
Targeting Persistent Human Papillomavirus Infection



Abstract

{kind=link}
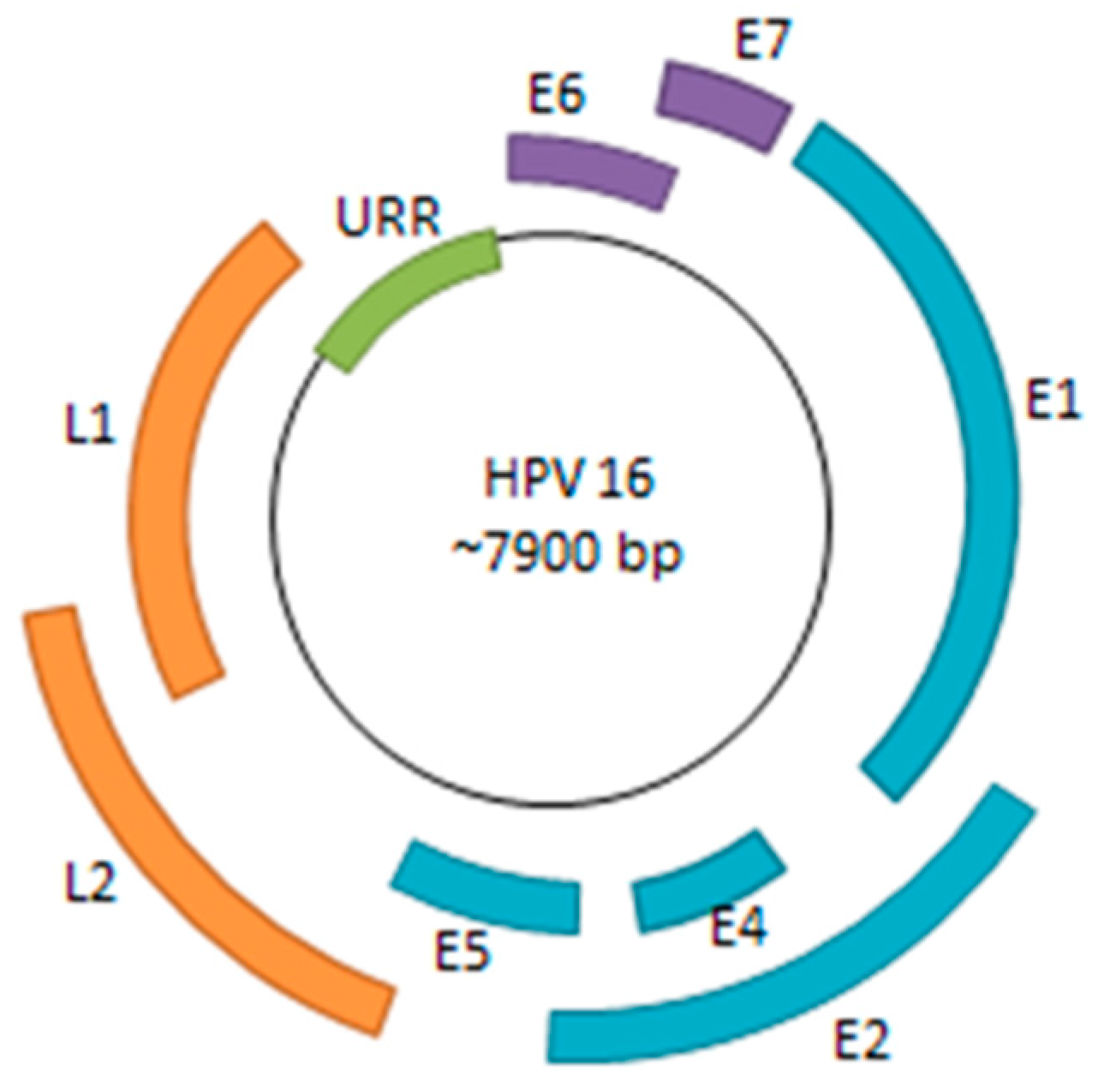{kind=link}
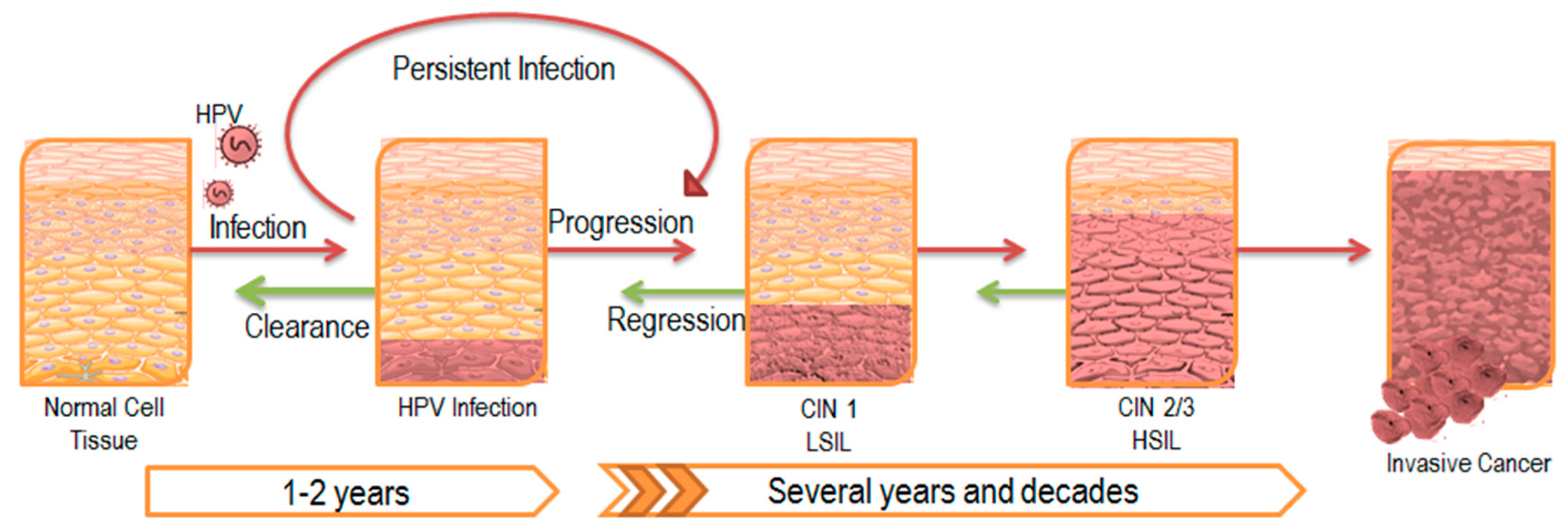{kind=link}
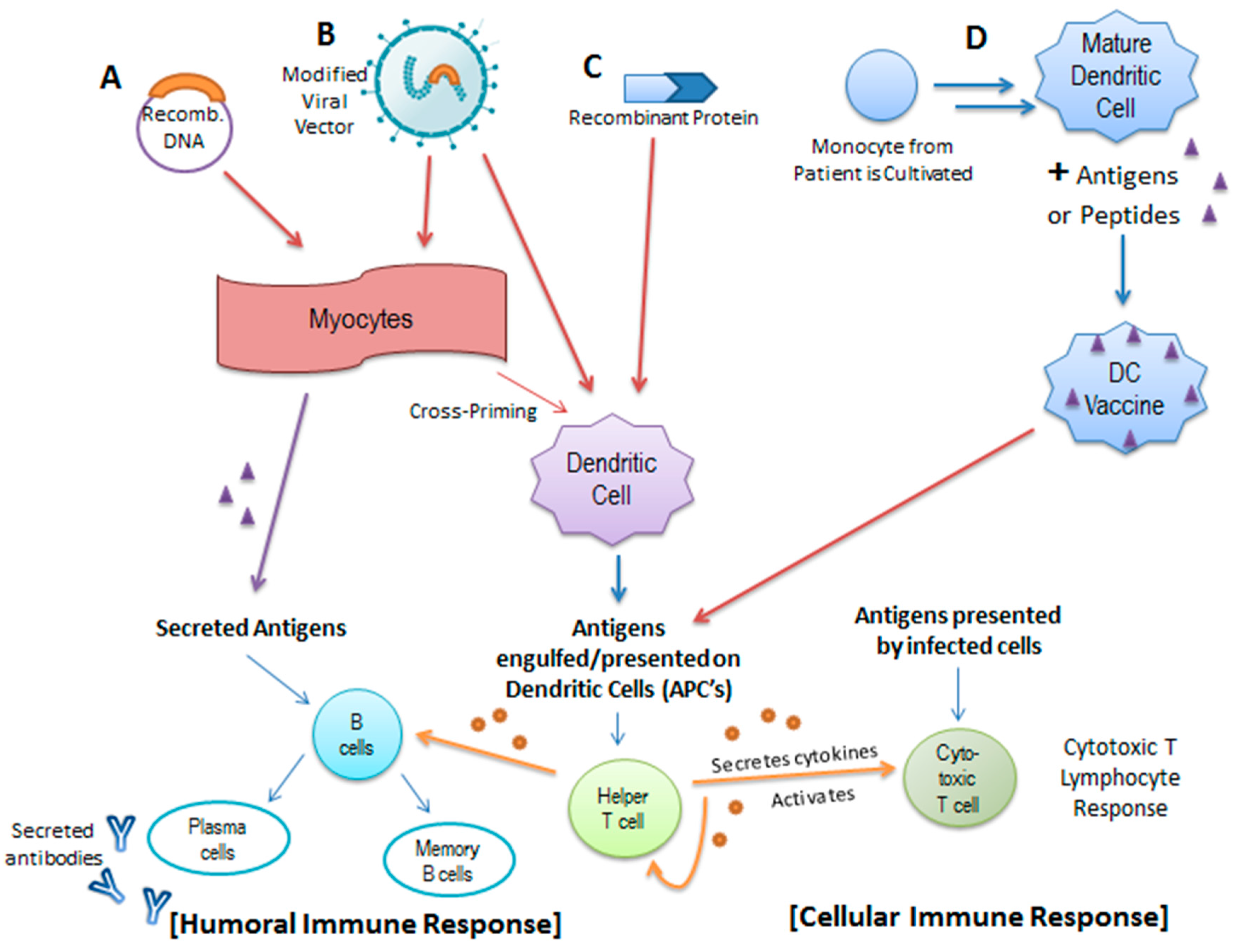{kind=link}
1. Persistent HPV Infection
2. The Impact of Persistent Infection on Cancer
3. Molecular Mechanisms Underlying Persistent HPV Infection
3.1. Viral Life Cycle and Immune System Evasion
3.2. Viral Episome Hitchhiking on Host Mitotic Chromosomes
3.3. E2 Interaction with Host Receptors
4. Current Therapeutic Strategies for Clearing Persistent HPV Infection
4.1. HPV Prophylactic Vaccines
4.2. The Promise of Therapeutic Vaccines
4.3. Chemopreventive Strategies
4.4. Small Molecular Inhibitors
5. Perspectives and Future Direction
Acknowledgments
Author Contributions
Conflicts of Interest
References
- McLaughlin-Drubin, M.E.; Münger, K. Oncogenic activities of human papillomaviruses. Virus Res. 2009, 143, 195–208. [Google Scholar] [CrossRef] [PubMed]
- Galloway, D.A.; Laimins, L.A. Human papillomaviruses: Shared and distinct pathways for pathogenesis. Curr. Opin. Virol. 2015, 14, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Radley, D.; Saah, A.; Stanley, M. Persistent infection with human papillomavirus 16 or 18 is strongly linked with high-grade cervical disease. Hum. Vaccines Immunother. 2015, 12, 768–772. [Google Scholar] [CrossRef] [PubMed]
- Gillison, M.L.; Koch, W.M.; Capone, R.B.; Spafford, M.; Westra, W.H.; Wu, L.; Zahurak, M.L.; Daniel, R.W.; Viglione, M.; Symer, D.E.; et al. Evidence for a causal association between human papillomavirus and a subset of head and neck cancers. J. Natl. Cancer Inst. 2000, 92, 709–720. [Google Scholar] [CrossRef] [PubMed]
- Frazer, I.H. Interaction of human papillomaviruses with the host immune system: A well evolved relationship. Virology 2009, 384, 410–414. [Google Scholar] [CrossRef] [PubMed]
- Boldogh, I.; Albrecht, T.; Porter, D.D. Persistent viral infections. In Medical Microbiology, 4th ed.; Baron, S., Ed.; University of Texas Medical Branch at Galveston: Galveston, TX, USA, 1996. [Google Scholar]
- Haukioja, A.; Asunta, M.; Söderling, E.; Syrjänen, S. Persistent oral human papillomavirus infection is associated with smoking and elevated salivary immunoglobulin g concentration. J. Clin. Virol. 2014, 61, 101–106. [Google Scholar] [CrossRef] [PubMed]
- Rositch, A.F.; Koshiol, J.; Hudgens, M.; Razzaghi, H.; Backes, D.M.; Pimenta, J.M.; Franco, E.L.; Poole, C.; Smith, J.S. Patterns of persistent genital human papillomavirus infection among women worldwide: A literature review and meta-analysis. Int. J. Cancer 2013, 133, 1271–1285. [Google Scholar] [CrossRef] [PubMed]
- Oh, H.Y.; Seo, S.-S.; Kim, M.K.; Lee, D.O.; Chung, Y.K.; Lim, M.C.; Kim, J.-Y.; Lee, C.W.; Park, S.-Y. Synergistic effect of viral load and alcohol consumption on the risk of persistent high-risk human papillomavirus infection. PLoS ONE 2014, 9, e104374. [Google Scholar] [CrossRef] [PubMed]
