Mechanisms by which HPV Induces a Replication Competent Environment in Differentiating Keratinocytes

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
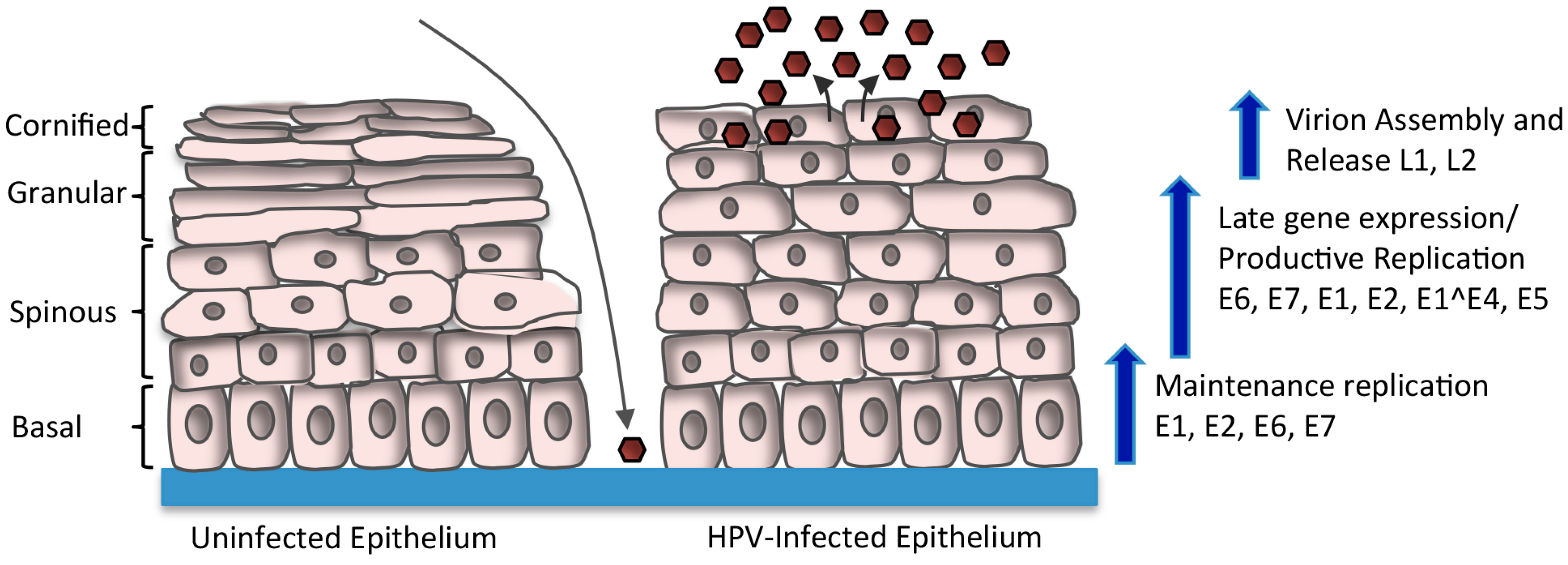
2. HPV Life Cycle
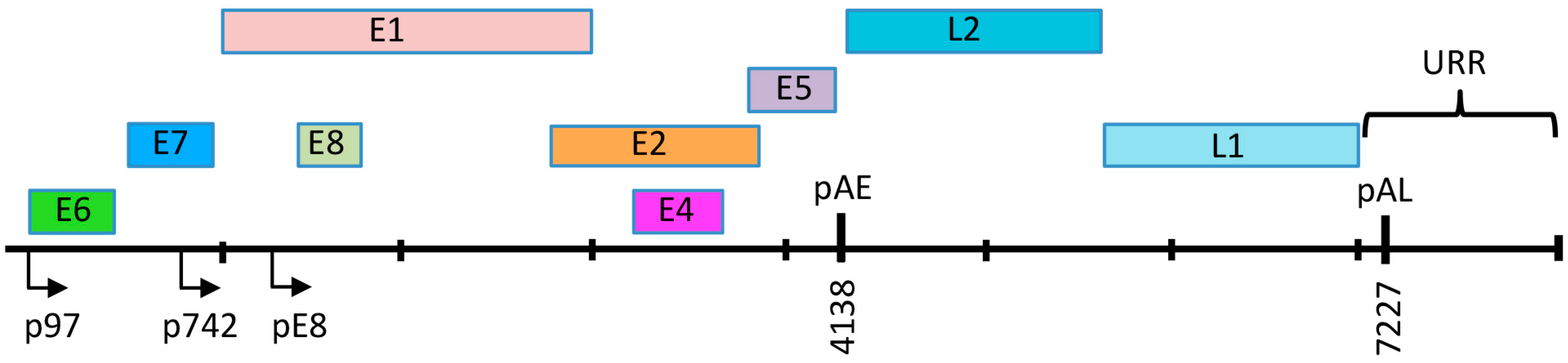
3. HPV Genome Organization
4. Regulation of Viral Gene Expression upon Keratinocyte Differentiation
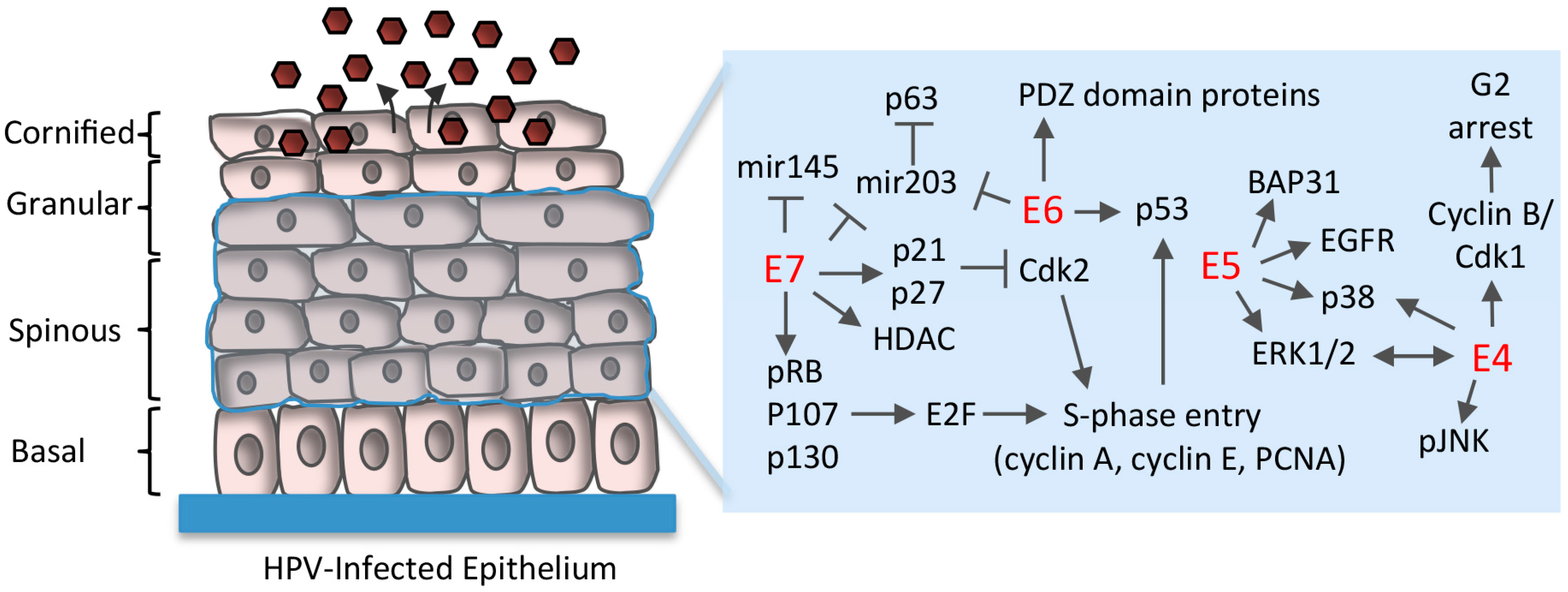
5. Maintenance of Proliferative Potential in Differentiating Cells
5.1. Disruption of Rb/E2F Complexes
5.2. Uncoupling of Differentiation From Proliferation
5.3. E6 Abrogation of p53 and Targeting of PDZ (PSD95/DLG1/ZO-1) Domain-Containing Proteins
5.4. Regulation of Differentiation-Induced microRNA Expression
6. Establishment of a G2-Arrested Environment
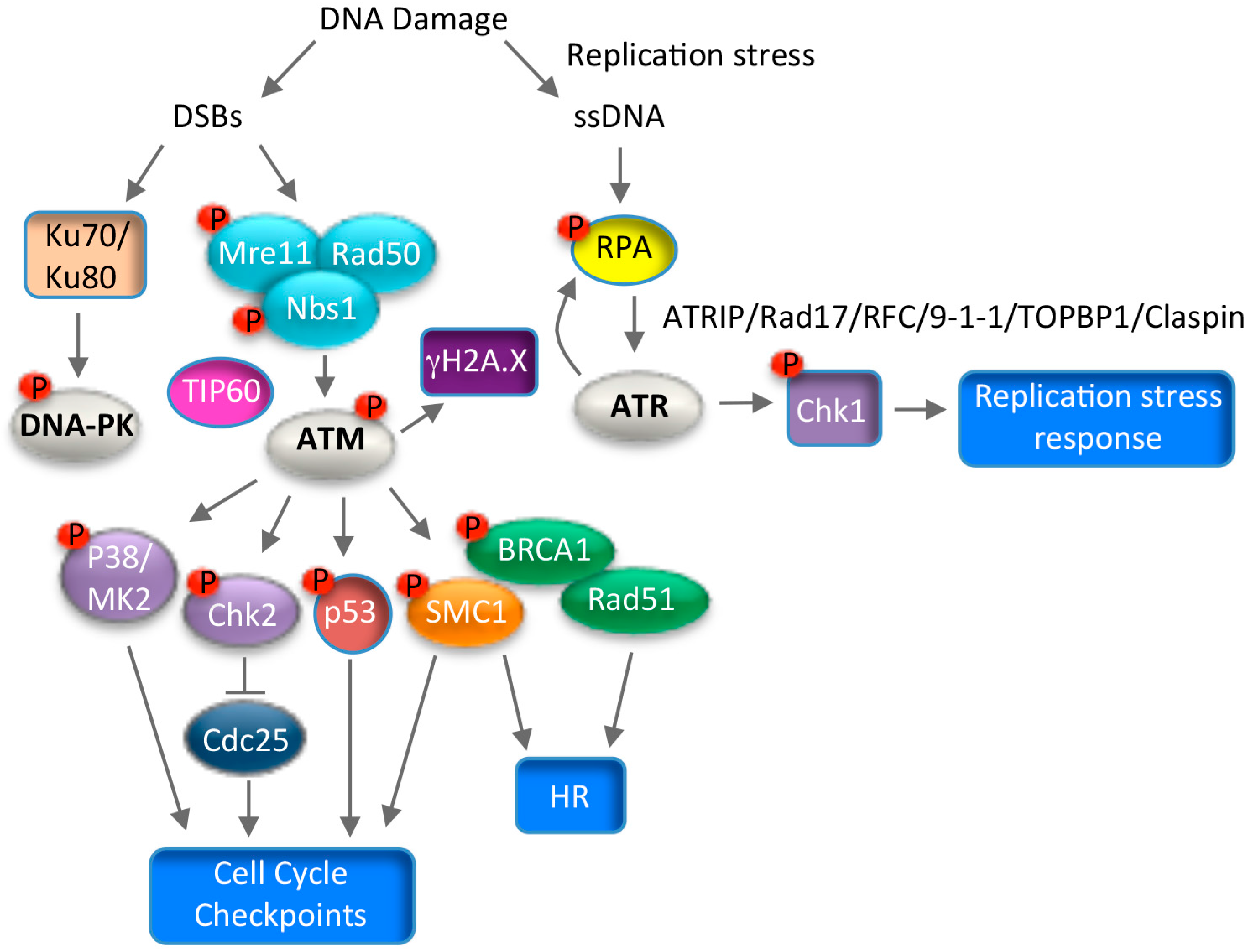
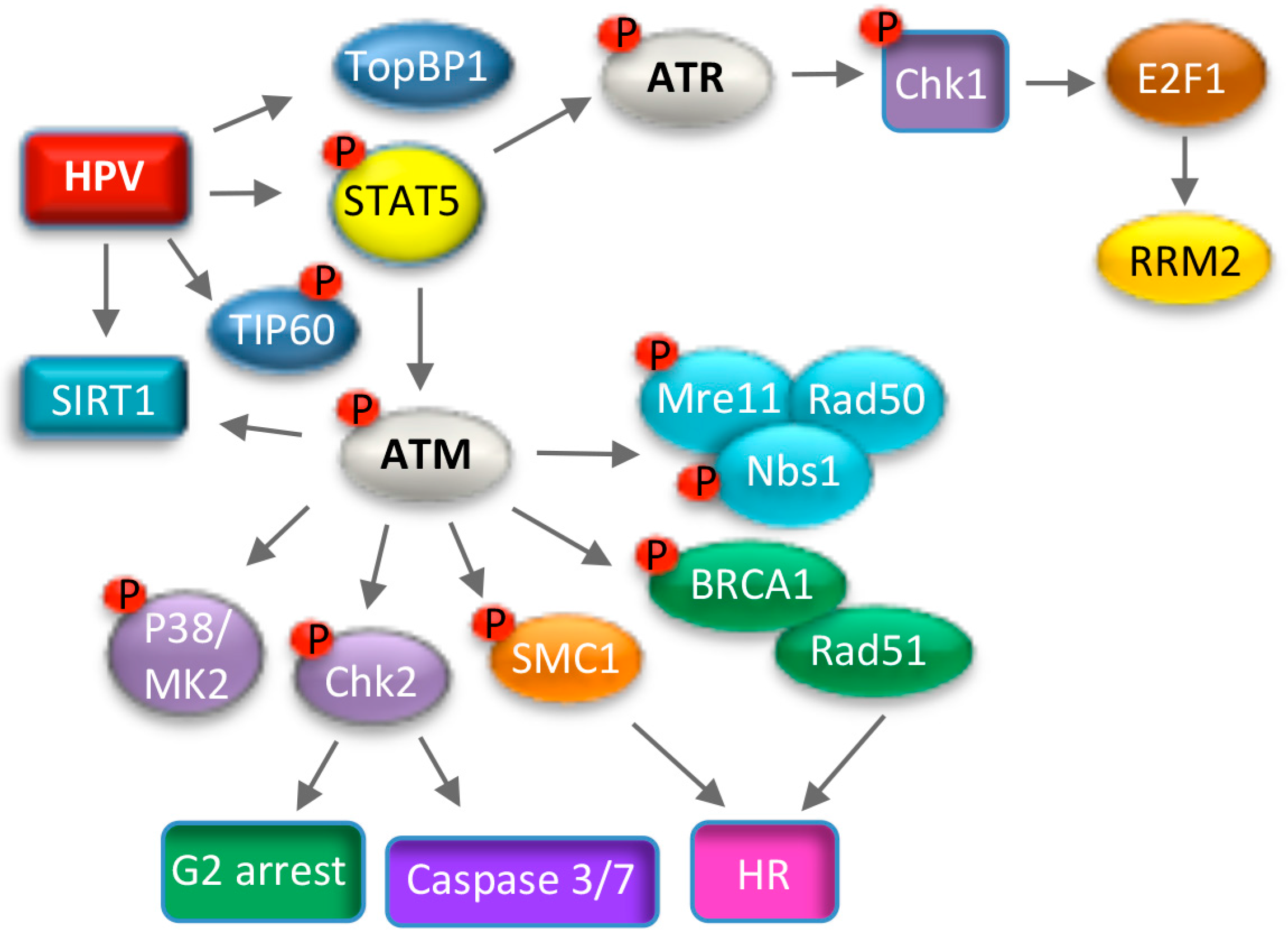
7. Use of DNA Damage Response Pathways for Productive Replication
7.1. ATM Signaling and Productive Viral Replication
7.2. ATR Signaling and Productive Viral Replication
7.3. Consequences of Utilizing the DNA Damage Response for Replication
8. Conclusions
Acknowledgments
Conflicts of Interest
References
- Van Doorslaer, K.; Tan, Q.; Xirasagar, S.; Bandaru, S.; Gopalan, V.; Mohamoud, Y.; Huyen, Y.; McBride, A.A. The papillomavirus episteme: A central resource for papillomavirus sequence data and analysis. Nucleic Acids Res. 2013, 41, 571–578. [Google Scholar] [CrossRef] [PubMed]
- Bernard, H.U.; Burk, R.D.; Chen, Z.; van Doorslaer, K.; zur Hausen, H.; de Villiers, E.M. Classification of papillomaviruses (PVS) based on 189 PV types and proposal of taxonomic amendments. Virology 2010, 401, 70–79. [Google Scholar] [CrossRef] [PubMed]
- Walboomers, J.M.; Jacobs, M.V.; Manos, M.M.; Bosch, F.X.; Kummer, J.A.; Shah, K.V.; Snijders, P.J.; Peto, J.; Meijer, C.J.; Munoz, N. Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. J. Pathol. 1999, 189, 12–19. [Google Scholar] [CrossRef]
- Stanley, M. Pathology and epidemiology of HPV infection in females. Gynecol. Oncol. 2010, 117, S5–S10. [Google Scholar] [CrossRef] [PubMed]
- Gillison, M.L.; Chaturvedi, A.K.; Anderson, W.F.; Fakhry, C. Epidemiology of human papillomavirus-positive head and neck squamous cell carcinoma. J. Clin. Oncol. 2015, 33, 3235–3242. [Google Scholar] [CrossRef] [PubMed]
- Tommasino, M. The biology of β human papillomaviruses. Virus Res. 2017, 231, 128–138. [Google Scholar] [CrossRef] [PubMed]
- Howley, P.M.; Pfister, H.J. β genus papillomaviruses and skin cancer. Virology 2015, 479–480, 290–296. [Google Scholar] [CrossRef] [PubMed]
- Egawa, N.; Egawa, K.; Griffin, H.; Doorbar, J. Human papillomaviruses; epithelial tropisms, and the development of neoplasia. Viruses 2015, 7, 3863–3890. [Google Scholar] [CrossRef] [PubMed]
- McBride, A.A. Mechanisms and strategies of papillomavirus replication. Biol. Chem. 2017, 398, 919–927. [Google Scholar] [CrossRef] [PubMed]
- Longworth, M.S.; Laimins, L.A. Pathogenesis of human papillomaviruses in differentiating epithelia. Microbiol. Mol. Biol. Rev. 2004, 68, 362–372. [Google Scholar] [CrossRef] [PubMed]
- Pyeon, D.; Pearce, S.M.; Lank, S.M.; Ahlquist, P.; Lambert, P.F. Establishment of human papillomavirus infection requires cell cycle progression. PLoS Pathog. 2009, 5, e1000318. [Google Scholar] [CrossRef] [PubMed]
- Maglennon, G.A.; McIntosh, P.; Doorbar, J. Persistence of viral DNA in the epithelial basal layer suggests a model for papillomavirus latency following immune regression. Virology 2011, 414, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Koster, M.I.; Roop, D.R. Mechanisms regulating epithelial stratification. Annu. Rev. Cell Dev. Biol. 2007, 23, 93–113. [Google Scholar] [CrossRef] [PubMed]
- Favre, M.; Breitburd, F.; Croissant, O.; Orth, G. Chromatin-like structures obtained after alkaline disruption of bovine and human papillomaviruses. J. Virol. 1977, 21, 1205–1209. [Google Scholar] [PubMed]
- Stunkel, W.; Bernard, H.U. The chromatin structure of the long control region of human papillomavirus type 16 represses viral oncoprotein expression. J. Virol. 1999, 73, 1918–1930. [Google Scholar] [PubMed]
- Graham, S.V.; Faizo, A.A. Control of human papillomavirus gene expression by alternative splicing. Virus Res. 2017, 231, 83–95. [Google Scholar] [CrossRef] [PubMed]
- Van Doorslaer, K.; Li, Z.; Xirasagar, S.; Maes, P.; Kaminsky, D.; Liou, D.; Sun, Q.; Kaur, R.; Huyen, Y.; McBride, A.A. The papillomavirus episteme: A major update to the papillomavirus sequence database. Nucleic Acids Res. 2017, 45, D499–D506. [Google Scholar] [CrossRef] [PubMed]
- Geisen, C.; Kahn, T. Promoter activity of sequences located upstream of the human papillomavirus types of 16 and 18 late regions. J. Gen. Virol. 1996, 77, 2193–2200. [Google Scholar] [CrossRef] [PubMed]
- Ozbun, M.A.; Meyers, C. Temporal usage of multiple promoters during the life cycle of human papillomavirus type 31b. J. Virol. 1998, 72, 2715–2722. [Google Scholar] [PubMed]
- Braunstein, T.H.; Madsen, B.S.; Gavnholt, B.; Rosenstierne, M.W.; Johnsen, C.K.; Norrild, B. Identification of a new promoter in the early region of the human papillomavirus type 16 genome. J. Gen. Virol. 1999, 80, 3241–3250. [Google Scholar] [CrossRef] [PubMed]
- Bergvall, M.; Melendy, T.; Archambault, J. The E1 proteins. Virology 2013, 445, 35–56. [Google Scholar] [CrossRef] [PubMed]
