PERK Signal-Modulated Protein Translation Promotes the Survivability of Dengue 2 Virus-Infected Mosquito Cells and Extends Viral Replication


Abstract
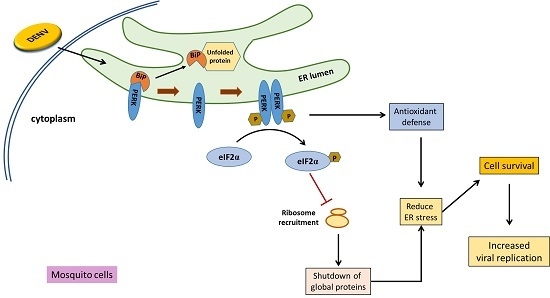
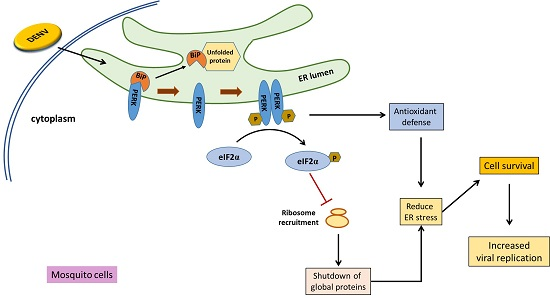
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Virus Propagation and Cell Culture
2.2. RNA Extraction and Reverse Transcription
2.3. Quantitative Real-Time PCR
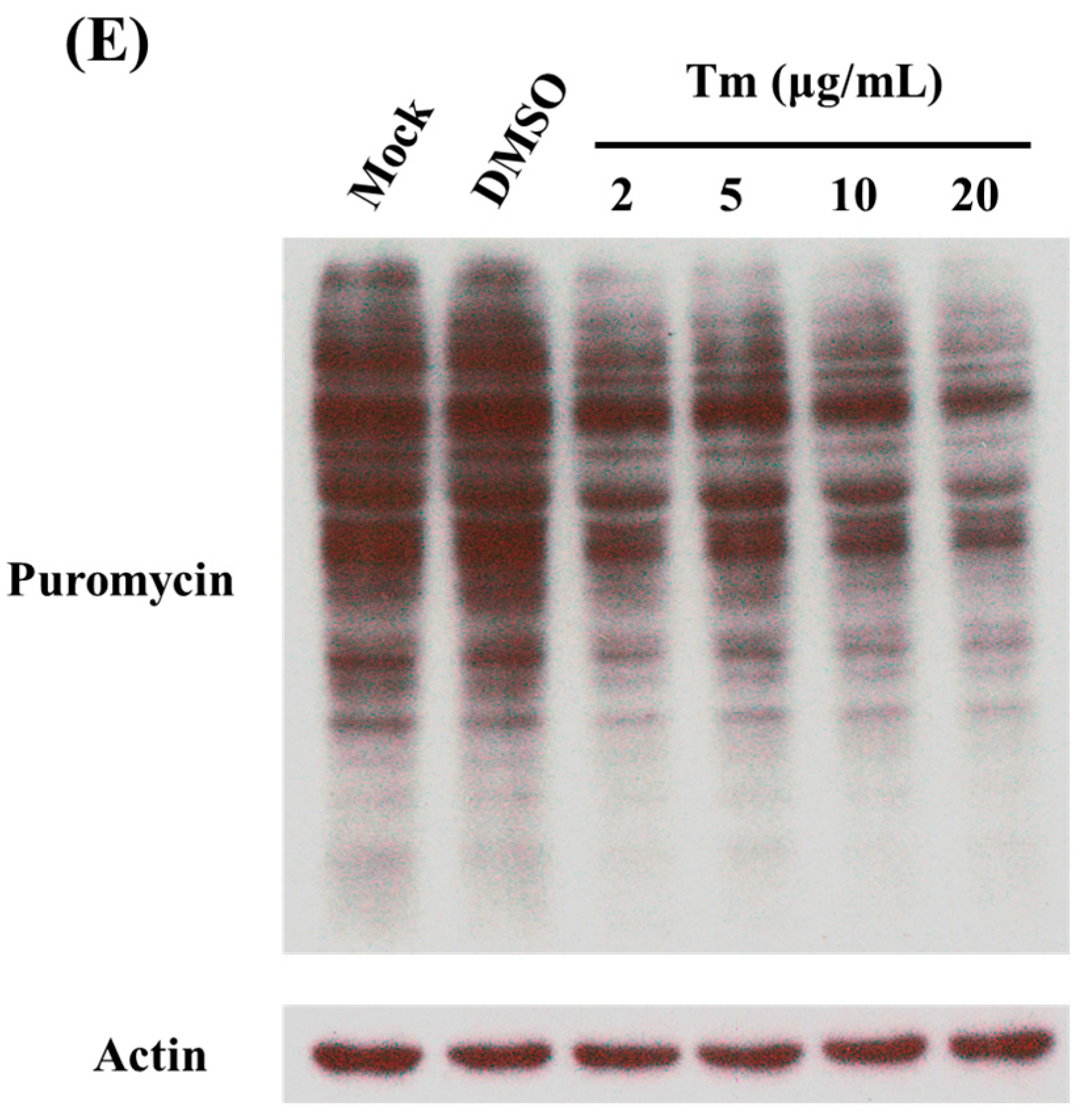
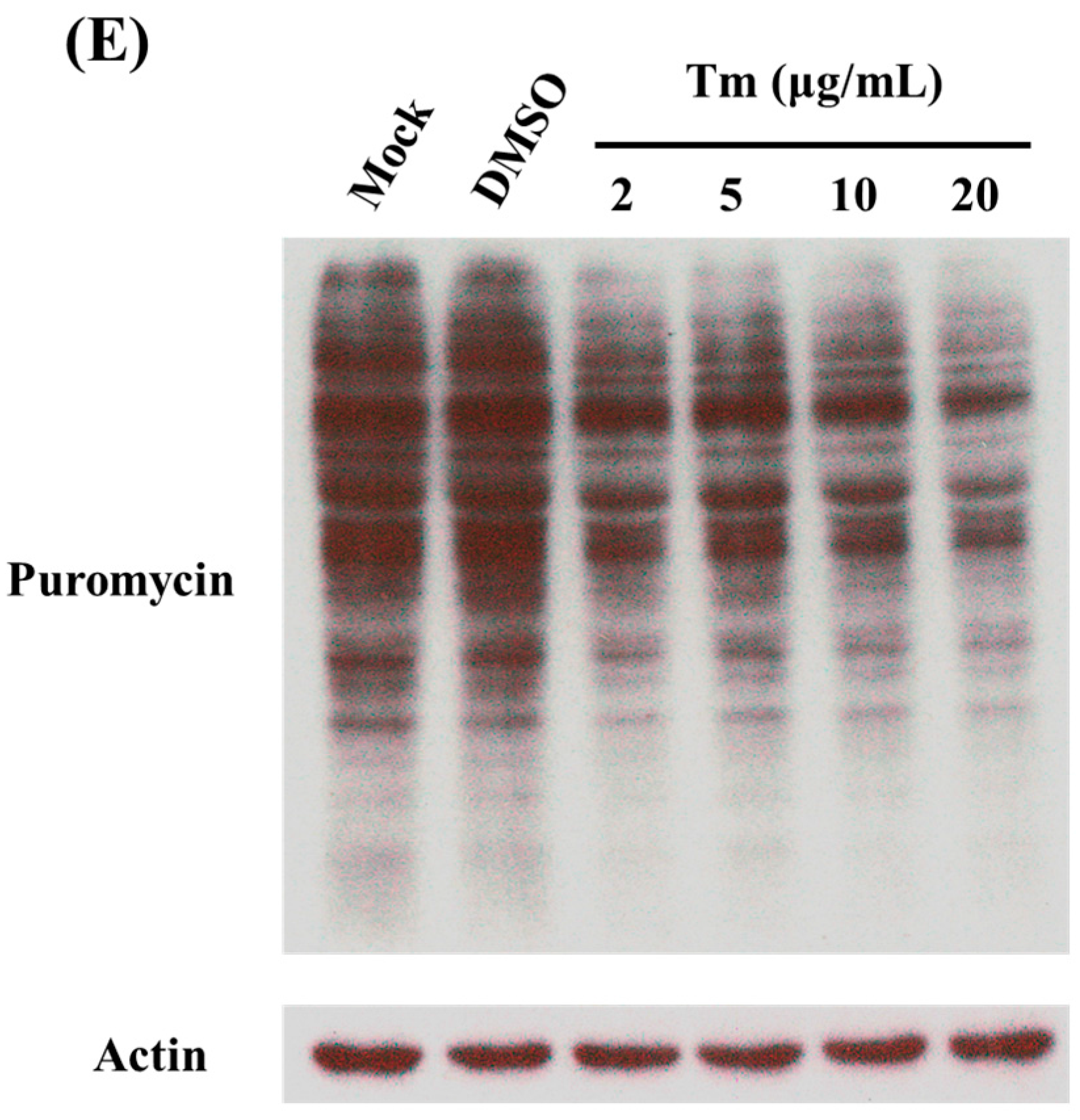
2.4. Treatment of C6/36 Cells with Tunicamycin
2.5. Surface Sensing of Translation Used to Measure Newly Synthesized Proteins
2.6. Cap Pull-Down Assay
2.7. Western Blotting
2.8. Treatment of C6/36 Cells with a PERK Inhibitor
2.9. Analysis of the Mitochondrial Membrane Potential
2.10. Measurement of Intracellular Reactive Oxygen Species
2.11. Assay for Measuring the Apoptosis Rate
2.12. Measurement of Caspase-3 and -9 Activities
2.13. PI Staining to Detect Cell Death
2.14. Double Luciferase Reporter Assay
2.15. Statistical Analysis
3. Results
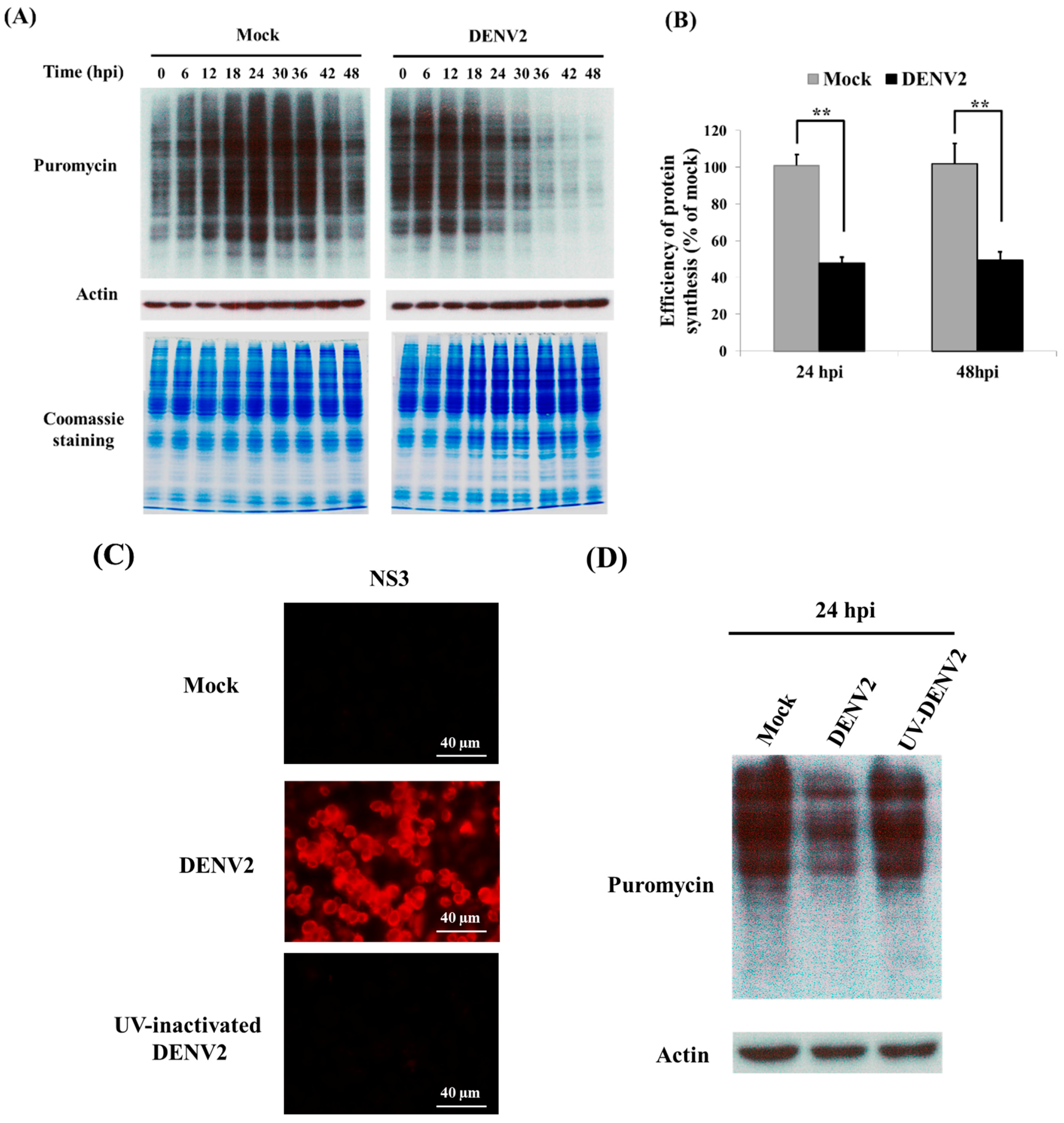
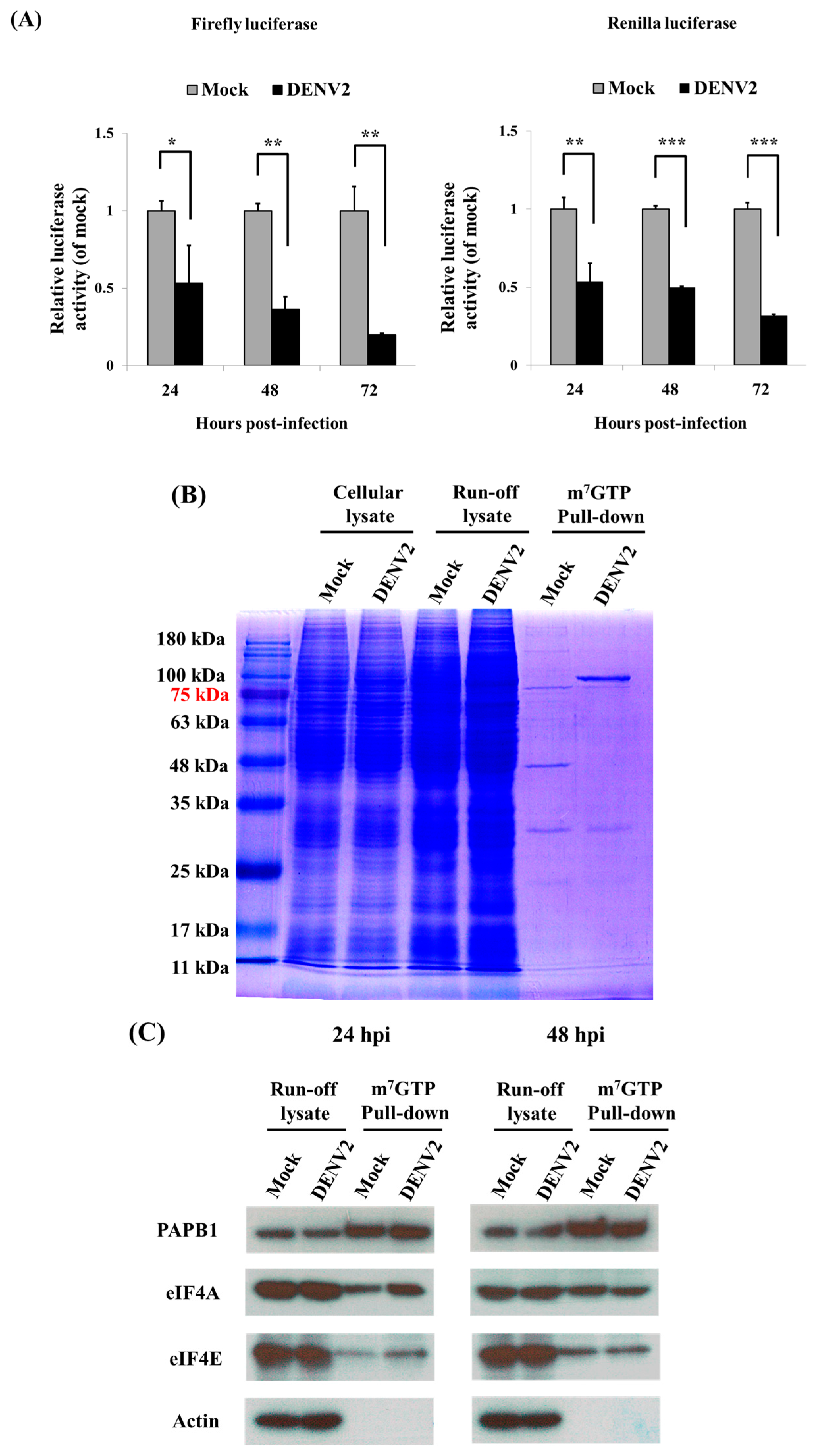
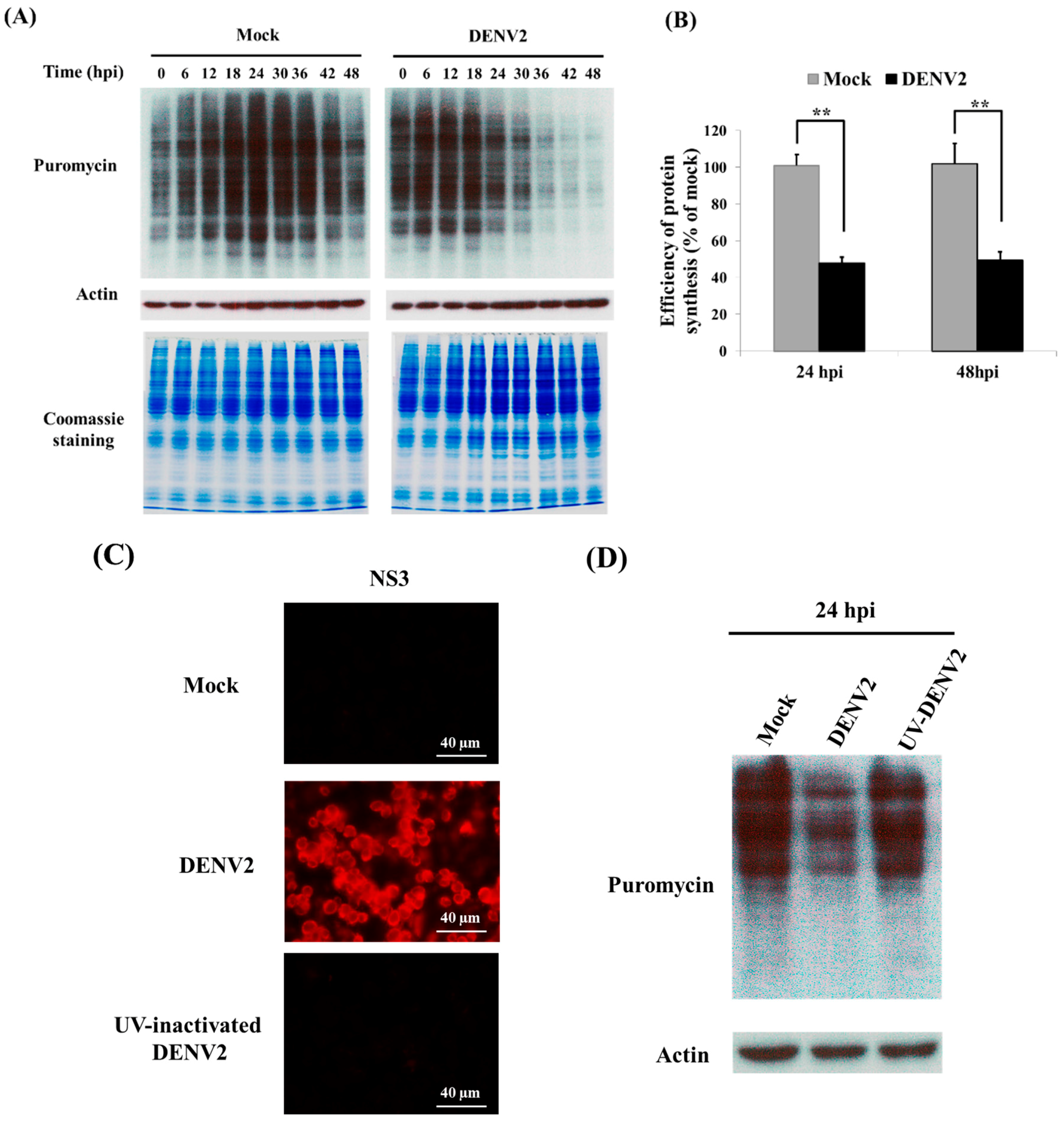
3.1. Shutdown of Protein Synthesis in C6/36 Cells with DENV2 Infection
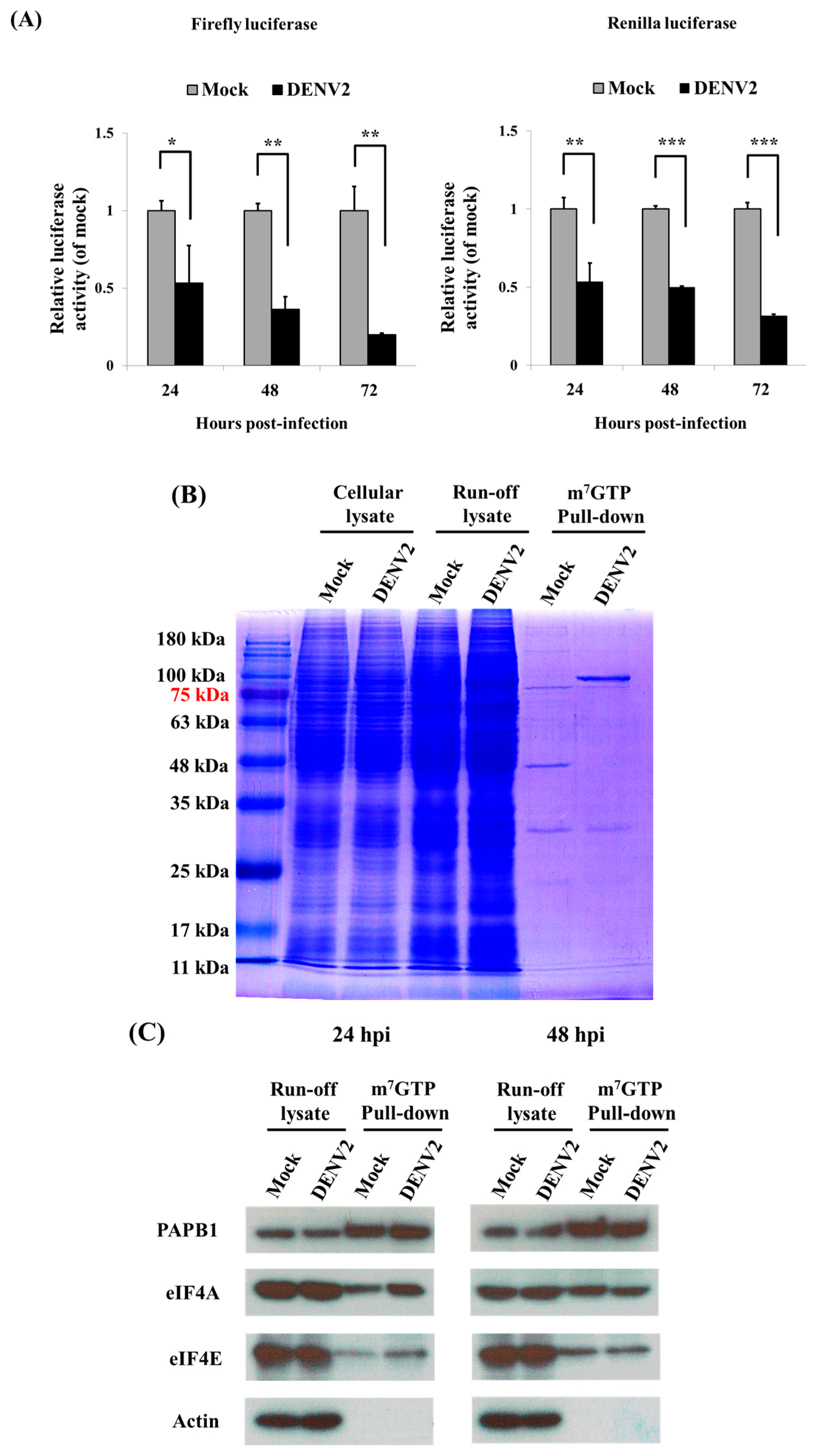
3.2. Cap-Dependent Protein Translation in DENV2-Infected C6/36 Cells
3.3. Activation of the TOR Signaling Pathway in C6/36 Cells Infected with the DENV2
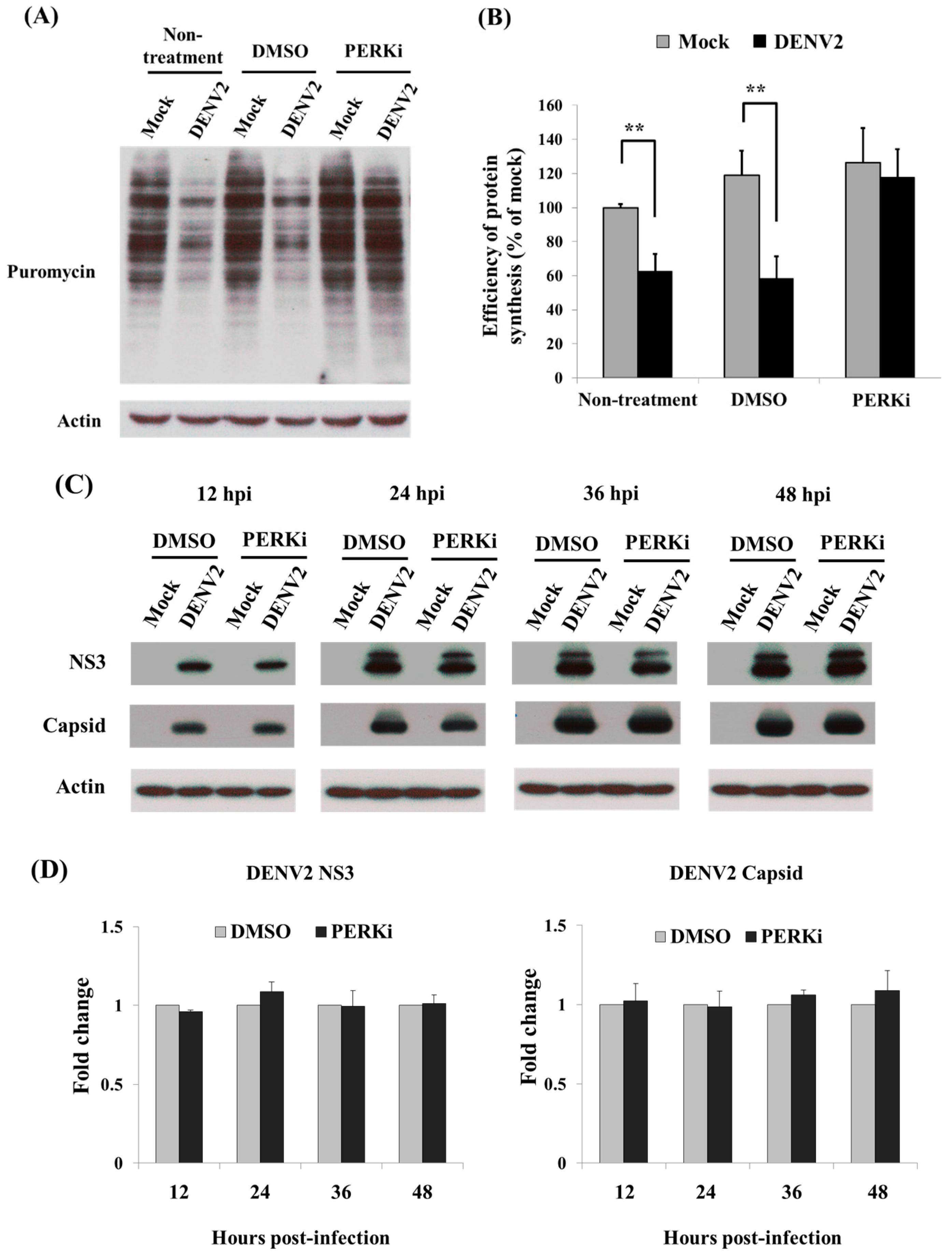
3.4. Recovery of Cellular Protein Synthesis in DENV2-Infected C6/36 Cells Treated with a PERK Inhibitor
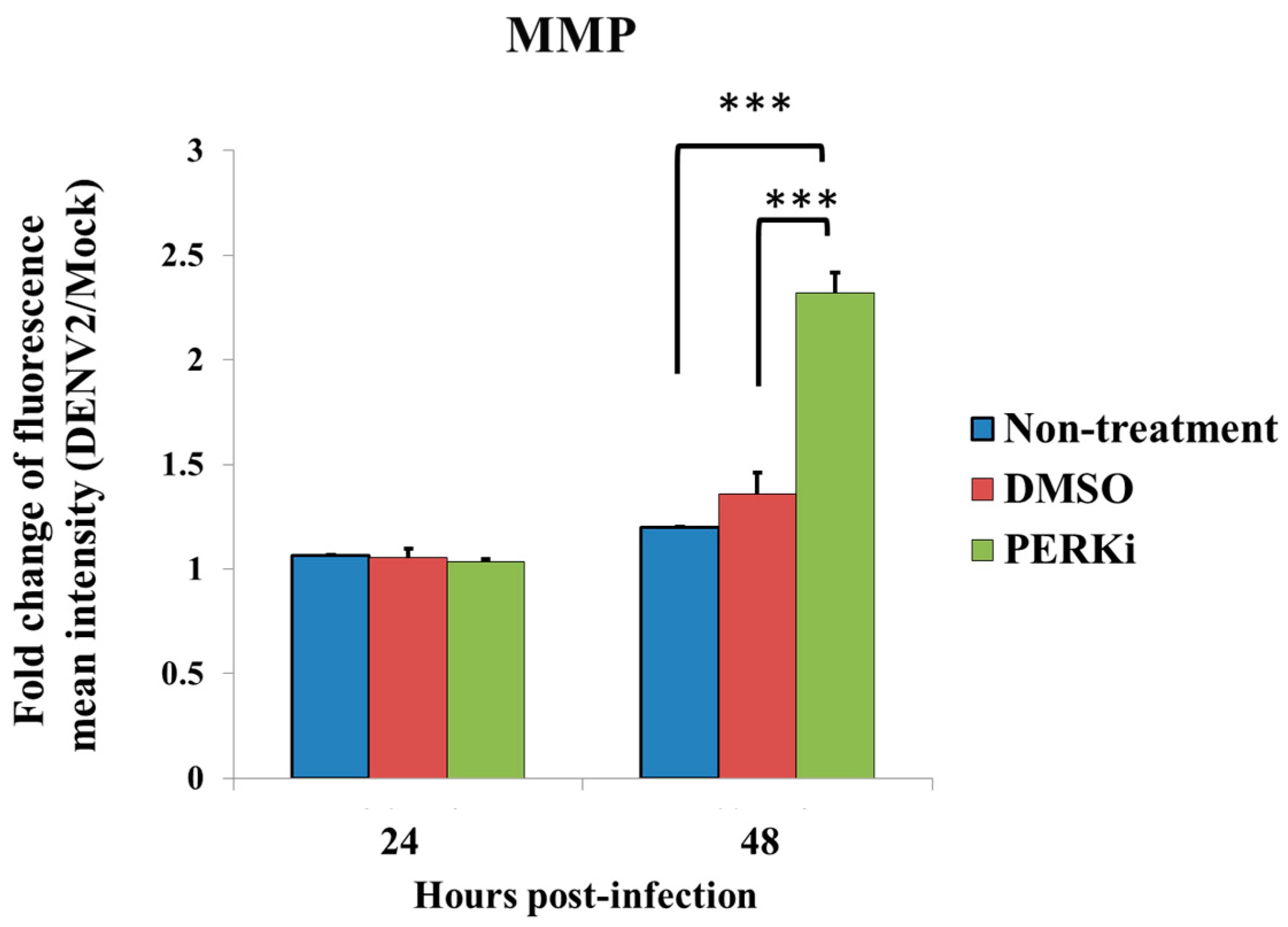
3.5. Evaluation of ER Stress by Measuring the MMP
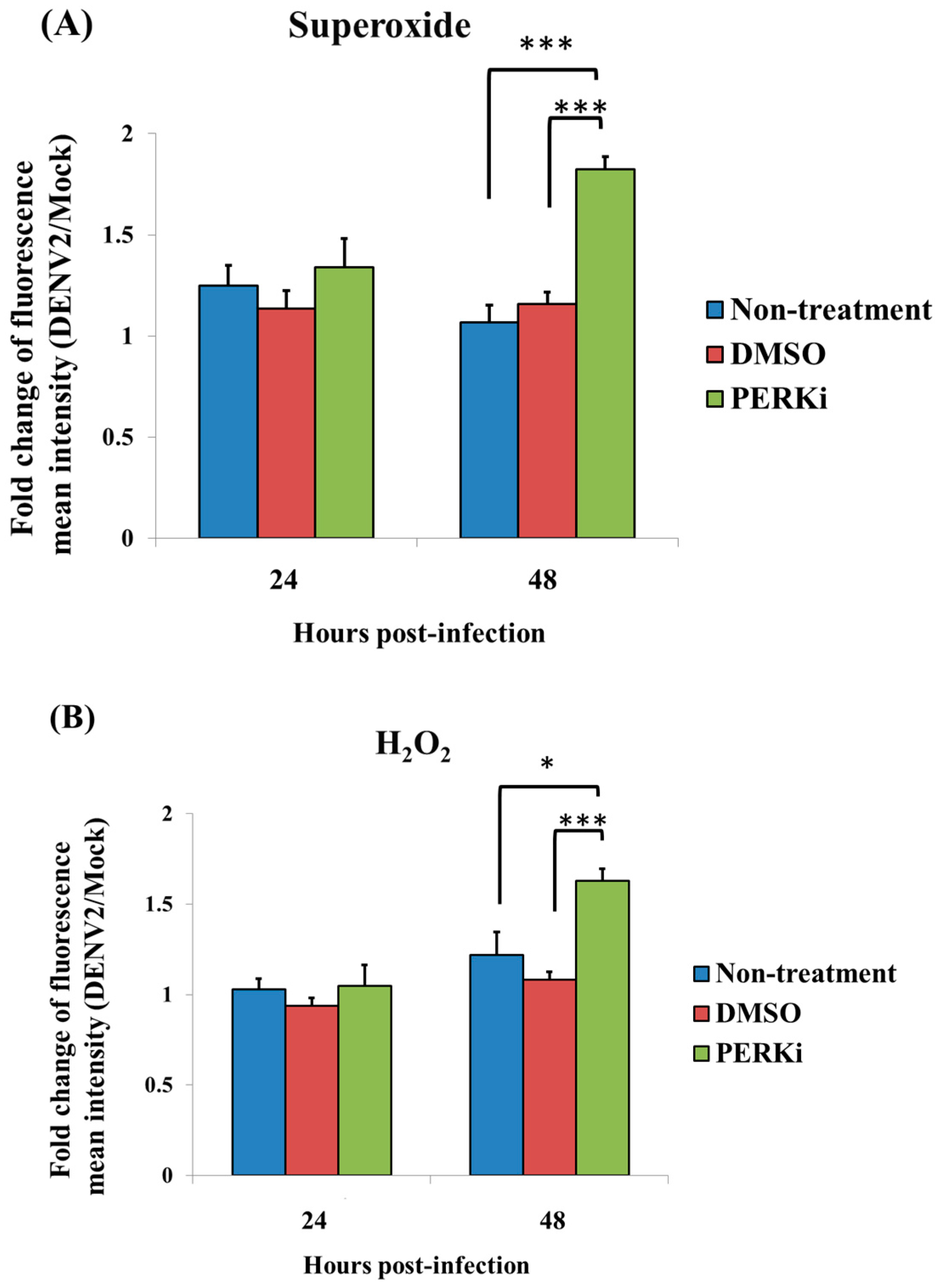
3.6. Involvement of the PERK Signaling Pathway in Generating ROS in C6/36 Cells with DENV2 Infection
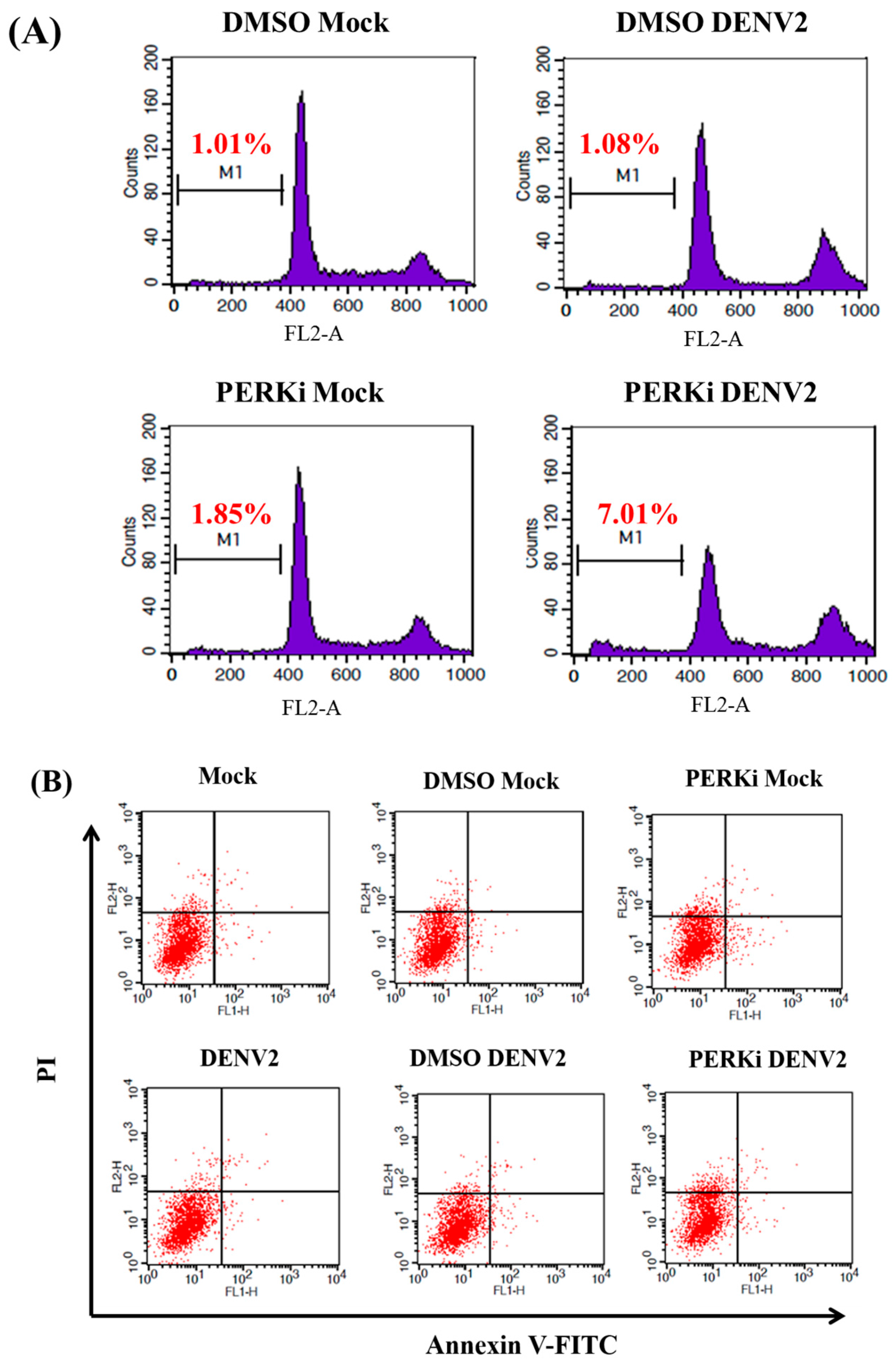
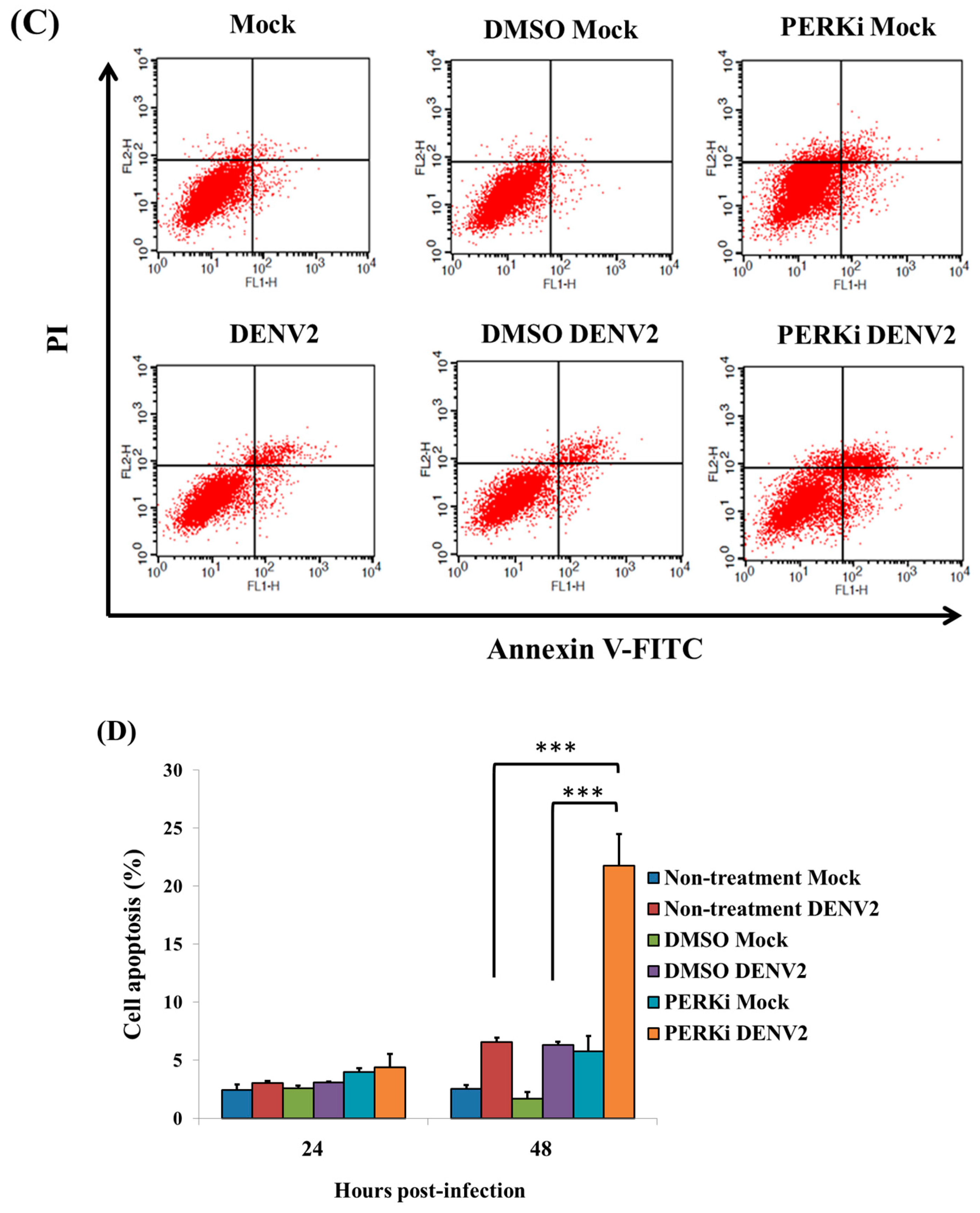