- Oh, H.Y.; Kim, M.K.; Seo, S.; Lee, D.O.; Chung, Y.K.; Lim, M.C.; Kim, J.; Lee, C.W.; Park, S. Alcohol consumption and persistent infection of high-risk human papillomavirus. Epidemiol. Infect. 2015, 143, 1442–1450. [Google Scholar] [CrossRef] [PubMed]
- Gunnell, A.S.; Tran, T.N.; Torrång, A.; Dickman, P.W.; Sparén, P.; Palmgren, J.; Ylitalo, N. Synergy between cigarette smoking and human papillomavirus type 16 in cervical cancer in situ development. Cancer Epidemiol. Biomark. Prev. 2006, 15, 2141–2147. [Google Scholar] [CrossRef] [PubMed]
- Xi, L.F.; Koutsky, L.A.; Castle, P.E.; Edelstein, Z.R.; Meyers, C.; Ho, J.; Schiffman, M. Relationship between cigarette smoking and human papilloma virus types 16 and 18 DNA load. Cancer Epidemiol. Biomark. Prev. 2009, 18, 3490–3496. [Google Scholar] [CrossRef] [PubMed]
- Peng, S.; Trimble, C.; Wu, L.; Pardoll, D.; Roden, R.; Hung, C.-F.; Wu, T.C. HLA-DQB1*02-restricted HPV-16 E7 peptide-specific CD4+ T-cell immune responses correlate with regression of HPV-16-associated high-grade squamous intraepithelial lesions. Clin. Cancer Res. 2007, 13, 2479–2487. [Google Scholar] [CrossRef] [PubMed]
- Wank, R.; Thomssen, C. High risk of squamous cell carcinoma of the cervix for women with HLA-DQw3. Nature 1991, 352, 723–725. [Google Scholar] [CrossRef] [PubMed]
- Zoodsma, M.; Nolte, I.M.; Schipper, M.; Oosterom, E.; van der Steege, G.; de Vries, E.G.E.; Te Meerman, G.J.; van der Zee, A.G.J. Analysis of the entire hla region in susceptibility for cervical cancer: A comprehensive study. J. Med. Genet. 2005, 42, e49. [Google Scholar] [CrossRef] [PubMed]
- Bernal-Silva, S.; Granados, J.; Gorodezky, C.; Aláez, C.; Flores-Aguilar, H.; Cerda-Flores, R.M.; Guerrero-González, G.; Valdez-Chapa, L.D.; Morales-Casas, J.; González-Guerrero, J.F.; et al. HLA-DRB1 class II antigen level alleles are associated with persistent HPV infection in mexican women; a pilot study. Infect. Agent Cancer 2013, 8, 31. [Google Scholar] [CrossRef] [PubMed]
- Liaw, K.L.; Hildesheim, A.; Burk, R.D.; Gravitt, P.; Wacholder, S.; Manos, M.M.; Scott, D.R.; Sherman, M.E.; Kurman, R.J.; Glass, A.G.; et al. A prospective study of human papillomavirus (HPV) type 16 DNA detection by polymerase chain reaction and its association with acquisition and persistence of other HPV types. J. Infect. Dis. 2001, 183, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Rousseau, M.C.; Pereira, J.S.; Prado, J.C.; Villa, L.L.; Rohan, T.E.; Franco, E.L. Cervical coinfection with human papillomavirus (HPV) types as a predictor of acquisition and persistence of HPV infection. J. Infect. Dis. 2001, 184, 1508–1517. [Google Scholar] [CrossRef] [PubMed]
- Thomas, K.K.; Hughes, J.P.; Kuypers, J.M.; Kiviat, N.B.; Lee, S.K.; Adam, D.E.; Koutsky, L.A. Concurrent and sequential acquisition of different genital human papillomavirus types. J. Infect. Dis. 2000, 182, 1097–1102. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Liao, H.; Yang, B.; Geffre, C.P.; Zhang, A.; Zhou, A.; Cao, H.; Wang, J.; Zhang, Z.; Zheng, W. Variants of human papillomavirus type 16 predispose toward persistent infection. Int. J. Clin. Exp. Pathol. 2015, 8, 8453–8459. [Google Scholar] [PubMed]
