Hijacking of the Ubiquitin/Proteasome Pathway by the HIV Auxiliary Proteins



Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
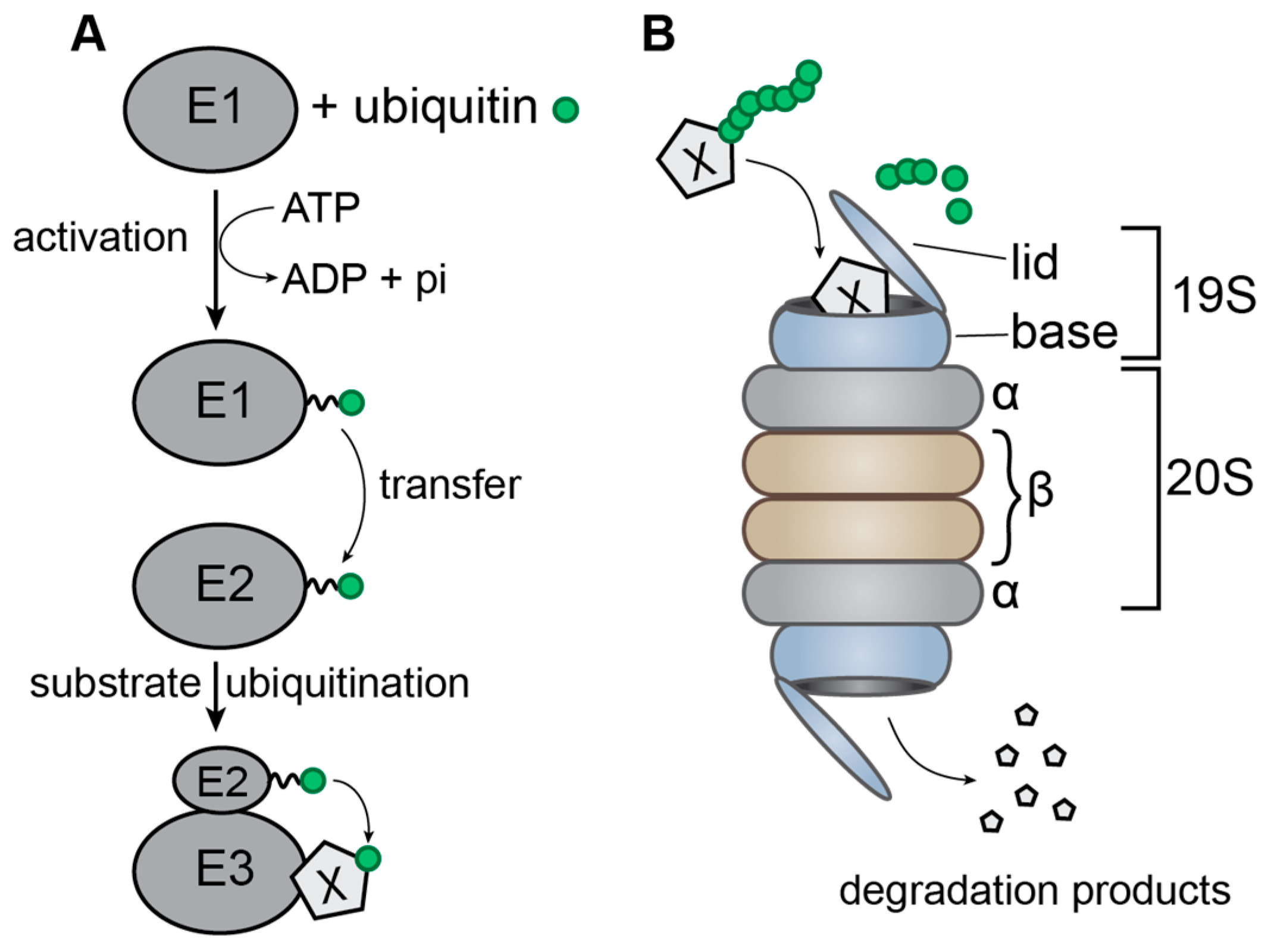
2. The Ubiquitin-Proteasome System
- (1)
- Some cellular E3 ubiquitin ligases recognize viral proteins and induce their ubiquitination, which can have a positive effect on viral replication. For instance, ubiquitination of the p6 domain of the HIV-1 Gag polyprotein is important for the interaction of p6 with the ESCRT machinery. However, the mono-ubiquitination of lysine residues within the p6 domain (K27 and K33) does not seem to be sufficient to facilitate budding of new virions, the latter being also dependent on the cumulative ubiquitination of NC-p2 (NucleoCapsid-peptide 2) domain [21,22,23,24]. Ubiquitination of the HIV-1 accessory protein Tat by cellular E3 ligases stimulates transcription of viral RNA [25,26].