- McBride, A.A. The papillomavirus E2 proteins. Virology 2013, 445, 57–79. [Google Scholar] [CrossRef] [PubMed]
- Roman, A.; Munger, K. The papillomavirus E7 proteins. Virology 2013, 445, 138–168. [Google Scholar] [CrossRef] [PubMed]
- Vande Pol, S.B.; Klingelhutz, A.J. Papillomavirus E6 oncoproteins. Virology 2013, 445, 115–137. [Google Scholar] [CrossRef] [PubMed]
- Anacker, D.C.; Moody, C.A. Modulation of the DNA damage response during the life cycle of human papillomaviruses. Virus Res. 2017, 231, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Moody, C.A.; Laimins, L.A. Human papillomavirus oncoproteins: Pathways to transformation. Nat. Rev. Cancer 2010, 10, 550–560. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Laimins, L.A. Manipulation of the innate immune response by human papillomaviruses. Virus Res. 2017, 231, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Doorbar, J. The E4 protein; structure, function and patterns of expression. Virology 2013, 445, 80–98. [Google Scholar] [CrossRef] [PubMed]
- Egawa, N.; Wang, Q.; Griffin, H.M.; Murakami, I.; Jackson, D.; Mahmood, R.; Doorbar, J. HPV16 and 18 genome amplification show different e4-dependence, with 16E4 enhancing E1 nuclear accumulation and replicative efficiency via its cell cycle arrest and kinase activation functions. PLoS Pathog. 2017, 13, e1006282. [Google Scholar] [CrossRef] [PubMed]
- Fang, L.; Budgeon, L.R.; Doorbar, J.; Briggs, E.R.; Howett, M.K. The human papillomavirus type 11 E1^E4 protein is not essential for viral genome amplification. Virology 2006, 351, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Wilson, R.; Fehrmann, F.; Laimins, L.A. Role of the E1^E4 protein in the differentiation-dependent life cycle of human papillomavirus type 31. J. Virol. 2005, 79, 6732–6740. [Google Scholar] [CrossRef] [PubMed]
- Wilson, R.; Ryan, G.B.; Knight, G.L.; Laimins, L.A.; Roberts, S. The full-length E1E4 protein of human papillomavirus type 18 modulates differentiation-dependent viral DNA amplification and late gene expression. Virology 2007, 362, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Fehrmann, F.; Klumpp, D.J.; Laimins, L.A. Human papillomavirus type 31 E5 protein supports cell cycle progression and activates late viral functions upon epithelial differentiation. J. Virol. 2003, 77, 2819–2831. [Google Scholar] [CrossRef] [PubMed]
- Dreer, M.; van de Poel, S.; Stubenrauch, F. Control of viral replication and transcription by the papillomavirus E8^E2 protein. Virus Res. 2017, 231, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Grassmann, K.; Rapp, B.; Maschek, H.; Petry, K.U.; Iftner, T. Identification of a differentiation-inducible promoter in the E7 open reading frame of human papillomavirus type 16 (HPV-16) in raft cultures of a new cell line containing high copy numbers of episomal HPV-16 DNA. J. Virol. 1996, 70, 2339–2349. [Google Scholar] [PubMed]
- Hummel, M.; Hudson, J.B.; Laimins, L.A. Differentiation-induced and constitutive transcription of human papillomavirus type 31B in cell lines containing viral episomes. J. Virol. 1992, 66, 6070–6080. [Google Scholar] [PubMed]
- Klumpp, D.J.; Laimins, L.A. Differentiation-induced changes in promoter usage for transcripts encoding the human papillomavirus type 31 replication protein E1. Virology 1999, 257, 239–246. [Google Scholar] [CrossRef] [PubMed]
- Wooldridge, T.R.; Laimins, L.A. Regulation of human papillomavirus type 31 gene expression during the differentiation-dependent life cycle through histone modifications and transcription factor binding. Virology 2008, 374, 371–380. [Google Scholar] [CrossRef] [PubMed]
- Del Mar Pena, L.M.; Laimins, L.A. Differentiation-dependent chromatin rearrangement coincides with activation of human papillomavirus type 31 late gene expression. J. Virol. 2001, 75, 10005–10013. [Google Scholar] [CrossRef] [PubMed]
- Carson, A.; Khan, S.A. Characterization of transcription factor binding to human papillomavirus type 16 DNA during cellular differentiation. J. Virol. 2006, 80, 4356–4362. [Google Scholar] [CrossRef] [PubMed]
- Gunasekharan, V.; Hache, G.; Laimins, L. Differentiation-dependent changes in levels of C/EBPβ repressors and activators regulate human papillomavirus type 31 late gene expression. J. Virol. 2012, 86, 5393–5398. [Google Scholar] [CrossRef] [PubMed]
- Songock, W.K.; Scott, M.L.; Bodily, J.M. Regulation of the human papillomavirus type 16 late promoter by transcriptional elongation. Virology 2017, 507, 179–191. [Google Scholar] [CrossRef] [PubMed]
- Johansson, C.; Somberg, M.; Li, X.; Backstrom Winquist, E.; Fay, J.; Ryan, F.; Pim, D.; Banks, L.; Schwartz, S. HPV-16 E2 contributes to induction of HPV-16 late gene expression by inhibiting early polyadenylation. EMBO J. 2012, 31, 3212–3227. [Google Scholar] [CrossRef] [PubMed]
- Maitland, N.J.; Conway, S.; Wilkinson, N.S.; Ramsdale, J.; Morris, J.R.; Sanders, C.M.; Burns, J.E.; Stern, P.L.; Wells, M. Expression patterns of the human papillomavirus type 16 transcription factor E2 in low- and high-grade cervical intraepithelial neoplasia. J. Pathol. 1998, 186, 275–280. [Google Scholar] [CrossRef]