- La Torre, G.; de Waure, C.; Chiaradia, G.; Mannocci, A.; Ricciardi, W. HPV vaccine efficacy in preventing persistent cervical HPV infection: A systematic review and meta-analysis. Vaccine 2007, 25, 8352–8358. [Google Scholar] [CrossRef] [PubMed]
- Clifford, G.M.; Smith, J.S.; Aguado, T.; Franceschi, S. Comparison of HPV type distribution in high-grade cervical lesions and cervical cancer: A meta-analysis. Br. J. Cancer 2003, 89, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Zur Hausen, H. Papillomaviruses in the causation of human cancers—A brief historical account. Virology 2009, 384, 260–265. [Google Scholar] [CrossRef] [PubMed]
- Lizano, M.; Berumen, J.; García-Carrancá, A. HPV-related carcinogenesis: Basic concepts, viral types and variants. Arch. Med. Res. 2009, 40, 428–434. [Google Scholar] [CrossRef] [PubMed]
- Katki, H.A.; Cheung, L.C.; Fetterman, B.; Castle, P.E.; Sundaram, R. A joint model of persistent human papillomavirus infection and cervical cancer risk: Implications for cervical cancer screening. J. R Stat. Soc. Ser. A Stat. Soc. 2015, 178, 903–923. [Google Scholar] [CrossRef] [PubMed]
- Lowy, D.R.; Schiller, J.T. Reducing HPV-associated cancer globally. Cancer Prev. Res. 2012, 5, 18–23. [Google Scholar] [CrossRef] [PubMed]
- Gillison, M.L.; D’Souza, G.; Westra, W.; Sugar, E.; Xiao, W.; Begum, S.; Viscidi, R. Distinct risk factor profiles for human papillomavirus type 16-positive and human papillomavirus type 16-negative head and neck cancers. J. Natl. Cancer Inst. 2008, 100, 407–420. [Google Scholar] [CrossRef] [PubMed]
- Weinberger, P.M.; Yu, Z.; Haffty, B.G.; Kowalski, D.; Harigopal, M.; Brandsma, J.; Sasaki, C.; Joe, J.; Camp, R.L.; Rimm, D.L.; et al. Molecular classification identifies a subset of human papillomavirus—Associated oropharyngeal cancers with favorable prognosis. J. Clin. Oncol. 2006, 24, 736–747. [Google Scholar] [CrossRef] [PubMed]
- Newell, G.R.; Krementz, E.T.; Roberts, J.D. Excess occurrence of cancer of the oral cavity, lung, and bladder following cancer of the cervix. Cancer 1975, 36, 2155–2158. [Google Scholar] [CrossRef] [PubMed]
- Ciesielska, U.; Nowińska, K.; Podhorska-Okołów, M.; Dziegiel, P. The role of human papillomavirus in the malignant transformation of cervix epithelial cells and the importance of vaccination against this virus. Adv. Clin. Exp. Med. 2012, 21, 235–244. [Google Scholar] [PubMed]
- D’Souza, G.; Kreimer, A.R.; Viscidi, R.; Pawlita, M.; Fakhry, C.; Koch, W.M.; Westra, W.H.; Gillison, M.L. Case-control study of human papillomavirus and oropharyngeal cancer. N. Engl. J. Med. 2007, 356, 1944–1956. [Google Scholar] [CrossRef] [PubMed]
- Akagi, K.; Li, J.; Broutian, T.R.; Padilla-Nash, H.; Xiao, W.; Jiang, B.; Rocco, J.W.; Teknos, T.N.; Kumar, B.; Wangsa, D.; et al. Genome-wide analysis of HPV integration in human cancers reveals recurrent, focal genomic instability. Genome Res. 2014, 24, 185–199. [Google Scholar] [CrossRef] [PubMed]
