
Reliable Detection of Herpes Simplex Virus Sequence Variation by High-Throughput Resequencing

Abstract
1. Introduction
2. Materials and Methods
2.1. Viruses
2.2. DNA Extraction, Illumina Library Preparation and Sequencing
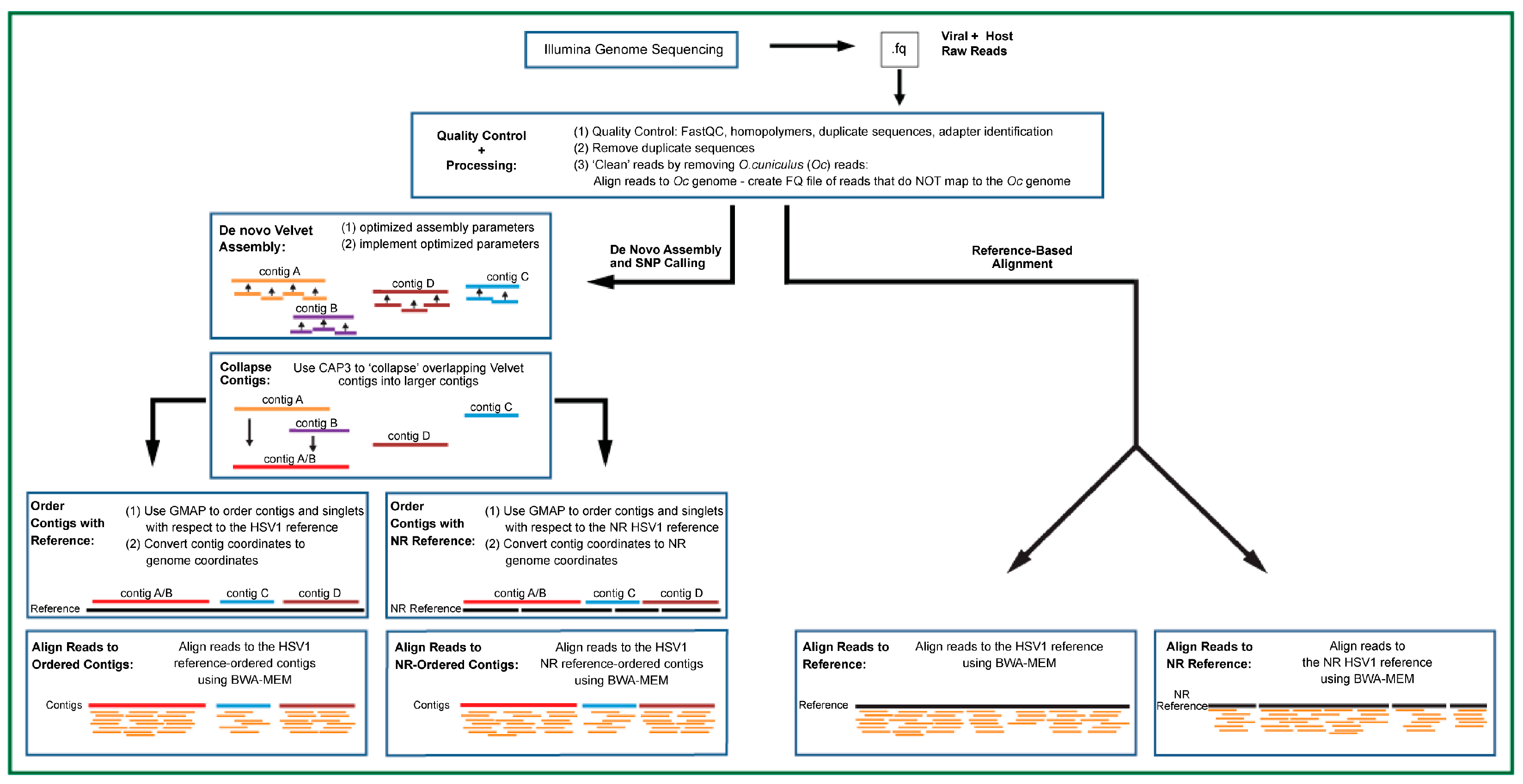
2.3. Illumina Raw Read QC and Processing
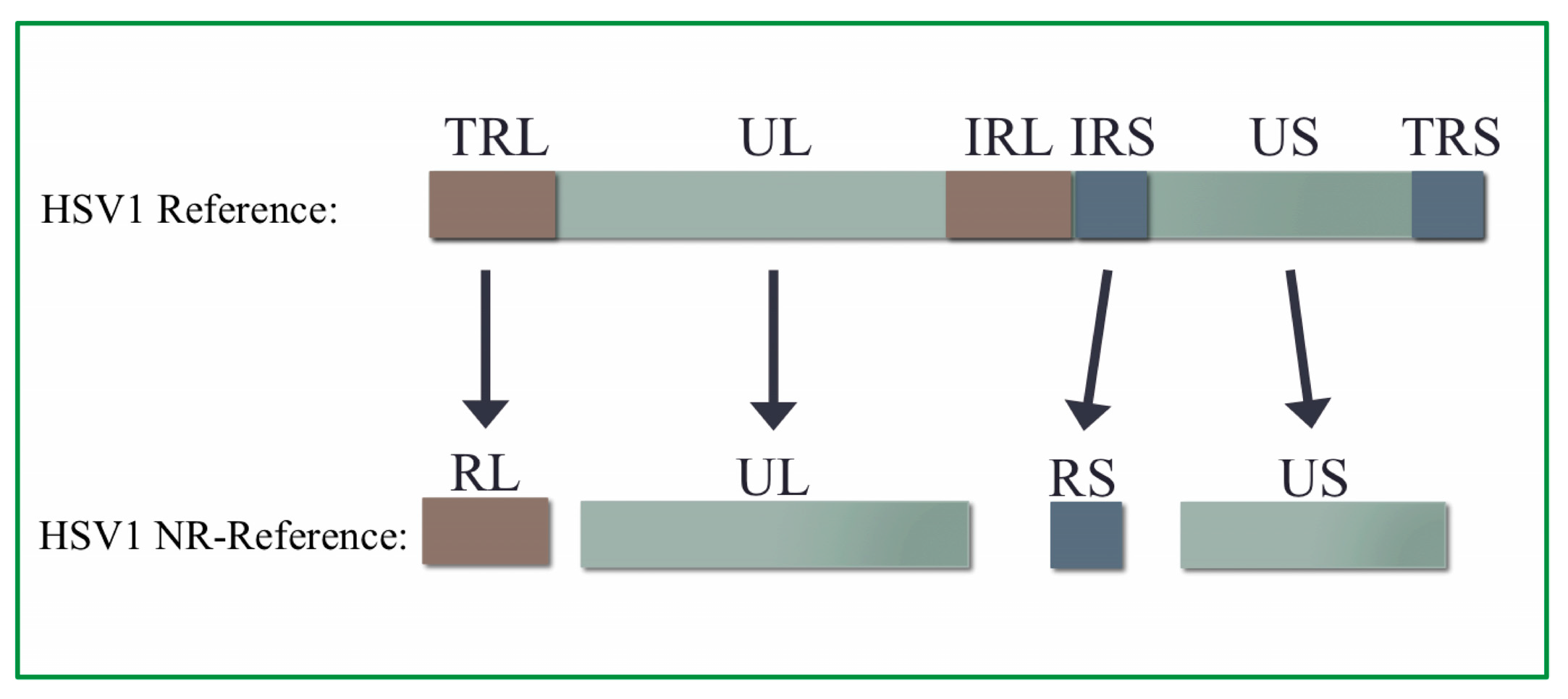
2.4. Creation of HSV1 Non-Redundant Genome
2.5. De Novo Contig Assembly
2.6. Alignment of Host-Processed Reads
2.7. Variant Discovery
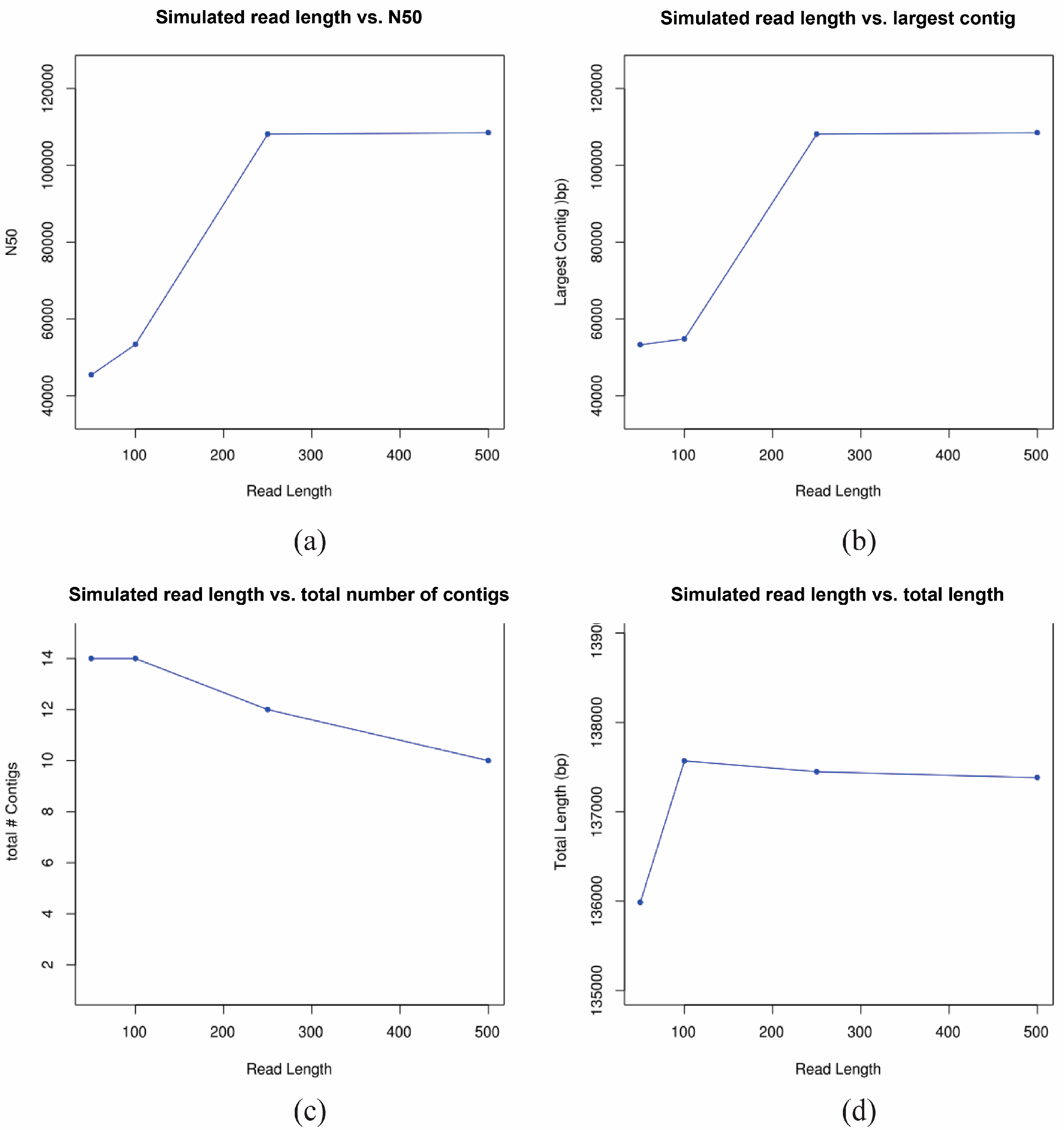
2.8. Simulation Study
3. Results
3.1. Quality Control and Processing of High-Throughput Sequencing Reads
3.2. Non-Redundant HSV1 Genome
3.3. De Novo Assembly of 17syn+
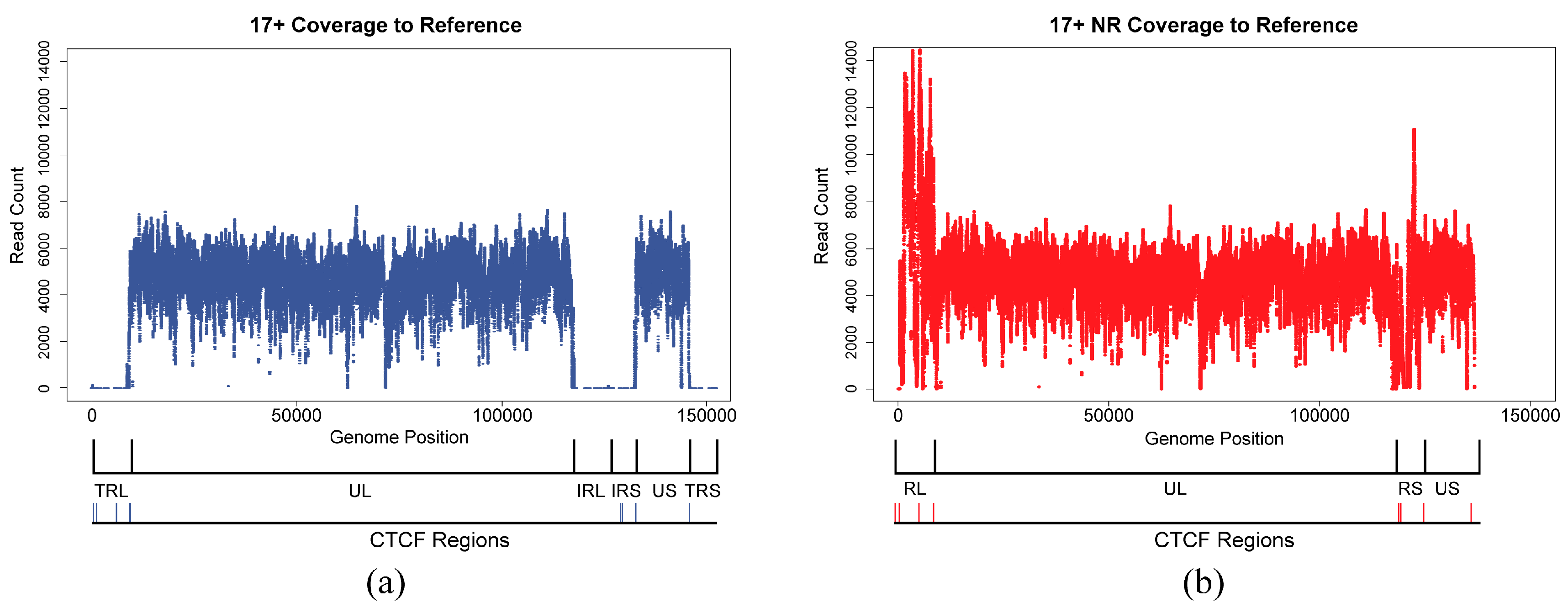
3.4. Alignments and Variant Calling
4. Discussion
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Macdonald, S.J.; Mostafa, H.H.; Morrison, L.A.; Davido, D.J. Genome sequence of herpes simplex virus 1 strain KOS. J. Virol. 2012, 86, 6371–6372. [Google Scholar] [CrossRef] [PubMed]
- Macdonald, S.J.; Mostafa, H.H.; Morrison, L.A.; Davido, D.J. Genome sequence of herpes simplex virus 1 strain McKrae. J. Virol. 2012, 86, 9540–9541. [Google Scholar] [CrossRef] [PubMed]
- Szpara, M.L.; Parsons, L.; Enquist, L.W. Sequence variability in clinical and laboratory isolates of herpes simplex virus 1 reveals new mutations. J. Virol. 2010, 84, 5303–5313. [Google Scholar] [CrossRef] [PubMed]
- Cunha, C.W.; Taylor, K.E.; Pritchard, S.M.; Delboy, M.G.; Sari, T.K.; Aguilar, H.C.; Mossman, K.L.; Nicola, A.V. Widely used herpes simplex virus 1 ICP0 deletion mutant strain dl1403 and its derivative viruses do not express glycoprotein c due to a secondary mutation in the gC gene. PLoS ONE 2015, 10, e0131129. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, C.; Gatherer, D.; Hilfrich, B.; Baluchova, K.; Dargan, D.J.; Thomson, M.; Griffiths, P.D.; Wilkinson, G.W.G.; Schulz, T.F.; Davison, A.J. Sequences of complete human cytomegalovirus genomes from infected cell cultures and clinical specimens. J. Gen. Virol. 2010, 91, 605–615. [Google Scholar] [CrossRef] [PubMed]
- Szpara, M.L.; Gatherer, D.; Ochoa, A.; Greenbaum, B.; Dolan, A.; Bowden, R.J.; Enquist, L.W.; Legendre, M.; Davison, A.J. Evolution and diversity in human herpes simplex virus genomes. J. Virol. 2014, 88, 1209–1227. [Google Scholar] [CrossRef] [PubMed]
- Colgrove, R.; Diaz, F.; Newman, R.; Saif, S.; Shea, T.; Young, S.; Henn, M.; Knipe, D.M. Genomic sequences of a low passage herpes simplex virus 2 clinical isolate and its plaque-purified derivative strain. Virology 2014, 450–451, 140–145. [Google Scholar] [CrossRef] [PubMed]
- Gnerre, S.; Lander, E.S.; Lindblad-Toh, K.; Jaffe, D.B. Assisted assembly: How to improve a de novo genome assembly by using related species. Genome Biol. 2009, 10, R88. [Google Scholar] [CrossRef] [PubMed]
- Van Gurp, T.P.; McIntyre, L.M.; Verhoeven, K.J.F. Consistent errors in first strand cDNA due to random hexamer mispriming. PLoS ONE 2013, 8, e85583. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Speed, T.P. Summarizing and correcting the GC content bias in high-throughput sequencing. Nucleic Acids Res. 2012, 40, e72. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, R.; Paul, J.S.; Albrechtsen, A.; Song, Y.S. Genotype and SNP calling from next-generation sequencing data. Nat. Rev. Genet. 2011, 12, 443–451. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Ruan, J.; Durbin, R. Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res. 2008, 18, 1851–1858. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Exploring single-sample SNP and INDEL calling with whole-genome de novo assembly. Bioinformatics 2012, 28, 1838–1844. [Google Scholar] [CrossRef] [PubMed]