- Xue, Y.; Bellanger, S.; Zhang, W.; Lim, D.; Low, J.; Lunny, D.; Thierry, F. HPV16 E2 is an immediate early marker of viral infection, preceding E7 expression in precursor structures of cervical carcinoma. Cancer Res. 2010, 70, 5316–5325. [Google Scholar] [CrossRef] [PubMed]
- Graham, S.V. Keratinocyte differentiation-dependent human papillomavirus gene regulation. Viruses 2017, 9, E245. [Google Scholar] [CrossRef] [PubMed]
- Bentley, D.L. Coupling mRNA processing with transcription in time and space. Nat. Rev. Genet. 2014, 15, 163–175. [Google Scholar] [CrossRef] [PubMed]
- Klymenko, T.; Hernandez-Lopez, H.; MacDonald, A.I.; Bodily, J.M.; Graham, S.V. Human papillomavirus E2 regulates SRSF3 (SRP20) to promote capsid protein expression in infected differentiated keratinocytes. J. Virol. 2016, 90, 5047–5058. [Google Scholar] [CrossRef] [PubMed]
- Somberg, M.; Li, X.; Johansson, C.; Orru, B.; Chang, R.; Rush, M.; Fay, J.; Ryan, F.; Schwartz, S. Serine/arginine-rich protein 30c activates human papillomavirus type 16 L1 mRNA expression via a bimodal mechanism. J. Gen. Virol. 2011, 92, 2411–2421. [Google Scholar] [CrossRef] [PubMed]
- Paris, C.; Pentland, I.; Groves, I.; Roberts, D.C.; Powis, S.J.; Coleman, N.; Roberts, S.; Parish, J.L. CCCTC-binding factor recruitment to the early region of the human papillomavirus 18 genome regulates viral oncogene expression. J. Virol. 2015, 89, 4770–4785. [Google Scholar] [CrossRef] [PubMed]
- Dyson, N.; Howley, P.M.; Munger, K.; Harlow, E. The human papilloma virus-16 E7 oncoprotein is able to bind to the retinoblastoma gene product. Science 1989, 243, 934–937. [Google Scholar] [CrossRef] [PubMed]
- Dyson, N. The regulation of E2F by pRB-family proteins. Genes Dev. 1998, 12, 2245–2262. [Google Scholar] [CrossRef] [PubMed]
- Munger, K.; Werness, B.A.; Dyson, N.; Phelps, W.C.; Harlow, E.; Howley, P.M. Complex formation of human papillomavirus E7 proteins with the retinoblastoma tumor suppressor gene product. EMBO J. 1989, 8, 4099–4105. [Google Scholar] [PubMed]
- Chellappan, S.; Kraus, V.B.; Kroger, B.; Munger, K.; Howley, P.M.; Phelps, W.C.; Nevins, J.R. Adenovirus E1A, simian virus 40 tumor antigen, and human papillomavirus E7 protein share the capacity to disrupt the interaction between transcription factor E2F and the retinoblastoma gene product. Proc. Natl. Acad. Sci. USA 1992, 89, 4549–4553. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.; Schmidt-Grimminger, D.C.; Murant, T.; Broker, T.R.; Chow, L.T. Differentiation-dependent up-regulation of the human papillomavirus E7 gene reactivates cellular DNA replication in suprabasal differentiated keratinocytes. Genes Dev. 1995, 9, 2335–2349. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.Z.; Tsai, S.Y.; Leone, G. Emerging roles of E2Fs in cancer: An exit from cell cycle control. Nat. Rev. Cancer 2009, 9, 785–797. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.L.; Alani, R.M.; Munger, K. The human papillomavirus E7 oncoprotein can uncouple cellular differentiation and proliferation in human keratinocytes by abrogating p21CIP1-mediated inhibition of CDK2. Genes Dev. 1997, 11, 2101–2111. [Google Scholar] [CrossRef] [PubMed]
- Demers, G.W.; Espling, E.; Harry, J.B.; Etscheid, B.G.; Galloway, D.A. Abrogation of growth arrest signals by human papillomavirus type 16 E7 is mediated by sequences required for transformation. J. Virol. 1996, 70, 6862–6869. [Google Scholar] [PubMed]
- Demeter, L.M.; Stoler, M.H.; Broker, T.R.; Chow, L.T. Induction of proliferating cell nuclear antigen in differentiated keratinocytes of human papillomavirus-infected lesions. Hum. Pathol. 1994, 25, 343–348. [Google Scholar] [CrossRef]
- Barrow-Laing, L.; Chen, W.; Roman, A. Low- and high-risk human papillomavirus E7 proteins regulate p130 differently. Virology 2010, 400, 233–239. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Chen, W.; Roman, A. The E7 proteins of low- and high-risk human papillomaviruses share the ability to target the pRB family member p130 for degradation. Proc. Natl. Acad. Sci. USA 2006, 103, 437–442. [Google Scholar] [CrossRef] [PubMed]
- Wilson, R.; Laimins, L.A. Differentiation of HPV-containing cells using organotypic "raft" culture or methylcellulose. Methods Mol. Med. 2005, 119, 157–169. [Google Scholar] [PubMed]
- Flores, E.R.; Allen-Hoffmann, B.L.; Lee, D.; Lambert, P.F. The human papillomavirus type 16 E7 oncogene is required for the productive stage of the viral life cycle. J. Virol. 2000, 74, 6622–6631. [Google Scholar] [CrossRef] [PubMed]
- Longworth, M.S.; Laimins, L.A. The binding of histone deacetylases and the integrity of zinc finger-like motifs of the E7 protein are essential for the life cycle of human papillomavirus type 31. J. Virol. 2004, 78, 3533–3541. [Google Scholar] [CrossRef] [PubMed]
- Longworth, M.S.; Wilson, R.; Laimins, L.A. HPV31 E7 facilitates replication by activating E2F2 transcription through its interaction with HDACs. EMBO J. 2005, 24, 1821–1830. [Google Scholar] [CrossRef] [PubMed]
- Brehm, A.; Nielsen, S.J.; Miska, E.A.; McCance, D.J.; Reid, J.L.; Bannister, A.J.; Kouzarides, T. The E7 oncoprotein associates with MI2 and histone deacetylase activity to promote cell growth. EMBO J. 1999, 18, 2449–2458. [Google Scholar] [CrossRef] [PubMed]
- Missero, C.; Calautti, E.; Eckner, R.; Chin, J.; Tsai, L.H.; Livingston, D.M.; Dotto, G.P. Involvement of the cell-cycle inhibitor CIP1/WAF1 and the E1A-associated p300 protein in terminal differentiation. Proc. Natl. Acad. Sci. USA 1995, 92, 5451–5455. [Google Scholar] [CrossRef] [PubMed]