- (2)
- Ubiquitination of viral proteins can also induce their degradation, thereby blocking the viral life cycle. This is a strategy used by certain restriction factors. The polymerase PB1 (Protein Binding 1) of the Influenza A virus (IAV) for example is ubiquitinated (K48-linked ubiquitin) by the cellular E3 ubiquitin ligase TRIM32 (TRIpartite Motif-containing protein 32), followed by its degradation by the proteasome [27]. This seems to be a general mechanism as PB1 proteins derived from various IAV serotypes (H1N1 (Hemagglutinin 1 Neuraminidase 1), H3N2, H5N1 or H7N9) associate with TRIM32 in multiple cell types and this suggests that PB1 has not yet adapted to avoid TRIM32 targeting [28]. The Human herpesvirus type I (HSV-1) capsid protein Vp5 has also been shown to be degraded by the ubiquitin proteasome system, leaving the viral genome exposed to innate immune sensors [29]. Interestingly, TRIM5α was reported to inhibit HSV-1 and -2 replication at an early stage of the infection cycle [30], suggesting a role for this or related protein in cytosolic sensing of herpesvirus capsids.
- (3)
- Certain viruses have evolved to recruit the cellular E3 ligases to induce the degradation of cellular proteins that might have harmful effects on the viral life cycle. For instance, the protein E6 of Human papillomavirus (HPV) recruits the cellular E3 ubiquitin ligase E6-AP to induce ubiquitination and degradation of p53, thereby allowing viral replication [31,32]. The NSP1 (Non-Structural RNA binding protein 1) protein of Rotaviruses subverts the Skp1-Cul1-Fbox (SCF) E3 ligase to induce the ubiquitination and degradation of β-TrCP (β-Transducin repeat Containing Protein). β-TrCP is by itself a substrate adaptor of an E3 ligase and its degradation leads to accumulation of the NF-ϰB inhibitor IϰB, resulting in inhibition of the NF-ϰB induced antiviral responses [33,34]. These mechanisms are important for HIV replication and will be detailed in Section 5.
- (4)
- Other viruses directly encode their own E3 ligases. Kaposi sarcoma herpesvirus (KSHV) protein K3 and K5 (RING-CH family of ligases) ubiquitinate MHC-I (Major Histocompatibility Complex I), resulting in its down-regulation from the cell surface through a clathrin-dependent sorting pathway to an endolysosomal compartment [35,36]. This endolysosomal sorting requires K63-linked instead of K48-linked polyubiquitin chains [19]. Another well-known example is the ICP0 protein (Infected Cell Protein 0) of HSV-1, an E3 ubiquitin ligase which induces the degradation of the ND10 (Nuclear Domain 10) nuclear body components PML (Promyelocytic Leukemia Protein) and Sp100 through the UPS, thereby avoiding antiviral sensing [37,38]. ICP0 has also been shown to have a RING-independent E3 ligase activity that polyubiquitinates the E2 enzyme cdc34. ICP0 influences many cellular pathways and is required for the activation of most viral and many cellular genes, for reactivation from latency and suppression of innate immunity [19].
- (5)
- Finally, ubiquitin modifications can be reversed by the isopeptide-bond specific proteolytic activity of DUBs. In addition to cellular DUBs, it has been reported that various virus families code their own DUBs (Coronavirus, Herpesvirus etc.) to evade host antiviral immune response and promote virus replication (for a recent review see [1]). For instance, in the herpesviridae family, a variety of DUBs play an important role in the virus life cycle (e.g., UL36USP (Ubiquitin Ligase 36 Ubiquitin Specific Protease) of HSV-1, tegument protein pUL48 of human cytomegalovirus (HCMV)). Regarding HIV-1, a recent study reported that several cellular DUBs (USP7 and USP47, Ubiquitin Specific Protease family) play an important role in its replication by regulating Gag processing and thus the infectivity of released virions and simultaneously the entry of Gag into the UPS and MHC-I pathway [39]. Moreover, this study showed that treatment with DUB inhibitors targeting USP47 causes a general Gag processing defect, indicating that USP47 interacts with Gag and prevents its entry into the UPS. Similarly, proteasome inhibitors have been shown to impact HIV-1 replication by reducing the release and maturation of infectious particles [40,41] or by suppressing its transcription [42]. Taken together, these studies suggest a potential antiretroviral activity of DUB and proteasome inhibitors.