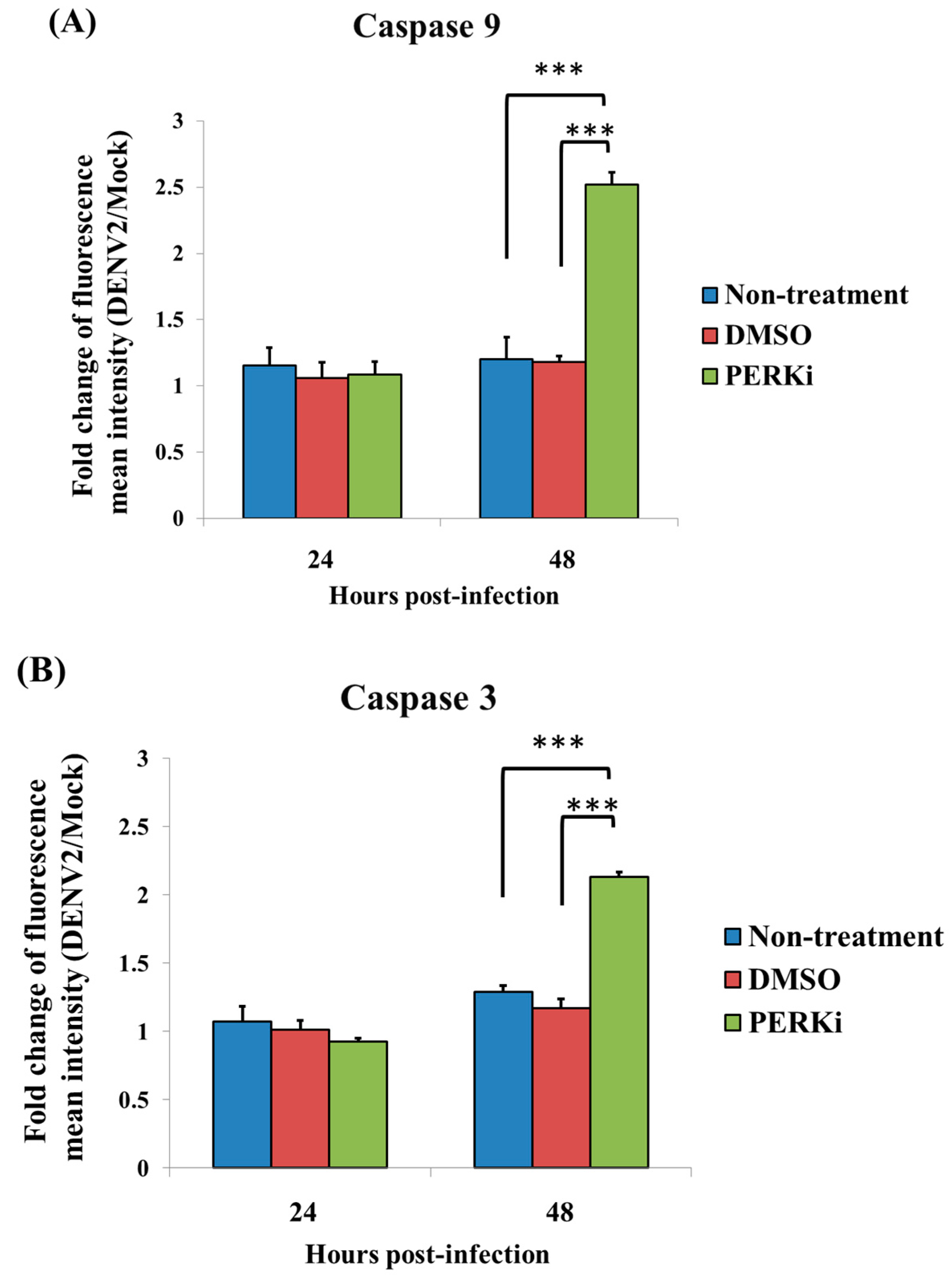
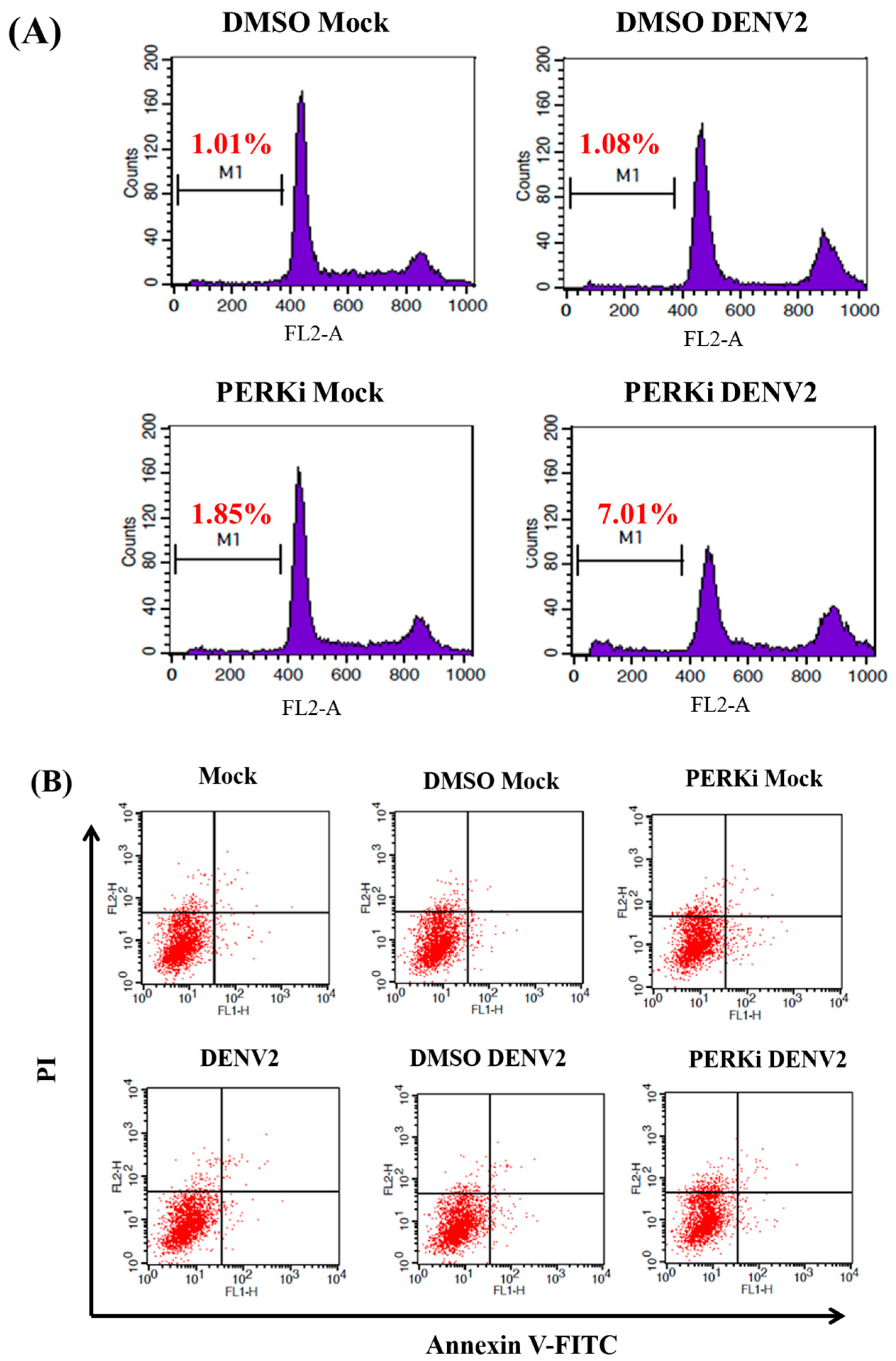
3.7. The PERK Signaling Pathway Is Involved in Reducing Cell Death of C6/36 Cells Infected with the DENV2
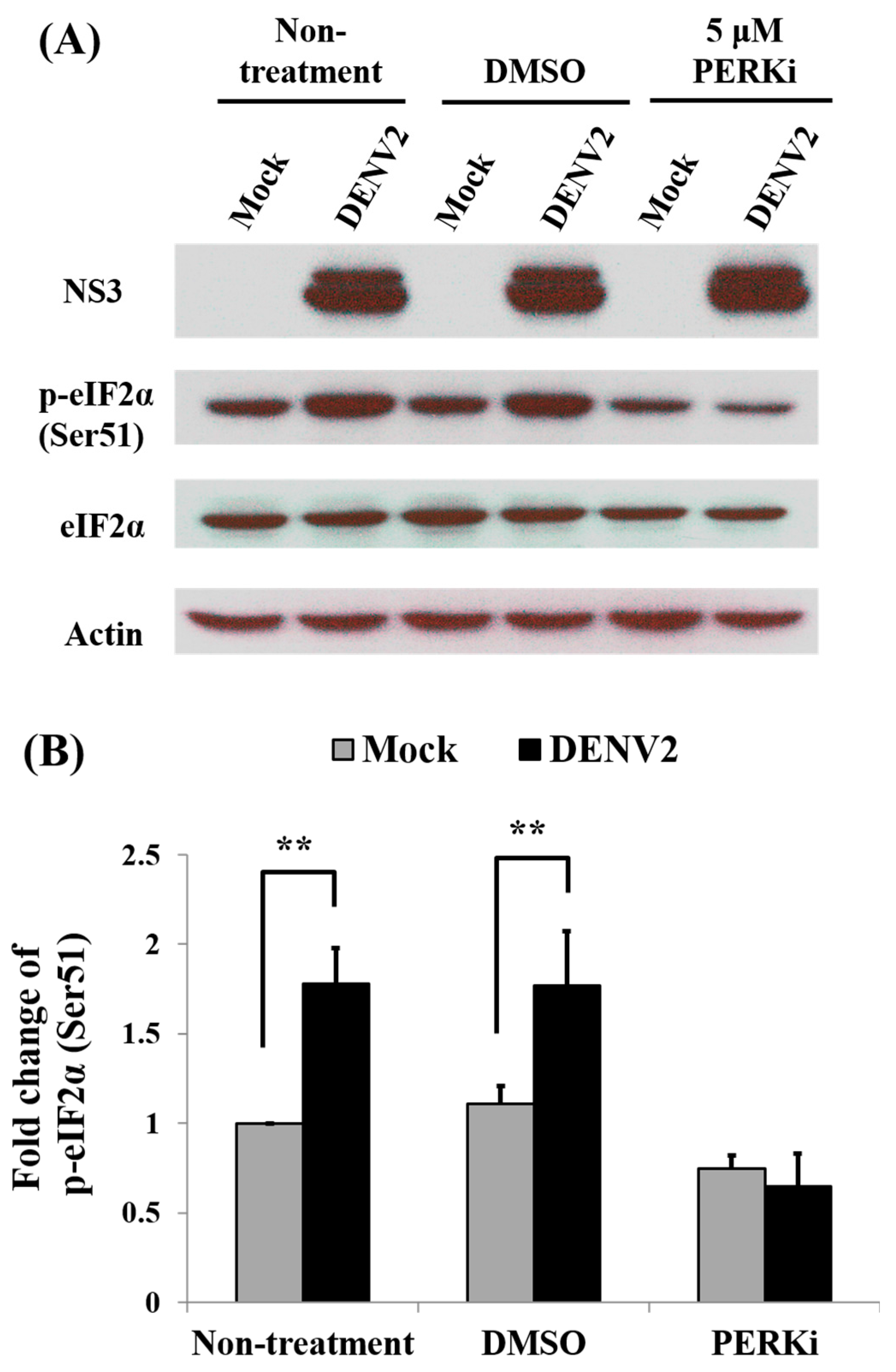
3.8. Activity of eIF2α in the PERK Signaling Pathway during Protein Translation of C6/36 Cells with DENV2 Infection
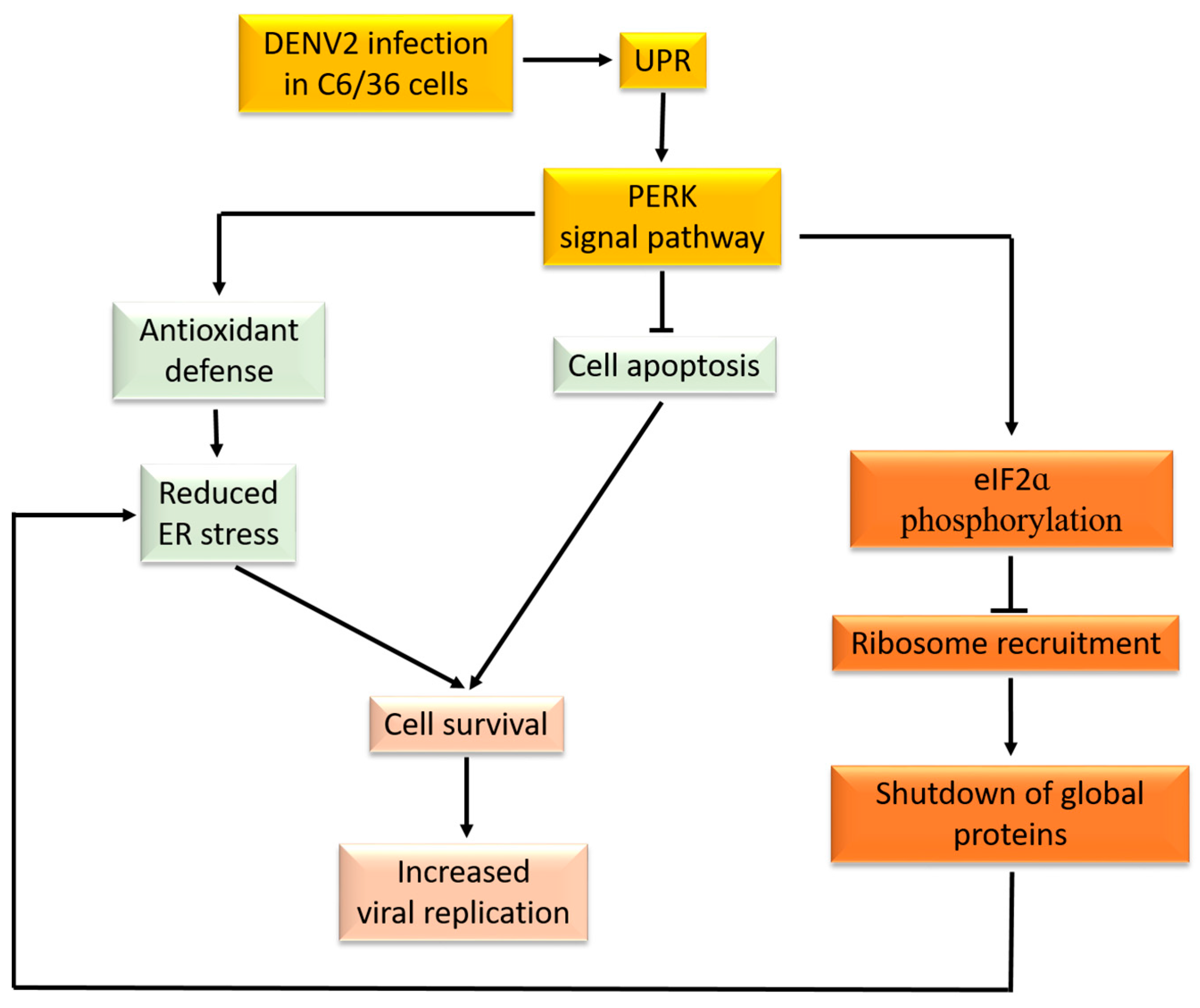
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Halstead, S.B. Dengue. Lancet 2007, 370, 1644–1652. [Google Scholar] [CrossRef]
- Yang, C.F.; Hou, J.N.; Chen, T.H.; Chen, W.J. Discriminable roles of Aedes aegypti and Aedes albopictus in establishment of dengue outbreaks in Taiwan. Acta Trop. 2014, 130, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Lindenbach, B.D.; Thiel, H.J.; Rice, C.M. Flaviviridae: The viruses and their replication. In Fields Virology, 5th ed.; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams and Wilkins: Ambler, PA, USA, 2007; pp. 1101–1152. [Google Scholar]
- Umareddy, I.; Pluquet, O.; Wang, Q.Y.; Vasudevan, S.G.; Chevet, E.; Gu, F. Dengue virus serotype infection specifies the activation of the unfolded protein response. Virol. J. 2007, 4, 91. [Google Scholar] [CrossRef] [PubMed]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Sriurairatna, S.; Bhamarapravati, N.; Diwan, A.R.; Halstead, S.B. Ultrastructural studies on dengue virus infection of human lymphoblasts. Infect. Immun. 1978, 20, 173–179. [Google Scholar] [PubMed]
- Marianneau, P.; Flamand, M.; Deubel, V.; Despres, P. Apoptotic cell death in response to dengue virus infection: The pathogenesis of dengue haemorrhagic fever revisited. Clin. Diagn. Virol. 1998, 10, 113–119. [Google Scholar] [CrossRef]