- Deshpande, A.; Sicinski, P.; Hinds, P.W. Cyclins and CDKs in development and cancer: A perspective. Oncogene 2005, 24, 2909–2915. [Google Scholar] [CrossRef] [PubMed]
- Funk, J.O.; Waga, S.; Harry, J.B.; Espling, E.; Stillman, B.; Galloway, D.A. Inhibition of CDK activity and pCNA-dependent DNA replication by p21 is blocked by interaction with the HPV-16 E7 oncoprotein. Genes Dev. 1997, 11, 2090–2100. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, C.L.; Munger, K. Direct association of the HPV16 E7 oncoprotein with cyclin A/CDK2 and cyclin E/CDK2 complexes. Virology 2008, 380, 21–25. [Google Scholar] [CrossRef] [PubMed]
- Katich, S.C.; Zerfass-Thome, K.; Hoffmann, I. Regulation of the CDC25A gene by the human papillomavirus type 16 E7 oncogene. Oncogene 2001, 20, 543–550. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.X.; Westbrook, T.F.; McCance, D.J. Human papillomavirus type 16 E7 maintains elevated levels of the CDC25A tyrosine phosphatase during deregulation of cell cycle arrest. J. Virol. 2002, 76, 619–632. [Google Scholar] [CrossRef] [PubMed]
- Deng, W.; Lin, B.Y.; Jin, G.; Wheeler, C.G.; Ma, T.; Harper, J.W.; Broker, T.R.; Chow, L.T. Cyclin/CDK regulates the nucleocytoplasmic localization of the human papillomavirus E1 DNA helicase. J. Virol. 2004, 78, 13954–13965. [Google Scholar] [CrossRef] [PubMed]
- Fradet-Turcotte, A.; Moody, C.; Laimins, L.A.; Archambault, J. Nuclear export of human papillomavirus type 31 E1 is regulated by CDK2 phosphorylation and required for viral genome maintenance. J. Virol. 2010, 84, 11747–11760. [Google Scholar] [CrossRef] [PubMed]
- Genther, S.M.; Sterling, S.; Duensing, S.; Munger, K.; Sattler, C.; Lambert, P.F. Quantitative role of the human papillomavirus type 16 E5 gene during the productive stage of the viral life cycle. J. Virol. 2003, 77, 2832–2842. [Google Scholar] [CrossRef] [PubMed]
- Regan, J.A.; Laimins, L.A. Bap31 is a novel target of the human papillomavirus E5 protein. J. Virol. 2008, 82, 10042–10051. [Google Scholar] [CrossRef] [PubMed]
- DiMaio, D.; Petti, L.M. The E5 proteins. Virology 2013, 445, 99–114. [Google Scholar] [CrossRef] [PubMed]
- Huibregtse, J.M.; Scheffner, M.; Howley, P.M. A cellular protein mediates association of p53 with the E6 oncoprotein of human papillomavirus types 16 or 18. EMBO J. 1991, 10, 4129–4135. [Google Scholar] [PubMed]
- Scheffner, M.; Werness, B.A.; Huibregtse, J.M.; Levine, A.J.; Howley, P.M. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell 1990, 63, 1129–1136. [Google Scholar] [CrossRef]
- Scheffner, M.; Huibregtse, J.M.; Vierstra, R.D.; Howley, P.M. The HPV-16 E6 and E6-AP complex functions as a ubiquitin-protein ligase in the ubiquitination of p53. Cell 1993, 75, 495–505. [Google Scholar] [CrossRef]
- Kho, E.Y.; Wang, H.K.; Banerjee, N.S.; Broker, T.R.; Chow, L.T. HPV-18 E6 mutants reveal p53 modulation of viral DNA amplification in organotypic cultures. Proc. Natl. Acad. Sci. USA 2013, 110, 7542–7549. [Google Scholar] [CrossRef] [PubMed]
- Massimi, P.; Pim, D.; Bertoli, C.; Bouvard, V.; Banks, L. Interaction between the HPV-16 E2 transcriptional activator and p53. Oncogene 1999, 18, 7748–7754. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.; Kowalczyk, A.M.; Taylor, E.R.; Morgan, I.M.; Gaston, K. p53 represses human papillomavirus type 16 DNA replication via the viral E2 protein. Virol. J. 2008, 5, 5. [Google Scholar] [CrossRef] [PubMed]
- Ganti, K.; Broniarczyk, J.; Manoubi, W.; Massimi, P.; Mittal, S.; Pim, D.; Szalmas, A.; Thatte, J.; Thomas, M.; Tomaic, V.; et al. The human papillomavirus E6 PDZ binding motif: From life cycle to malignancy. Viruses 2015, 7, 3530–3551. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.S.; Weiss, R.S.; Javier, R.T. Binding of human virus oncoproteins to HDLG/Sap97, a mammalian homolog of the drosophila discs large tumor suppressor protein. Proc. Natl. Acad. Sci. USA 1997, 94, 6670–6675. [Google Scholar] [CrossRef] [PubMed]
- Kiyono, T.; Hiraiwa, A.; Fujita, M.; Hayashi, Y.; Akiyama, T.; Ishibashi, M. Binding of high-risk human papillomavirus E6 oncoproteins to the human homologue of the drosophila discs large tumor suppressor protein. Proc. Natl. Acad. Sci. USA 1997, 94, 11612–11616. [Google Scholar] [CrossRef] [PubMed]
- Delury, C.P.; Marsh, E.K.; James, C.D.; Boon, S.S.; Banks, L.; Knight, G.L.; Roberts, S. The role of protein kinase a regulation of the E6 PDZ-binding domain during the differentiation-dependent life cycle of human papillomavirus type 18. J. Virol. 2013, 87, 9463–9472. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Laimins, L.A. Role of the PDZ domain-binding motif of the oncoprotein E6 in the pathogenesis of human papillomavirus type 31. J. Virol. 2004, 78, 12366–12377. [Google Scholar] [CrossRef] [PubMed]