- Cullen, A.P.; Reid, R.; Campion, M.; Lörincz, A.T. Analysis of the physical state of different human papillomavirus DNAs in intraepithelial and invasive cervical neoplasm. J. Virol. 1991, 65, 606–612. [Google Scholar] [PubMed]
- Schneider-Maunoury, S.; Croissant, O.; Orth, G. Integration of human papillomavirus type 16 DNA sequences: A possible early event in the progression of genital tumors. J. Virol. 1987, 61, 3295–3298. [Google Scholar] [PubMed]
- Parfenov, M.; Pedamallu, C.S.; Gehlenborg, N.; Freeman, S.S.; Danilova, L.; Bristow, C.A.; Lee, S.; Hadjipanayis, A.G.; Ivanova, E.V.; Wilkerson, M.D.; et al. Characterization of HPV and host genome interactions in primary head and neck cancers. Proc. Natl. Acad. Sci. USA 2014, 111, 15544–15549. [Google Scholar] [CrossRef] [PubMed]
- Steger, G.; Corbach, S. Dose-dependent regulation of the early promoter of human papillomavirus type 18 by the viral E2 protein. J. Virol. 1997, 71, 50–58. [Google Scholar] [PubMed]
- Dong, G.; Broker, T.R.; Chow, L.T. Human papillomavirus type 11 E2 proteins repress the homologous E6 promoter by interfering with the binding of host transcription factors to adjacent elements. J. Virol. 1994, 68, 1115–1127. [Google Scholar] [PubMed]
- Dyson, N.; Howley, P.M.; Munger, K.; Harlow, E. The human papilloma virus-16 E7 oncoprotein is able to bind to the retinoblastoma gene product. Science 1989, 243, 934–937. [Google Scholar] [CrossRef] [PubMed]
- Huibregtse, J.M.; Scheffner, M.; Howley, P.M. A cellular protein mediates association of p53 with the E6 oncoprotein of human papillomavirus types 16 or 18. EMBO J. 1991, 10, 4129–4135. [Google Scholar] [PubMed]
- Jeon, S.; Lambert, P.F. Integration of human papillomavirus type 16 DNA into the human genome leads to increased stability of E6 and E7 mRNAs: Implications for cervical carcinogenesis. Proc. Natl. Acad. Sci. USA 1995, 92, 1654–1658. [Google Scholar] [CrossRef] [PubMed]
- Bastien, N.; McBride, A.A. Interaction of the papillomavirus E2 protein with mitotic chromosomes. Virology 2000, 270, 124–134. [Google Scholar] [CrossRef] [PubMed]
- Groves, I.J.; Coleman, N. Pathogenesis of human papillomavirus-associated mucosal disease. J. Pathol. 2015, 235, 527–538. [Google Scholar] [CrossRef] [PubMed]
- Skiadopoulos, M.H.; McBride, A.A. Bovine papillomavirus type 1 genomes and the E2 transactivator protein are closely associated with mitotic chromatin. J. Virol. 1998, 72, 2079–2088. [Google Scholar] [PubMed]
- Ballestas, M.E.; Chatis, P.A.; Kaye, K.M. Efficient persistence of extrachromosomal KSHV DNA mediated by latency-associated nuclear antigen. Science 1999, 284, 641–644. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, P.; Lavoie, B.D.; Frappier, L. EBP2 plays a key role in Epstein–Barr virus mitotic segregation and is regulated by aurora family kinases. Mol. Cell. Biol. 2005, 25, 4934–4945. [Google Scholar] [CrossRef] [PubMed]
- Shire, K.; Ceccarelli, D.F.; Avolio-Hunter, T.M.; Frappier, L. EBP2, a human protein that interacts with sequences of the Epstein–Barr virus nuclear antigen 1 important for plasmid maintenance. J. Virol. 1999, 73, 2587–2595. [Google Scholar] [PubMed]