- Chen, T.H.; Tang, P.; Yang, C.F.; Kao, L.H.; Lo, Y.P.; Chuang, C.K.; Shih, Y.T.; Chen, W.J. Antioxidant defense is one of the mechanisms by which mosquito cells survive dengue 2 viral infection. Virology 2011, 410, 410–417. [Google Scholar] [CrossRef] [PubMed]
- Walsh, D.; Mathews, M.B.; Mohr, I. Tinkering with translation: Protein synthesis in virus-infected cells. Cold Spring Harb. Perspect. Biol. 2013, 5, a012351. [Google Scholar] [CrossRef] [PubMed]
- Fung, T.S.; Liao, Y.; Liu, D.X. Regulation of stress responses and translational control by coronavirus. Viruses 2016, 8. [Google Scholar] [CrossRef] [PubMed]
- Harding, H.P.; Novoa, I.; Zhang, Y.; Zeng, H.; Wek, R.; Schapira, M.; Ron, D. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol. Cell 2000, 6, 1099–1108. [Google Scholar] [CrossRef]
- Peña, J.; Harris, E. Dengue virus modulates the unfolded protein response in a time-dependent manner. J. Biol. Chem. 2011, 286, 14226–14236. [Google Scholar] [CrossRef] [PubMed]
- Sriurairatna, S.; Bhamarapravati, N. Replication of dengue-2 virus in Aedes albopictus mosquitoes. An electron microscopic study. Am. J. Trop. Med. Hyg. 1977, 26, 1199–1205. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.J.; Chen, S.L.; Fang, A.H. Phenotypic characteristics of dengue 2 virus persistently infected in a C6/36 clone of Aedes albopictus cells. Intervirology 1994, 37, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Barth, O.M.; Cortes, L.M.; Farias Filho, J.; Schatzmayr, H.G. Ultrastructural aspects of the replication of dengue virus type 2 isolated in Brazil. Mem. Inst. Oswaldo Cruz 1996, 91, 255–256. [Google Scholar] [CrossRef] [PubMed]
- Buchkovich, N.J.; Yu, Y.; Zampieri, C.A.; Alwine, J.C. The torrid affairs of viruses: Effects of mammalian DNA viruses on the PI3K-AKT-mTOR signalling pathway. Nat. Rev. Microbiol. 2008, 6, 266–275. [Google Scholar] [CrossRef] [PubMed]
- Holcik, M.; Sonenberg, N. Translational control in stress and apoptosis. Nat. Rev. Mol. Cell. Biol. 2005, 6, 318–327. [Google Scholar] [CrossRef] [PubMed]