- Sohn, J.; Nam, J.-W. The present and future of de novo whole-genome assembly. Brief. Bioinform. 2016. [Google Scholar] [CrossRef] [PubMed]
- Willerth, S.M.; Pedro, H.A.M.; Pachter, L.; Humeau, L.M.; Arkin, A.P.; Schaffer, D.V. Development of a low bias method for characterizing viral populations using next generation sequencing technology. PLoS ONE 2010, 5, e13564. [Google Scholar] [CrossRef] [PubMed]
- Olson, N.D.; Lund, S.P.; Colman, R.E.; Foster, J.T.; Sahl, J.W.; Schupp, J.M.; Keim, P.; Morrow, J.B.; Salit, M.L.; Zook, J.M. Best practices for evaluating single nucleotide variant calling methods for microbial genomics. Front. Genet. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Zerbino, D.R.; Birney, E. Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008, 18, 821–829. [Google Scholar] [CrossRef] [PubMed]
- Hage, E.; Wilkie, G.S.; Linnenweber-Held, S.; Dhingra, A.; Suárez, N.M.; Schmidt, J.J.; Kay-Fedorov, P.; Mischak-Weissinger, E.; Heim, A.; Schwarz, A.; et al. Characterization of human cytomegalovirus genome diversity in immunocompromised hosts by whole genomic sequencing directly from clinical specimens. J. Infect. Dis. 2017. [Google Scholar] [CrossRef] [PubMed]
- Amelio, A.L.; McAnany, P.K.; Bloom, D.C. A chromatin insulator-like element in the herpes simplex virus type 1 latency-associated transcript region binds CCCTC-binding factor and displays enhancer-blocking and silencing activities. J. Virol. 2006, 80, 2358–2368. [Google Scholar] [CrossRef] [PubMed]