- Nicolaides, L.; Davy, C.; Raj, K.; Kranjec, C.; Banks, L.; Doorbar, J. Stabilization of HPV16 E6 protein by PDZ proteins, and potential implications for genome maintenance. Virology 2011, 414, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Marsh, E.K.; Delury, C.P.; Davies, N.J.; Weston, C.J.; Miah, M.A.L.; Banks, L.; Parish, J.L.; Higgs, M.R.; Roberts, S. Mitotic control of human papillomavirus genome-containing cells is regulated by the function of the PDZ-binding motif of the E6 oncoprotein. Oncotarget 2017, 8, 19491–19506. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Li, G.; Laimins, L.A.; Cullen, B.R. Human papillomavirus genotype 31 does not express detectable microRNA levels during latent or productive virus replication. J. Virol. 2006, 80, 10890–10893. [Google Scholar] [CrossRef] [PubMed]
- Yi, R.; Poy, M.N.; Stoffel, M.; Fuchs, E. A skin microrna promotes differentiation by repressing ‘stemness’. Nature 2008, 452, 225–229. [Google Scholar] [CrossRef] [PubMed]
- Melino, G.; Memmi, E.M.; Pelicci, P.G.; Bernassola, F. Maintaining epithelial stemness with p63. Sci. Signal. 2015, 8, 9. [Google Scholar] [CrossRef] [PubMed]
- Mighty, K.K.; Laimins, L.A. P63 is necessary for the activation of human papillomavirus late viral functions upon epithelial differentiation. J. Virol. 2011, 85, 8863–8869. [Google Scholar] [CrossRef] [PubMed]
- Melar-New, M.; Laimins, L.A. Human papillomaviruses modulate expression of microRNA 203 upon epithelial differentiation to control levels of p63 proteins. J. Virol. 2010, 84, 5212–5221. [Google Scholar] [CrossRef] [PubMed]
- Gunasekharan, V.; Laimins, L.A. Human papillomaviruses modulate microrna 145 expression to directly control genome amplification. J. Virol. 2013, 87, 6037–6043. [Google Scholar] [CrossRef] [PubMed]
- Sen, G.L.; Boxer, L.D.; Webster, D.E.; Bussat, R.T.; Qu, K.; Zarnegar, B.J.; Johnston, D.; Siprashvili, Z.; Khavari, P.A. Znf750 is a p63 target gene that induces KLF4 to drive terminal epidermal differentiation. Dev. Cell. 2012, 22, 669–677. [Google Scholar] [CrossRef] [PubMed]
- Ghaleb, A.M.; Yang, V.W. Kruppel-like factor 4 (KLF4): What we currently know. Gene 2017, 611, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Gunasekharan, V.K.; Li, Y.; Andrade, J.; Laimins, L.A. Post-transcriptional regulation of KLF4 by high-risk human papillomaviruses is necessary for the differentiation-dependent viral life cycle. PLoS Pathog. 2016, 12, e1005747. [Google Scholar] [CrossRef] [PubMed]
- Moody, C.A.; Laimins, L.A. Human papillomaviruses activate the ATM DNA damage pathway for viral genome amplification upon differentiation. PLoS Pathog. 2009, 5, e1000605. [Google Scholar] [CrossRef] [PubMed]
- Chappell, W.H.; Gautam, D.; Ok, S.T.; Johnson, B.A.; Anacker, D.C.; Moody, C.A. Homologous recombination repair factors RAD51 and BRCA1 are necessary for productive replication of human papillomavirus 31. J. Virol. 2015, 90, 2639–2652. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, N.S.; Genovese, N.J.; Noya, F.; Chien, W.M.; Broker, T.R.; Chow, L.T. Conditionally activated E7 proteins of high-risk and low-risk human papillomaviruses induce S phase in postmitotic, differentiated human keratinocytes. J. Virol. 2006, 80, 6517–6524. [Google Scholar] [CrossRef] [PubMed]
- Genovese, N.J.; Banerjee, N.S.; Broker, T.R.; Chow, L.T. Casein kinase II motif-dependent phosphorylation of human papillomavirus E7 protein promotes p130 degradation and S-phase induction in differentiated human keratinocytes. J. Virol. 2008, 82, 4862–4873. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, N.S.; Wang, H.K.; Broker, T.R.; Chow, L.T. Human papillomavirus (HPV) E7 induces prolonged G2 following S phase reentry in differentiated human keratinocytes. J. Biol. Chem. 2011, 286, 15473–15482. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.K.; Duffy, A.A.; Broker, T.R.; Chow, L.T. Robust production and passaging of infectious HPV in squamous epithelium of primary human keratinocytes. Genes Dev. 2009, 23, 181–194. [Google Scholar] [CrossRef] [PubMed]
- Nakahara, T.; Peh, W.L.; Doorbar, J.; Lee, D.; Lambert, P.F. Human papillomavirus type 16 e1circumflexe4 contributes to multiple facets of the papillomavirus life cycle. J. Virol. 2005, 79, 13150–13165. [Google Scholar] [CrossRef] [PubMed]
- Stark, G.R.; Taylor, W.R. Control of the G2/M transition. Mol. Biotechnol. 2006, 32, 227–248. [Google Scholar] [CrossRef]
- Kousholt, A.N.; Menzel, T.; Sorensen, C.S. Pathways for genome integrity in G2 phase of the cell cycle. Biomolecules 2012, 2, 579–607. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.B.; Elledge, S.J. The DNA damage response: Putting checkpoints in perspective. Nature 2000, 408, 433–439. [Google Scholar] [PubMed]
- Hong, S.; Cheng, S.; Iovane, A.; Laimins, L.A. STAT-5 regulates transcription of the topoisomerase IIβ-binding protein 1 (TOPBP1) gene to activate the ATR pathway and promote human papillomavirus replication. MBio 2015, 6, e02006–02015. [Google Scholar] [CrossRef] [PubMed]
- Anacker, D.C.; Aloor, H.L.; Shepard, C.N.; Lenzi, G.M.; Johnson, B.A.; Kim, B.; Moody, C.A. HPV31 utilizes the ATR-CHK1 pathway to maintain elevated RRM2 levels and a replication-competent environment in differentiating keratinocytes. Virology 2016, 499, 383–396. [Google Scholar] [CrossRef] [PubMed]
- Davy, C.E.; Jackson, D.J.; Wang, Q.; Raj, K.; Masterson, P.J.; Fenner, N.F.; Southern, S.; Cuthill, S.; Millar, J.B.; Doorbar, J. Identification of a G(2) arrest domain in the E1 wedge E4 protein of human papillomavirus type 16. J. Virol. 2002, 76, 9806–9818. [Google Scholar] [CrossRef] [PubMed]