- Richter, J.D.; Sonenberg, N. Regulation of cap-dependent translation by eIF4E inhibitory proteins. Nature 2005, 433, 477–480. [Google Scholar] [CrossRef] [PubMed]
- Laplante, M.; Sabatini, D.M. mTOR signaling at a glance. J. Cell Sci. 2009, 122, 3589–3594. [Google Scholar] [CrossRef] [PubMed]
- Gingras, A.C.; Svitkin, Y.; Belsham, G.J.; Pause, A.; Sonenberg, N. Activation of the translational suppressor 4E-BP following infection with encephalomyocarditis virus and poliovirus. Proc. Natl. Acad. Sci. USA 1996, 93, 5578–5583. [Google Scholar] [CrossRef] [PubMed]
- Connor, J.H.; Lyles, D.S. Vesicular stomatitis virus infection alters the eIF4F translation initiation complex and causes dephosphorylation of the eIF4E binding protein 4E-BP. J. Virol. 2002, 76, 10177–10187. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.H.; Lo, Y.P.; Yang, C.F.; Chen, W.J. Additive protection by antioxidant and apoptosis-inhibiting effects on mosquito cells with dengue 2 virus infection. PLoS Negl. Trop. Dis. 2012, 6, e1613. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, E.K.; Clavarino, G.; Ceppi, M.; Pierre, P. SUnSET, a nonradioactive method to monitor protein synthesis. Nat. Methods 2009, 6, 275–277. [Google Scholar] [CrossRef] [PubMed]
- Mamane, Y.; Petroulakis, E.; LeBacquer, O.; Sonenberg, N. mTOR, translation initiation and cancer. Oncogene 2006, 25, 6416–6422. [Google Scholar] [CrossRef] [PubMed]
- Shives, K.D.; Beatman, E.L.; Chamanian, M.; O’Brien, C.; Hobson-Peters, J.; Beckham, J.D. West nile virus-induced activation of mammalian target of rapamycin complex 1 supports viral growth and viral protein expression. J. Virol. 2014, 88, 9458–9471. [Google Scholar] [CrossRef] [PubMed]