- Bloom, D.C. HSV Vectors for Gene Therapy. In Herpes Simplex Virus Protocols; Brown, S.M., MacLean, A.R., Eds.; Humana Press: Totowa, NJ, USA, 1998; pp. 369–386. ISBN 978-1-59259-594-5. [Google Scholar]
- Andrews, S. Babraham Bioinformatics—FastQC A Quality Control Tool for High Throughput Sequence Data. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 1 May 2017).
- Ewing, B.; Green, P. Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res. 1998, 8, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Ewing, B.; Hillier, L.; Wendl, M.C.; Green, P. Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res. 1998, 8, 175–185. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [PubMed]
- Zerbino, D.R. Using the Velvet de novo assembler for short-read sequencing technologies. Curr. Protoc. Bioinform. 2010. [Google Scholar] [CrossRef]
- Huang, X.; Madan, A. CAP3: A DNA sequence assembly program. Genome Res. 1999, 9, 868–877. [Google Scholar] [CrossRef] [PubMed]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef] [PubMed]
- Kent, W.J. BLAT—The BLAST-like alignment tool. Genome Res. 2002, 12, 656–664. [Google Scholar] [CrossRef] [PubMed]
- Frith, M.C.; Hamada, M.; Horton, P. Parameters for accurate genome alignment. BMC Bioinform. 2010, 11, 80. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.D.; Watanabe, C.K. GMAP: A genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics 2005, 21, 1859–1875. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [PubMed]
- Burrows-Wheeler Aligner. Available online: URL bio-bwa.sourceforge.net (accessed on 16 July 2014).
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [PubMed]
