printed Solid Dispersion Drug Products. Pharmaceutics. 2019; 11(12):672. https://doi.org/10.3390/pharmaceutics11120672
Chicago/Turabian StyleChew, Suet Li, Laura Modica de Mohac, and Bahijja Tolulope Raimi-Abraham. 2019. "3D-Printed Solid Dispersion Drug Products" Pharmaceutics 11, no. 12: 672. https://doi.org/10.3390/pharmaceutics11120672
APA StyleChew, S. L., Modica de Mohac, L., & Tolulope Raimi-Abraham, B. (2019). 3D-Printed Solid Dispersion Drug Products. Pharmaceutics, 11(12), 672. https://doi.org/10.3390/pharmaceutics11120672