- Oliveira, J.G.; Colf, L.A.; McBride, A.A. Variations in the association of papillomavirus E2 proteins with mitotic chromosomes. Proc. Natl. Acad. Sci. USA 2006, 103, 1047–1052. [Google Scholar] [CrossRef] [PubMed]
- McBride, A.A. The papillomavirus E2 proteins. Virology 2013, 445, 57–79. [Google Scholar] [CrossRef] [PubMed]
- Kurg, R. The Role of E2 Proteins in Papillomavirus DNA Replication; InTech: Rijeka, Croatia, 2011. [Google Scholar]
- McPhillips, M.G.; Ozato, K.; McBride, A.A. Interaction of bovine papillomavirus E2 protein with Brd4 stabilizes its association with chromatin. J. Virol. 2005, 79, 8920–8932. [Google Scholar] [CrossRef] [PubMed]
- Van Tine, B.A.; Dao, L.D.; Wu, S.-Y.; Sonbuchner, T.M.; Lin, B.Y.; Zou, N.; Chiang, C.-M.; Broker, T.R.; Chow, L.T. Human papillomavirus (HPV) origin-binding protein associates with mitotic spindles to enable viral DNA partitioning. Proc. Natl. Acad. Sci. USA 2004, 101, 4030–4035. [Google Scholar] [CrossRef] [PubMed]
- You, J.; Croyle, J.L.; Nishimura, A.; Ozato, K.; Howley, P.M. Interaction of the bovine papillomavirus E2 protein with Brd4 tethers the viral DNA to host mitotic chromosomes. Cell 2004, 117, 349–360. [Google Scholar] [CrossRef]
- Houzelstein, D.; Bullock, S.L.; Lynch, D.E.; Grigorieva, E.F.; Wilson, V.A.; Beddington, R.S.P. Growth and early postimplantation defects in mice deficient for the bromodomain-containing protein Brd4. Mol. Cell. Biol. 2002, 22, 3794–3802. [Google Scholar] [CrossRef] [PubMed]
- McBride, A.A.; McPhillips, M.G.; Oliveira, J.G. Brd4: Tethering, segregation and beyond. Trends Microbiol. 2004, 12, 527–529. [Google Scholar] [CrossRef] [PubMed]
- Abbate, E.A.; Voitenleitner, C.; Botchan, M.R. Structure of the papillomavirus DNA-tethering complex E2:Brd4 and a peptide that ablates HPV chromosomal association. Mol. Cell 2006, 24, 877–889. [Google Scholar] [CrossRef] [PubMed]
- Gauson, E.J.; Wang, X.; Dornan, E.S.; Herzyk, P.; Bristol, M.; Morgan, I.M. Failure to interact with Brd4 alters the ability of HPV16 E2 to regulate host genome expression and cellular movement. Virus Res. 2016, 211, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Helfer, C.M.; Wang, R.; You, J. Analysis of the papillomavirus E2 and bromodomain protein Brd4 interaction using bimolecular fluorescence complementation. PLoS ONE 2013, 8, e77994. [Google Scholar] [CrossRef] [PubMed]
- McPhillips, M.G.; Oliveira, J.G.; Spindler, J.E.; Mitra, R.; McBride, A.A. Brd4 is required for E2-mediated transcriptional activation but not genome partitioning of all papillomaviruses. J. Virol. 2006, 80, 9530–9543. [Google Scholar] [CrossRef] [PubMed]
- Jang, M.K.; Kwon, D.; McBride, A.A. Papillomavirus E2 proteins and the host Brd4 protein associate with transcriptionally active cellular chromatin. J. Virol. 2009, 83, 2592–2600. [Google Scholar] [CrossRef] [PubMed]