- McGeoch, D.J.; Dalrymple, M.A.; Davison, A.J.; Dolan, A.; Frame, M.C.; McNab, D.; Perry, L.J.; Scott, J.E.; Taylor, P. The complete DNA sequence of the long unique region in the genome of herpes simplex virus type 1. J. Gen. Virol. 1988, 69, 1531–1574. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Kolb, A.W.; Sverchkov, Y.; Cuellar, J.A.; Craven, M.; Brandt, C.R. Recombination analysis of herpes simplex virus 1 reveals a bias toward GC content and the inverted repeat regions. J. Virol. 2015, 89, 7214–7223. [Google Scholar] [CrossRef] [PubMed]
- Umene, K.; Yoshida, M.; Fukumaki, Y. Genetic variability in the region encompassing reiteration VII of herpes simplex virus type 1, including deletions and multiplications related to recombination between direct repeats. SpringerPlus 2015, 4, 200. [Google Scholar] [CrossRef] [PubMed]
- Hayward, G.S.; Jacob, R.J.; Wadsworth, S.C.; Roizman, B. Anatomy of herpes simplex virus DNA: Evidence for four populations of molecules that differ in the relative orientations of their long and short components. Proc. Natl. Acad. Sci. USA 1975, 72, 4243–4247. [Google Scholar] [CrossRef] [PubMed]
- Szpara, M.L.; Tafuri, Y.R.; Parsons, L.; Shamim, S.R.; Verstrepen, K.J.; Legendre, M.; Enquist, L.W. A wide extent of inter-strain diversity in virulent and vaccine strains of alphaherpesviruses. PLoS Pathog. 2011, 7, e1002282. [Google Scholar] [CrossRef] [PubMed]
- Lander, E.S.; Waterman, M.S. Genomic mapping by fingerprinting random clones: A mathematical analysis. Genomics 1988, 2, 231–239. [Google Scholar] [CrossRef]
- Chang, Z.; Wang, Z.; Li, G. The impacts of read length and transcriptome complexity for de novo assembly: A simulation study. PLoS ONE 2014, 9, e94825. [Google Scholar] [CrossRef] [PubMed]
- Ertel, M.K.; Cammarata, A.L.; Hron, R.J.; Neumann, D.M. CTCF occupation of the herpes simplex virus 1 genome is disrupted at early times postreactivation in a transcription-dependent manner. J. Virol. 2012, 86, 12741–12759. [Google Scholar] [CrossRef] [PubMed]