- Edgil, D.; Polacek, C.; Harris, E. Dengue virus utilizes a novel strategy for translation initiation when cap-dependent translation is inhibited. J. Virol. 2006, 80, 2976–2986. [Google Scholar] [CrossRef] [PubMed]
- Axten, J.M.; Medina, J.R.; Feng, Y.; Shu, A.; Romeril, S.P.; Grant, S.W.; Li, W.H.; Heerding, D.A.; Minthorn, E.; Mencken, T.; et al. Discovery of 7-methyl-5-(1-{[3-(trifluoromethyl)phenyl]acetyl}-2,3-dihydro-1h-indol-5-yl)-7h-p yrrolo[2,3-d]pyrimidin-4-amine (gsk2606414), a potent and selective first-in-class inhibitor of protein kinase r (PKR)-like endoplasmic reticulum kinase (PERK). J. Med. Chem. 2012, 55, 7193–7207. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Qi, B.; Gu, Y.; Xu, F.; Du, H.; Li, X.; Fang, W. Porcine circovirus 2 deploys PERK pathway and GRP78 for its enhanced replication in pk-15 cells. Viruses 2016, 8. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Chevet, E.; Harding, H.P. Targeting the unfolded protein response in disease. Nat. Rev. Drug Discov. 2013, 12, 703–719. [Google Scholar] [CrossRef] [PubMed]
- Boya, P.; Cohen, I.; Zamzami, N.; Vieira, H.L.; Kroemer, G. Endoplasmic reticulum stress-induced cell death requires mitochondrial membrane permeabilization. Cell Death Differ. 2002, 9, 465–467. [Google Scholar] [CrossRef] [PubMed]
- Zeeshan, H.M.; Lee, G.H.; Kim, H.R.; Chae, H.J. Endoplasmic reticulum stress and associated ROS. Int. J. Mol. Sci. 2016, 17, 327. [Google Scholar] [CrossRef] [PubMed]
- Kääriäinen, L.; Ranki, M. Inhibition of cell functions by RNA-virus infections. Annu. Rev. Microbiol. 1984, 38, 91–109. [Google Scholar] [CrossRef] [PubMed]
- Schneider, R.J.; Mohr, I. Translation initiation and viral tricks. Trends Biochem. Sci. 2003, 28, 130–136. [Google Scholar] [CrossRef]
- Roth, H.; Magg, V.; Uch, F.; Mutz, P.; Klein, P.; Haneke, K.; Lohmann, V.; Bartenschlager, R.; Fackler, O.T.; Locker, N.; et al. Flavivirus infection uncouples translation suppression from cellular stress responses. MBio 2017, 8, e02150-16. [Google Scholar] [CrossRef] [PubMed]