- Nakahara, T.; Nishimura, A.; Tanaka, M.; Ueno, T.; Ishimoto, A.; Sakai, H. Modulation of the cell division cycle by human papillomavirus type 18 E4. J. Virol. 2002, 76, 10914–10920. [Google Scholar] [CrossRef] [PubMed]
- Knight, G.L.; Turnell, A.S.; Roberts, S. Role for wee1 in inhibition of G2-to-M transition through the cooperation of distinct human papillomavirus type 1 E4 proteins. J. Virol. 2006, 80, 7416–7426. [Google Scholar] [CrossRef] [PubMed]
- Davy, C.E.; Jackson, D.J.; Raj, K.; Peh, W.L.; Southern, S.A.; Das, P.; Sorathia, R.; Laskey, P.; Middleton, K.; Nakahara, T.; et al. Human papillomavirus type 16 E1 E4-induced G2 arrest is associated with cytoplasmic retention of active CDK1/cyclin B1 complexes. J. Virol. 2005, 79, 3998–4011. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Kennedy, A.; Das, P.; McIntosh, P.B.; Howell, S.A.; Isaacson, E.R.; Hinz, S.A.; Davy, C.; Doorbar, J. Phosphorylation of the human papillomavirus type 16 E1-E4 protein at t57 by ERK triggers a structural change that enhances keratin binding and protein stability. J. Virol. 2009, 83, 3668–3683. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.H.; Lin, B.Y.; Deng, W.; Broker, T.R.; Chow, L.T. Mitogen-activated protein kinases activate the nuclear localization sequence of human papillomavirus type L1 E1 DNA helicase to promote efficient nuclear import. J. Virol. 2007, 81, 5066–5078. [Google Scholar] [CrossRef] [PubMed]
- McIntosh, P.B.; Laskey, P.; Sullivan, K.; Davy, C.; Wang, Q.; Jackson, D.J.; Griffin, H.M.; Doorbar, J. E1-E4-mediated keratin phosphorylation and ubiquitylation: A mechanism for keratin depletion in HPV16-infected epithelium. J. Cell. Sci. 2010, 123, 2810–2822. [Google Scholar] [CrossRef] [PubMed]
- DeSmet, M.; Kanginakudru, S.; Rietz, A.; Wu, W.H.; Roden, R.; Androphy, E.J. The replicative consequences of papillomavirus E2 protein binding to the origin replication factor ORC2. PLoS Pathog. 2016, 12, e1005934. [Google Scholar] [CrossRef] [PubMed]
- Sakakibara, N.; Chen, D.; McBride, A.A. Papillomaviruses use recombination-dependent replication to vegetatively amplify their genomes in differentiated cells. PLoS Pathog. 2013, 9, e1003321. [Google Scholar] [CrossRef] [PubMed]
- Ceccaldi, R.; Rondinelli, B.; D’Andrea, A.D. Repair pathway choices and consequences at the double-strand break. Trends Cell Biol. 2016, 26, 52–64. [Google Scholar] [CrossRef] [PubMed]
- Ciccia, A.; Elledge, S.J. The DNA damage response: Making it safe to play with knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef] [PubMed]
- Cimprich, K.A.; Cortez, D. ATR: An essential regulator of genome integrity. Nat. Rev. Mol. Cell Biol. 2008, 9, 616–627. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Paull, T.T. ATM activation by DNA double-strand breaks through the MRE11-RAD50-NBS1 complex. Science 2005, 308, 551–554. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Jiang, X.; Chen, S.; Fernandes, N.; Price, B.D. A role for the TIP60 histone acetyltransferase in the acetylation and activation of ATM. Proc. Natl. Acad. Sci. USA 2005, 102, 13182–13187. [Google Scholar] [CrossRef] [PubMed]
- Williams, R.S.; Williams, J.S.; Tainer, J.A. MRE11-RAD50-NBS1 is a keystone complex connecting DNA repair machinery, double-strand break signaling, and the chromatin template. Biochem. Cell Biol. 2007, 85, 509–520. [Google Scholar] [CrossRef] [PubMed]
- Bakkenist, C.J.; Kastan, M.B. Chromatin perturbations during the DNA damage response in higher eukaryotes. DNA Repair (Amst) 2015, 36, 8–12. [Google Scholar] [CrossRef] [PubMed]
- Reinhardt, H.C.; Yaffe, M.B. Kinases that control the cell cycle in response to DNA damage: CHK1, CHK2, and MK2. Curr. Opin. Cell Biol. 2009, 21, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Donzelli, M.; Draetta, G.F. Regulating mammalian checkpoints through CDC25 inactivation. EMBO Rep. 2003, 4, 671–677. [Google Scholar] [CrossRef] [PubMed]
- Yazdi, P.T.; Wang, Y.; Zhao, S.; Patel, N.; Lee, E.Y.; Qin, J. SMC1 is a downstream effector in the ATM/NBS1 branch of the human S-phase checkpoint. Genes Dev. 2002, 16, 571–582. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, A.R. The role of SMC proteins in the responses to DNA damage. DNA Repair (Amst) 2005, 4, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Satsuka, A.; Mehta, K.; Laimins, L. p38MAPK and MK2 pathways are important for the differentiation-dependent human papillomavirus life cycle. J. Virol. 2015, 89, 1919–1924. [Google Scholar] [CrossRef] [PubMed]
- Mehta, K.; Gunasekharan, V.; Satsuka, A.; Laimins, L.A. Human papillomaviruses activate and recruit SMC1 cohesin proteins for the differentiation-dependent life cycle through association with CTCF insulators. PLoS Pathog. 2015, 11, e1004763. [Google Scholar] [CrossRef] [PubMed]
- Sakakibara, N.; Mitra, R.; McBride, A.A. The papillomavirus E1 helicase activates a cellular DNA damage response in viral replication foci. J. Virol. 2011, 85, 8981–8995. [Google Scholar] [CrossRef] [PubMed]
- Anacker, D.C.; Gautam, D.; Gillespie, K.A.; Chappell, W.H.; Moody, C.A. Productive replication of human papillomavirus 31 requires DNA repair factor NBS1. J. Virol. 2014, 88, 8528–8544. [Google Scholar] [CrossRef] [PubMed]