- Garrison, E.; Marth, G. Haplotype-based variant detection from short-read sequencing. arXiv, 2012; arXiv:12073907. [Google Scholar]
- Alkan, C.; Coe, B.P.; Eichler, E.E. Genome structural variation discovery and genotyping. Nat. Rev. Genet. 2011, 12, 363–376. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, A.; Golicz, A.; Hackett, C.A.; Milne, I.; Stephen, G.; Marshall, D.; Flavell, A.J.; Bayer, M. An investigation of causes of false positive single nucleotide polymorphisms using simulated reads from a small eukaryote genome. BMC Bioinform. 2015, 16, 382. [Google Scholar] [CrossRef] [PubMed]
- Karamitros, T.; Harrison, I.; Piorkowska, R.; Katzourakis, A.; Magiorkinis, G.; Mbisa, J.L. De novo assembly of human herpes virus type 1 (HHV-1) genome, mining of non-canonical structures and detection of novel drug-resistance mutations using short- and long-read next generation sequencing technologies. PLoS ONE 2016, 11, e0157600. [Google Scholar] [CrossRef] [PubMed]
- Pirooznia, M.; Goes, F.S.; Zandi, P.P. Whole-genome CNV analysis: Advances in computational approaches. Front. Genet. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Smiley, J.R.; Fong, B.S.; Leung, W.-C. Construction of a double-jointed herpes simplex viral DNA molecule: Inverted repeats are required for segment inversion, and direct repeats promote deletions. Virology 1981, 113, 345–362. [Google Scholar] [CrossRef]
- Smiley, J.R.; Duncan, J.; Howes, M. Sequence requirements for DNA rearrangements induced by the terminal repeat of herpes simplex virus type 1 KOS DNA. J. Virol. 1990, 64, 5036–5050. [Google Scholar] [PubMed]