- Schweiger, M.-R.; You, J.; Howley, P.M. Bromodomain protein 4 mediates the papillomavirus E2 transcriptional activation function. J. Virol. 2006, 80, 4276–4285. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Helfer, C.M.; Pancholi, N.; Bradner, J.E.; You, J. Recruitment of Brd4 to the human papillomavirus type 16 DNA replication complex is essential for replication of viral DNA. J. Virol. 2013, 87, 3871–3884. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.Y.; Nin, D.S.; Lee, A.Y.; Simanski, S.; Kodadek, T.; Chiang, C.M. Brd4 phosphorylation regulates HPV E2-mediated viral transcription, origin replication, and cellular MMP-9 expression. Cell Rep. 2016, 16, 1733–1748. [Google Scholar] [CrossRef] [PubMed]
- Sakakibara, N.; Chen, D.; Jang, M.K.; Kang, D.W.; Luecke, H.F.; Wu, S.-Y.; Chiang, C.-M.; McBride, A.A. Brd4 is displaced from HPV replication factories as they expand and amplify viral DNA. PLoS Pathog. 2013, 9, e1003777. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, M.M.; Mackintosh, L.J.; Bodily, J.M.; Dornan, E.S.; Laimins, L.A.; Morgan, I.M. An interaction between human papillomavirus 16 E2 and TopBP1 is required for optimum viral DNA replication and episomal genome establishment. J. Virol. 2012, 86, 12806–12815. [Google Scholar] [CrossRef] [PubMed]
- Parish, J.L.; Bean, A.M.; Park, R.B.; Androphy, E.J. ChlR1 is required for loading papillomavirus E2 onto mitotic chromosomes and viral genome maintenance. Mol. Cell 2006, 24, 867–876. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, M.M.; Boner, W.; Morgan, I.M. TopBP1 regulates human papillomavirus type 16 E2 interaction with chromatin. J. Virol. 2007, 81, 4338–4342. [Google Scholar] [CrossRef] [PubMed]
- Bang, S.W.; Ko, M.J.; Kang, S.; Kim, G.S.; Kang, D.; Lee, J.; Hwang, D.S. Human TopBP1 localization to the mitotic centrosome mediates mitotic progression. Exp. Cell Res. 2011, 317, 994–1004. [Google Scholar] [CrossRef] [PubMed]
- Hirota, Y.; Lahti, J.M. Characterization of the enzymatic activity of hChlR1, a novel human DNA helicase. Nucleic Acids Res. 2000, 28, 917–924. [Google Scholar] [CrossRef] [PubMed]
- Christensen, N.D.; Budgeon, L.R. Vaccines and immunization against human papillomavirus. Curr. Probl. Dermatol. 2014, 45, 252–264. [Google Scholar] [PubMed]
- Sankaranarayanan, R. HPV vaccination: The most pragmatic cervical cancer primary prevention strategy. Int. J. Gynecol. Obstet. 2015, 131, S33–S35. [Google Scholar] [CrossRef] [PubMed]
- Pils, S.; Joura, E.A. From the monovalent to the nine-valent HPV vaccine. Clin. Microbiol. Infect. 2015, 21, 827–833. [Google Scholar] [CrossRef] [PubMed]
- Mollers, M.; King, A.J.; Knol, M.J.; Scherpenisse, M.; Meijer, C.J.L.M.; van der Klis, F.R.M.; de Melker, H.E. Effectiveness of human papillomavirus vaccine against incident and persistent infections among young girls: Results from a longitudinal dutch cohort study. Vaccine 2015, 33, 2678–2683. [Google Scholar] [CrossRef] [PubMed]