- López-Lastra, M.; Rivas, A.; Barria, M.I. Protein synthesis in eukaryotes: The growing biological relevance of cap-independent translation initiation. Biol. Res. 2005, 38, 121–146. [Google Scholar] [CrossRef] [PubMed]
- Showkat, M.; Beigh, M.A.; Andrabi, K.I. mTOR signaling in protein translation regulation: Implications in cancer genesis and therapeutic interventions. Mol. Biol. Int. 2014, 2014, 686984. [Google Scholar] [CrossRef] [PubMed]
- Svitkin, Y.V.; Herdy, B.; Costa-Mattioli, M.; Gingras, A.C.; Raught, B.; Sonenberg, N. Eukaryotic translation initiation factor 4E availability controls the switch between cap-dependent and internal ribosomal entry site-mediated translation. Mol. Cell Biol. 2005, 25, 10556–10565. [Google Scholar] [CrossRef] [PubMed]
- Magnuson, B.; Ekim, B.; Fingar, D.C. Regulation and function of ribosomal protein S6 kinase (S6K) within mtor signalling networks. Biochem. J. 2012, 441, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Harding, H.P.; Zhang, Y.; Zeng, H.; Novoa, I.; Lu, P.D.; Calfon, M.; Sadri, N.; Yun, C.; Popko, B.; Paules, R.; et al. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol. Cell 2003, 11, 619–633. [Google Scholar] [CrossRef]
- Liu, Z.; Lv, Y.; Zhao, N.; Guan, G.; Wang, J. Protein kinase R-like ER kinase and its role in endoplasmic reticulum stress-decided cell fate. Cell Death Dis. 2015, 6, e1822. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.H.; Chiang, Y.H.; Hou, J.N.; Cheng, C.C.; Sofiyatun, E.; Chiu, C.H.; Chiang-Ni, I.; Chen, W.J. Xbp1-mediated Bip/GRP78 upregulation copes with oxidative stress in mosquito cells during dengue 2 virus infection. BioMed Res. Int. 2017. Available online: https://www.hindawi.com/journals/bmri/aip/3519158/ (accessed on 3 July 2017).