- Perry, L.J.; McGeoch, D.J. The DNA sequences of the long repeat region and adjoining parts of the long unique region in the genome of herpes simplex virus type 1. J. Gen. Virol. 1988, 69, 2831–2846. [Google Scholar] [CrossRef] [PubMed]
- Chou, J.; Roizman, B. The herpes simplex virus 1 gene for ICP34.5, which maps in inverted repeats, is conserved in several limited-passage isolates but not in strain 17syn+. J. Virol. 1990, 64, 1014–1020. [Google Scholar] [PubMed]
- Parsons, L.R.; Tafuri, Y.R.; Shreve, J.T.; Bowen, C.D.; Shipley, M.M.; Enquist, L.W.; Szpara, M.L. Rapid genome assembly and comparison decode intrastrain variation in human alphaherpesviruses. mBio 2015, 6. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reads | 17syn+ | 17∆CTRL2 |
|---|---|---|
| total # paired end reads | 49,967,108 | 43,635,207 |
| # paired end aligning (unique + ambig) to Oc (%) | 23,418,917 (46.9%) | 254,356 (6.9%) |
| # paired end non-host reads (unaln + ambig) | 30,149,765 (60.3%) | 3,414,197 (94.0%) |
| # paired end host-processed reads | 28,822,236 | 34,263,711 |
| # reads aligning uniquely to HSV1 reference | 18,802,764 | 49,774,582 |
| # reads aligning uniquely to HSV1 NR-reference | 21,418,760 | 55,660,042 |
| Metric | Velvet Contigs | Velvet-CAP3 Contigs | HSV1_Mapped_Contigs |
|---|---|---|---|
| Total # of Contigs | 157 | 105 | 15 |
| # Contigs ≥ 1000 bp | 27 | 27 | 10 |
| Largest Contig (bp) | 53,719 | 53,719 | 53,719 |
| Total Length ≥ 0 bp | 188,444 | 183,313 | 135,829 |
| Total Length ≥ 1000 bp | 162,124 | 162,124 | 131,913 |
| N50 | 45,694 | 45,694 | 45,694 |
| L50 | 2 | 2 | 2 |
| Genome Fraction (%) | 97.37 | 97.44 | 97.44 |
| Duplication Ratio | 1.018 | 1.018 | 1.018 |
| % GC | 64.34 | 64.32 | 67.41 |
| NR-Reference Length (bp) | 136,770 | 136,770 | 136,770 |