- Moody, C.A.; Fradet-Turcotte, A.; Archambault, J.; Laimins, L.A. Human papillomaviruses activate caspases upon epithelial differentiation to induce viral genome amplification. Proc. Natl. Acad. Sci. USA 2007, 104, 19541–19546. [Google Scholar] [CrossRef] [PubMed]
- Sakakibara, N.; Chen, D.; Jang, M.K.; Kang, D.W.; Luecke, H.F.; Wu, S.Y.; Chiang, C.M.; McBride, A.A. Brd4 is displaced from HPV replication factories as they expand and amplify viral DNA. PLoS Pathog. 2013, 9, e1003777. [Google Scholar] [CrossRef] [PubMed]
- Gillespie, K.A.; Mehta, K.P.; Laimins, L.A.; Moody, C.A. Human papillomaviruses recruit cellular DNA repair and homologous recombination factors to viral replication centers. J. Virol. 2012, 86, 9520–9526. [Google Scholar] [CrossRef] [PubMed]
- Langsfeld, E.S.; Bodily, J.M.; Laimins, L.A. The deacetylase sirtuin 1 regulates human papillomavirus replication by modulating histone acetylation and recruitment of DNA damage factors NBS1 and RAD51 to viral genomes. PLoS Pathog. 2015, 11, e1005181. [Google Scholar] [CrossRef] [PubMed]
- Flores, E.R.; Lambert, P.F. Evidence for a switch in the mode of human papillomavirus type 16 DNA replication during the viral life cycle. J. Virol. 1997, 71, 7167–7179. [Google Scholar] [PubMed]
- Hirano, T. SMC proteins and chromosome mechanics: From bacteria to humans. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2005, 360, 507–514. [Google Scholar] [CrossRef] [PubMed]
- Johnson, B.A.; Aloor, H.L.; Moody, C.A. The RB binding domain of Hpv31 E7 is required to maintain high levels of DNA repair factors in infected cells. Virology 2017, 500, 22–34. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Laimins, L.A. The JAK-STAT transcriptional regulator, STAT-5, activates the ATM DNA damage pathway to induce Hpv 31 genome amplification upon epithelial differentiation. PLoS Pathog. 2013, 9, e1003295. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Dutta, A.; Laimins, L.A. The acetyltransferase tip60 is a critical regulator of the differentiation-dependent amplification of human papillomaviruses. J. Virol. 2015, 89, 4668–4675. [Google Scholar] [CrossRef] [PubMed]
- Reinson, T.; Toots, M.; Kadaja, M.; Pipitch, R.; Allik, M.; Ustav, E.; Ustav, M. Engagement of the ATR-dependent DNA damage response at the human papillomavirus 18 replication centers during the initial amplification. J. Virol. 2013, 87, 951–964. [Google Scholar] [CrossRef] [PubMed]
- Fradet-Turcotte, A.; Bergeron-Labrecque, F.; Moody, C.A.; Lehoux, M.; Laimins, L.A.; Archambault, J. Nuclear accumulation of the papillomavirus E1 helicase blocks S-phase progression and triggers an ATM-dependent DNA damage response. J. Virol. 2011, 85, 8996–9012. [Google Scholar] [CrossRef] [PubMed]
- Gauson, E.J.; Donaldson, M.M.; Dornan, E.S.; Wang, X.; Bristol, M.; Bodily, J.M.; Morgan, I.M. Evidence supporting a role for TOPBP1 and BRD4 in the initiation but not continuation of human papillomavirus 16 E1/E2-mediated DNA replication. J. Virol. 2015, 89, 4980–4991. [Google Scholar] [CrossRef] [PubMed]
- Galloway, D.A.; Laimins, L.A. Human papillomaviruses: Shared and distinct pathways for pathogenesis. Curr. Opin. Virol. 2015, 14, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Papamichos-Chronakis, M.; Peterson, C.L. Chromatin and the genome integrity network. Nat. Rev. Genet. 2013, 14, 62–75. [Google Scholar] [CrossRef] [PubMed]
- Polo, S.E.; Jackson, S.P. Dynamics of DNA damage response proteins at DNA breaks: A focus on protein modifications. Genes Dev. 2011, 25, 409–433. [Google Scholar] [CrossRef] [PubMed]
- Van Attikum, H.; Gasser, S.M. Crosstalk between histone modifications during the DNA damage response. Trends Cell Biol. 2009, 19, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Gautam, D.; Moody, C.A. Impact of the DNA damage response on human papillomavirus chromatin. PLoS Pathog. 2016, 12, e1005613. [Google Scholar] [CrossRef] [PubMed]
- Oberdoerffer, P.; Michan, S.; McVay, M.; Mostoslavsky, R.; Vann, J.; Park, S.K.; Hartlerode, A.; Stegmuller, J.; Hafner, A.; Loerch, P.; et al. SIRT1 redistribution on chromatin promotes genomic stability but alters gene expression during aging. Cell 2008, 135, 907–918. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Cho, N.W.; Cui, G.; Manion, E.M.; Shanbhag, N.M.; Botuyan, M.V.; Mer, G.; Greenberg, R.A. Acetylation limits 53BP1 association with damaged chromatin to promote homologous recombination. Nat. Struct. Mol. Biol. 2013, 20, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Saldivar, J.C.; Cortez, D.; Cimprich, K.A. The essential kinase ATR: Ensuring faithful duplication of a challenging genome. Nat. Rev. Mol. Cell Biol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Spardy, N.; Covella, K.; Cha, E.; Hoskins, E.E.; Wells, S.I.; Duensing, A.; Duensing, S. Human papillomavirus 16 E7 oncoprotein attenuates DNA damage checkpoint control by increasing the proteolytic turnover of claspin. Cancer Res. 2009, 69, 7022–7029. [Google Scholar] [CrossRef] [PubMed]
- Bester, A.C.; Roniger, M.; Oren, Y.S.; Im, M.M.; Sarni, D.; Chaoat, M.; Bensimon, A.; Zamir, G.; Shewach, D.S.; Kerem, B. Nucleotide deficiency promotes genomic instability in early stages of cancer development. Cell 2011, 145, 435–446. [Google Scholar] [CrossRef] [PubMed]
- Ellison, V.; Stillman, B. Biochemical characterization of DNA damage checkpoint complexes: Clamp loader and clamp complexes with specificity for 5’ recessed DNA. PLoS Biol. 2003, 1, E33. [Google Scholar] [CrossRef] [PubMed]