- Sano, R.; Reed, J.C. ER stress-induced cell death mechanisms. Biochim. Biophys. Acta 2013, 1833, 3460–3470. [Google Scholar] [CrossRef] [PubMed]
- Datan, E.; Roy, S.G.; Germain, G.; Zali, N.; McLean, J.E.; Golshan, G.; Harbajan, S.; Lockshin, R.A.; Zakeri, Z. Dengue-induced autophagy, virus replication and protection from cell death require ER stress (PERK) pathway activation. Cell Death Dis. 2016, 7, e2127. [Google Scholar] [CrossRef] [PubMed]
- Gale, M., Jr.; Tan, S.L.; Katze, M.G. Translational control of viral gene expression in eukaryotes. Microbiol. Mol. Biol. Rev. 2000, 64, 239–280. [Google Scholar] [CrossRef] [PubMed]
- Alcami, A.; Koszinowski, U.H. Viral mechanisms of immune evasion. Immunol. Today 2000, 21, 447–455. [Google Scholar] [CrossRef]
- Lindenbach, B.D.; Rice, C.M. Molecular biology of flaviviruses. Adv. Virus Res. 2003, 59, 23–61. [Google Scholar] [PubMed]
- Villas-Bôas, C.S.; Conceicão, T.M.; Ramírez, J.; Santoro, A.B.; Da Poian, A.T.; Montero-Lomelí, M. Dengue virus-induced regulation of the host cell translational machinery. Braz. J. Med. Biol. Res. 2009, 42, 1020–1026. [Google Scholar] [CrossRef] [PubMed]
- Khromykh, A.A.; Meka, H.; Guyatt, K.J.; Westaway, E.G. Essential role of cyclization sequences in flavivirus RNA replication. J. Virol. 2001, 75, 6719–6728. [Google Scholar] [CrossRef] [PubMed]
- Baird, T.D.; Palam, L.R.; Fusakio, M.E.; Willy, J.A.; Davis, C.M.; McClintick, J.N.; Anthony, T.G.; Wek, R.C. Selective mRNA translation during eIF2 phosphorylation induces expression of IBTKα. Mol. Biol. Cell. 2014, 25, 1686–1697. [Google Scholar] [CrossRef] [PubMed]
- Sarnow, P. Translation of glucose-regulated protein 78/immunoglobulin heavy-chain binding protein mRNA is increased in poliovirus-infected cells at a time when cap-dependent translation of cellular mRNA is inhibited. Proc. Natl. Acad. Sci. USA 1989, 86, 5795–5799. [Google Scholar] [CrossRef] [PubMed]