- Stern, P.L.; van der Burg, S.H.; Hampson, I.N.; Broker, T.; Fiander, A.; Lacey, C.J.; Kitchener, H.C.; Einstein, M.H. Therapy of human papillomavirus-related disease. Vaccine 2012, 30, F71–F82. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.J.; Jin, H.-T.; Hur, S.-Y.; Yang, H.G.; Seo, Y.B.; Hong, S.R.; Lee, C.-W.; Kim, S.; Woo, J.-W.; Park, K.S.; et al. Clearance of persistent HPV infection and cervical lesion by therapeutic DNA vaccine in CIN3 patients. Nat. Commun. 2014, 5, 5317. [Google Scholar] [CrossRef] [PubMed]
- Van der Burg, S.H.; Arens, R.; Melief, C.J.M. Immunotherapy for persistent viral infections and associated disease. Trends Immunol. 2011, 32, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Hus, I.; Gonet-Sebastianka, J.; Surdacka, A.; Bojarska-Junak, A.; Roliński, J. Analysis of peripheral blood immune cells after prophylactic immunization with HPV-16/18 ASO4-adjuvanted vaccine. Postep. Hig. Med. Doświadczalnej 2015, 69, 543–548. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-J.; Yang, A.; Wu, T.C.; Hung, C.-F. Immunotherapy for human papillomavirus-associated disease and cervical cancer: Review of clinical and translational research. J. Gynecol. Oncol. 2016, 27, e51. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.-X.; Li, D.; Guan, S.-S.; Zhao, C.; Li, Z.-L.; Zeng, Y. Immunotherapeutic effects of dendritic cells pulsed with a coden-optimized HPV 16 E6 and E7 fusion gene in vivo and in vitro. Asian Pac. J. Cancer Prev. 2015, 16, 3843–3847. [Google Scholar] [CrossRef] [PubMed]
- Mizuuchi, M.; Hirohashi, Y.; Torigoe, T.; Kuroda, T.; Yasuda, K.; Shimizu, Y.; Saito, T.; Sato, N. Novel oligomannose liposome-DNA complex DNA vaccination efficiently evokes anti-HPV E6 and E7 CTL responses. Exp. Mol. Pathol. 2012, 92, 185–190. [Google Scholar] [CrossRef] [PubMed]
- Van de Wall, S.; Nijman, H.W.; Daemen, T. HPV-specific immunotherapy: Key role for immunomodulators. Anticancer Agents Med. Chem. 2014, 14, 265–279. [Google Scholar] [CrossRef] [PubMed]
- Jindra, C.; Huber, B.; Shafti-Keramat, S.; Wolschek, M.; Ferko, B.; Muster, T.; Brandt, S.; Kirnbauer, R. Attenuated recombinant influenza a virus expressing HPV16 E6 and E7 as a novel therapeutic vaccine approach. PLoS ONE 2015, 10, e0138722. [Google Scholar]
- Rosales, R.; López-Contreras, M.; Rosales, C.; Magallanes-Molina, J.-R.; Gonzalez-Vergara, R.; Arroyo-Cazarez, J.M.; Ricardez-Arenas, A.; del Follo-Valencia, A.; Padilla-Arriaga, S.; Guerrero, M.V.; et al. Regression of human papillomavirus intraepithelial lesions is induced by MVA E2 therapeutic vaccine. Hum. Gene Ther. 2014, 25, 1035–1049. [Google Scholar] [CrossRef] [PubMed]
- Corona Gutierrez, C.M.; Tinoco, A.; Navarro, T.; Contreras, M.L.; Cortes, R.R.; Calzado, P.; Reyes, L.; Posternak, R.; Morosoli, G.; Verde, M.L.; et al. Therapeutic vaccination with MVA E2 can eliminate precancerous lesions (CIN 1, CIN 2, and CIN 3) associated with infection by oncogenic human papillomavirus. Hum. Gene Ther. 2004, 15, 421–431. [Google Scholar] [CrossRef] [PubMed]