- Zou, L.; Liu, D.; Elledge, S.J. Replication protein a-mediated recruitment and activation of RAD17 complexes. Proc. Natl. Acad. Sci. USA 2003, 100, 13827–13832. [Google Scholar] [CrossRef] [PubMed]
- Mordes, D.A.; Glick, G.G.; Zhao, R.; Cortez, D. TOPBP1 activates ATR through ATRIP and a PIKK regulatory domain. Genes Dev. 2008, 22, 1478–1489. [Google Scholar] [CrossRef] [PubMed]
- Edwards, T.G.; Helmus, M.J.; Koeller, K.; Bashkin, J.K.; Fisher, C. Human papillomavirus episome stability is reduced by aphidicolin and controlled by DNA damage response pathways. J. Virol. 2013, 87, 3979–3989. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Opiyo, S.O.; Manthey, K.; Glanzer, J.G.; Ashley, A.K.; Amerin, C.; Troksa, K.; Shrivastav, M.; Nickoloff, J.A.; Oakley, G.G. Distinct roles for DNA-PK, ATM and ATR in RPA phosphorylation and checkpoint activation in response to replication stress. Nucleic Acids Res. 2012, 40, 10780–10794. [Google Scholar] [CrossRef] [PubMed]
- Bertoli, C.; Klier, S.; McGowan, C.; Wittenberg, C.; de Bruin, R.A. CHK1 inhibits E2F6 repressor function in response to replication stress to maintain cell-cycle transcription. Curr. Biol. 2013, 23, 1629–1637. [Google Scholar] [CrossRef] [PubMed]
- Buisson, R.; Boisvert, J.L.; Benes, C.H.; Zou, L. Distinct but concerted roles of ATR, DNA-PK, and CHK1 in countering replication stress during S phase. Mol. Cell 2015, 59, 1011–1024. [Google Scholar] [CrossRef] [PubMed]
- Murga, M.; Campaner, S.; Lopez-Contreras, A.J.; Toledo, L.I.; Soria, R.; Montana, M.F.; D'Artista, L.; Schleker, T.; Guerra, C.; Garcia, E.; et al. Exploiting oncogene-induced replicative stress for the selective killing of Myc-driven tumors. Nat. Struct. Mol. Biol. 2011, 18, 1331–1335. [Google Scholar] [CrossRef] [PubMed]
- Toledo, L.I.; Murga, M.; Zur, R.; Soria, R.; Rodriguez, A.; Martinez, S.; Oyarzabal, J.; Pastor, J.; Bischoff, J.R.; Fernandez-Capetillo, O. A cell-based screen identifies ATR inhibitors with synthetic lethal properties for cancer-associated mutations. Nat. Struct. Mol. Biol. 2011, 18, 721–727. [Google Scholar] [CrossRef] [PubMed]
- Nordlund, P.; Reichard, P. Ribonucleotide reductases. Annu. Rev. Biochem. 2006, 75, 681–706. [Google Scholar] [CrossRef] [PubMed]
- Jang, M.K.; Shen, K.; McBride, A.A. Papillomavirus genomes associate with BRD4 to replicate at fragile sites in the host genome. PLoS Pathog. 2014, 10, e1004117. [Google Scholar] [CrossRef] [PubMed]
- Sarni, D.; Kerem, B. The complex nature of fragile site plasticity and its importance in cancer. Curr. Opin. Cell Biol. 2016, 40, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Gao, G.; Johnson, S.H.; Vasmatzis, G.; Pauley, C.E.; Tombers, N.M.; Kasperbauer, J.L.; Smith, D.I. Common fragile sites (CFS) and extremely large CFS genes are targets for human papillomavirus integrations and chromosome rearrangements in oropharyngeal squamous cell carcinoma. Genes Chromosomes Cancer 2017, 56, 59–74. [Google Scholar] [CrossRef] [PubMed]
- Bodelon, C.; Untereiner, M.E.; Machiela, M.J.; Vinokurova, S.; Wentzensen, N. Genomic characterization of viral integration sites in HPV-related cancers. Int. J. Cancer 2016, 139, 2001–2011. [Google Scholar] [CrossRef] [PubMed]
- Thorland, E.C.; Myers, S.L.; Persing, D.H.; Sarkar, G.; McGovern, R.M.; Gostout, B.S.; Smith, D.I. Human papillomavirus type 16 integrations in cervical tumors frequently occur in common fragile sites. Cancer Res. 2000, 60, 5916–5921. [Google Scholar] [PubMed]
- Choo, K.B.; Chen, C.M.; Han, C.P.; Cheng, W.T.; Au, L.C. Molecular analysis of cellular loci disrupted by papillomavirus 16 integration in cervical cancer: Frequent viral integration in topologically destabilized and transcriptionally active chromosomal regions. J. Med. Virol. 1996, 49, 15–22. [Google Scholar] [CrossRef]
- McBride, A.A.; Warburton, A. The role of integration in oncogenic progression of HPV-associated cancers. PLoS Pathog. 2017, 13, e1006211. [Google Scholar] [CrossRef] [PubMed]
- McBride, A.A. Playing with fire: Consequences of human papillomavirus DNA replication adjacent to genetically unstable regions of host chromatin. Curr. Opin. Virol. 2017, 26, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Wallace, N.A.; Khanal, S.; Robinson, K.L.; Wendel, S.O.; Messer, J.J.; Galloway, D.A. High risk α papillomavirus oncogenes impair the homologous recombination pathway. J. Virol. 2017. [Google Scholar] [CrossRef] [PubMed]





© 2017 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moody, C. Mechanisms by which HPV Induces a Replication Competent Environment in Differentiating Keratinocytes. Viruses 2017, 9, 261. https://doi.org/10.3390/v9090261
Moody C. Mechanisms by which HPV Induces a Replication Competent Environment in Differentiating Keratinocytes. Viruses. 2017; 9(9):261. https://doi.org/10.3390/v9090261
Chicago/Turabian StyleMoody, Cary. 2017. "Mechanisms by which HPV Induces a Replication Competent Environment in Differentiating Keratinocytes" Viruses 9, no. 9: 261. https://doi.org/10.3390/v9090261
APA StyleMoody, C. (2017). Mechanisms by which HPV Induces a Replication Competent Environment in Differentiating Keratinocytes. Viruses, 9(9), 261. https://doi.org/10.3390/v9090261