- Komar, A.A.; Hatzoglou, M. Cellular IRES-mediated translation: The war of itafs in pathophysiological states. Cell Cycle 2011, 10, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.J.; Chang Gung University, Kwei-San, Tao-Yuan, Taiwan. No change in translation of BiP/GRP78 in C6/36 cells treated with the PERK inhibitor. 2017. [Google Scholar]








© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hou, J.-N.; Chen, T.-H.; Chiang, Y.-H.; Peng, J.-Y.; Yang, T.-H.; Cheng, C.-C.; Sofiyatun, E.; Chiu, C.-H.; Chiang-Ni, C.; Chen, W.-J. PERK Signal-Modulated Protein Translation Promotes the Survivability of Dengue 2 Virus-Infected Mosquito Cells and Extends Viral Replication. Viruses 2017, 9, 262. https://doi.org/10.3390/v9090262
Hou J-N, Chen T-H, Chiang Y-H, Peng J-Y, Yang T-H, Cheng C-C, Sofiyatun E, Chiu C-H, Chiang-Ni C, Chen W-J. PERK Signal-Modulated Protein Translation Promotes the Survivability of Dengue 2 Virus-Infected Mosquito Cells and Extends Viral Replication. Viruses. 2017; 9(9):262. https://doi.org/10.3390/v9090262
Chicago/Turabian StyleHou, Jiun-Nan, Tien-Huang Chen, Yi-Hsuan Chiang, Jing-Yun Peng, Tsong-Han Yang, Chih-Chieh Cheng, Eny Sofiyatun, Cheng-Hsun Chiu, Chuan Chiang-Ni, and Wei-June Chen. 2017. "PERK Signal-Modulated Protein Translation Promotes the Survivability of Dengue 2 Virus-Infected Mosquito Cells and Extends Viral Replication" Viruses 9, no. 9: 262. https://doi.org/10.3390/v9090262
APA StyleHou, J.-N., Chen, T.-H., Chiang, Y.-H., Peng, J.-Y., Yang, T.-H., Cheng, C.-C., Sofiyatun, E., Chiu, C.-H., Chiang-Ni, C., & Chen, W.-J. (2017). PERK Signal-Modulated Protein Translation Promotes the Survivability of Dengue 2 Virus-Infected Mosquito Cells and Extends Viral Replication. Viruses, 9(9), 262. https://doi.org/10.3390/v9090262