| Reference % GC | 67.56 | 67.56 | 67.56 |
| # N’s | 8303 | 8303 | 1907 |
| # Mismatches + Indels | 82 | 83 | 83 |
| Reads | Reference | # Variants in RL | # Variants in RS | # Variants in US | # Variants in UL | Total # Variants |
|---|---|---|---|---|---|---|
| 17syn+ | HSV1 | 2 | 1 | 2 | 46 | 51 |
| 17syn+ | HSV1 NR | 9 | 3 | 2 | 46 | 60 |
| 17∆CTRL2 | HSV1 | 2 | 1 | 2 | 43 | 48 |
| 17∆CTRL2 | HSV1 NR | 8 | 3 | 2 | 43 | 56 |
| Alignment Strategy | # Variants in RL | # Variants in RS | # Variants in UL | # Variants in US | # Variants Total |
|---|---|---|---|---|---|
| 2a: segregating variants | 5 (0) | 1 (1) | 19 (19) | 1 (1) | 26 (21) |
| 2b: segregating variants | 3 (4) | 1 (3) | 24 (24) | 1 (1) | 29 (32) |
| 3a: divergent variants | 4 (2) | 2 (0) | 27 (27) | 1 (1) | 34 (30) |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morse, A.M.; Calabro, K.R.; Fear, J.M.; Bloom, D.C.; McIntyre, L.M. Reliable Detection of Herpes Simplex Virus Sequence Variation by High-Throughput Resequencing. Viruses 2017, 9, 226. https://doi.org/10.3390/v9080226
Morse AM, Calabro KR, Fear JM, Bloom DC, McIntyre LM. Reliable Detection of Herpes Simplex Virus Sequence Variation by High-Throughput Resequencing. Viruses. 2017; 9(8):226. https://doi.org/10.3390/v9080226
Chicago/Turabian StyleMorse, Alison M., Kaitlyn R. Calabro, Justin M. Fear, David C. Bloom, and Lauren M. McIntyre. 2017. "Reliable Detection of Herpes Simplex Virus Sequence Variation by High-Throughput Resequencing" Viruses 9, no. 8: 226. https://doi.org/10.3390/v9080226
APA StyleMorse, A. M., Calabro, K. R., Fear, J. M., Bloom, D. C., & McIntyre, L. M. (2017). Reliable Detection of Herpes Simplex Virus Sequence Variation by High-Throughput Resequencing. Viruses, 9(8), 226. https://doi.org/10.3390/v9080226