- García-Hernández, E.; González-Sánchez, J.L.; Andrade-Manzano, A.; Contreras, M.L.; Padilla, S.; Guzmán, C.C.; Jiménez, R.; Reyes, L.; Morosoli, G.; Verde, M.L.; et al. Regression of papilloma high-grade lesions (CIN 2 and CIN 3) is stimulated by therapeutic vaccination with MVA E2 recombinant vaccine. Cancer Gene Ther. 2006, 13, 592–597. [Google Scholar] [CrossRef] [PubMed]
- Adams, M.; Navabi, H.; Jasani, B.; Man, S.; Fiander, A.; Evans, A.S.; Donninger, C.; Mason, M. Dendritic cell (DC) based therapy for cervical cancer: Use of DC pulsed with tumour lysate and matured with a novel synthetic clinically non-toxic double stranded RNA analogue poly [I]:Poly [C12U] (Ampligen®). Vaccine 2003, 21, 787–790. [Google Scholar] [CrossRef]
- Santin, A.D.; Bellone, S.; Palmieri, M.; Zanolini, A.; Ravaggi, A.; Siegel, E.R.; Roman, J.J.; Pecorelli, S.; Cannon, M.J. Human papillomavirus type 16 and 18 E7-pulsed dendritic cell vaccination of stage IB or IIA cervical cancer patients: A phase I escalating-dose trial. J. Virol. 2008, 82, 1968–1979. [Google Scholar] [CrossRef] [PubMed]
- Nieto, K.; Gissmann, L.; Schädlich, L. Human papillomavirus-specific immune therapy: Failure and hope. Antivir. Ther. 2010, 15, 951–957. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Bae, S.N.; Lee, C.W.; Song, M.J.; Lee, S.J.; Yoon, J.H.; Lee, K.H.; Hur, S.Y.; Park, T.C.; Park, J.S. A pilot study to investigate the treatment of cervical human papillomavirus infection with zinc-citrate compound (cizar®). Gynecol. Oncol. 2011, 122, 303–306. [Google Scholar] [CrossRef] [PubMed]
- Carlos de Freitas, A.; da Conceicao Gomes Leitao, M.; Coimbra, E.C. Prospects of molecularly-targeted therapies for cervical cancer treatment. Curr. Drug Targets 2015, 16, 77–91. [Google Scholar] [CrossRef]
- Eyckerman, S.; Titeca, K.; van Quickelberghe, E.; Cloots, E.; Verhee, A.; Samyn, N.; de Ceuninck, L.; Timmerman, E.; de Sutter, D.; Lievens, S.; et al. Trapping mammalian protein complexes in viral particles. Nat. Commun. 2016, 7, 11416. [Google Scholar] [CrossRef] [PubMed]
- Lemmens, I.; Lievens, S.; Tavernier, J. Mappit, a mammalian two-hybrid method for in-cell detection of protein-protein interactions. Methods Mol. Biol. 2015, 1278, 447–455. [Google Scholar] [PubMed]
- Yan, J.; Li, Q.; Lievens, S.; Tavernier, J.; You, J. Abrogation of the Brd4-positive transcription elongation factor B complex by papillomavirus E2 protein contributes to viral oncogene repression. J. Virol. 2010, 84, 76–87. [Google Scholar] [CrossRef] [PubMed]



© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shanmugasundaram, S.; You, J. Targeting Persistent Human Papillomavirus Infection. Viruses 2017, 9, 229. https://doi.org/10.3390/v9080229
Shanmugasundaram S, You J. Targeting Persistent Human Papillomavirus Infection. Viruses. 2017; 9(8):229. https://doi.org/10.3390/v9080229
Chicago/Turabian StyleShanmugasundaram, Srinidhi, and Jianxin You. 2017. "Targeting Persistent Human Papillomavirus Infection" Viruses 9, no. 8: 229. https://doi.org/10.3390/v9080229
APA StyleShanmugasundaram, S., & You, J. (2017). Targeting Persistent Human Papillomavirus Infection. Viruses, 9(8), 229. https://doi.org/10.3390/v9080229