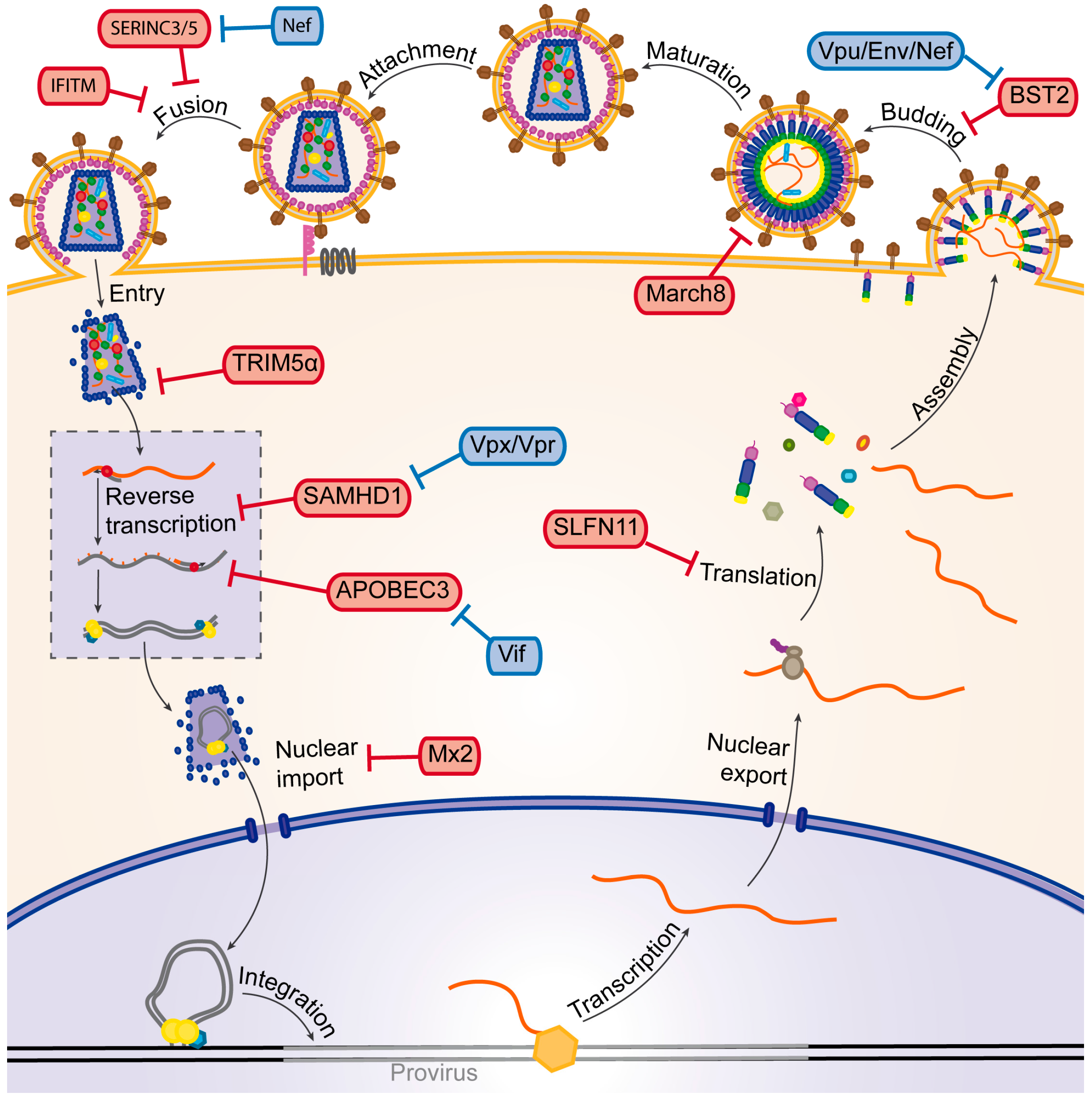
3. The HIV Life Cycle
4. Cellular Factors Mediating Viral Restriction Using the UPS
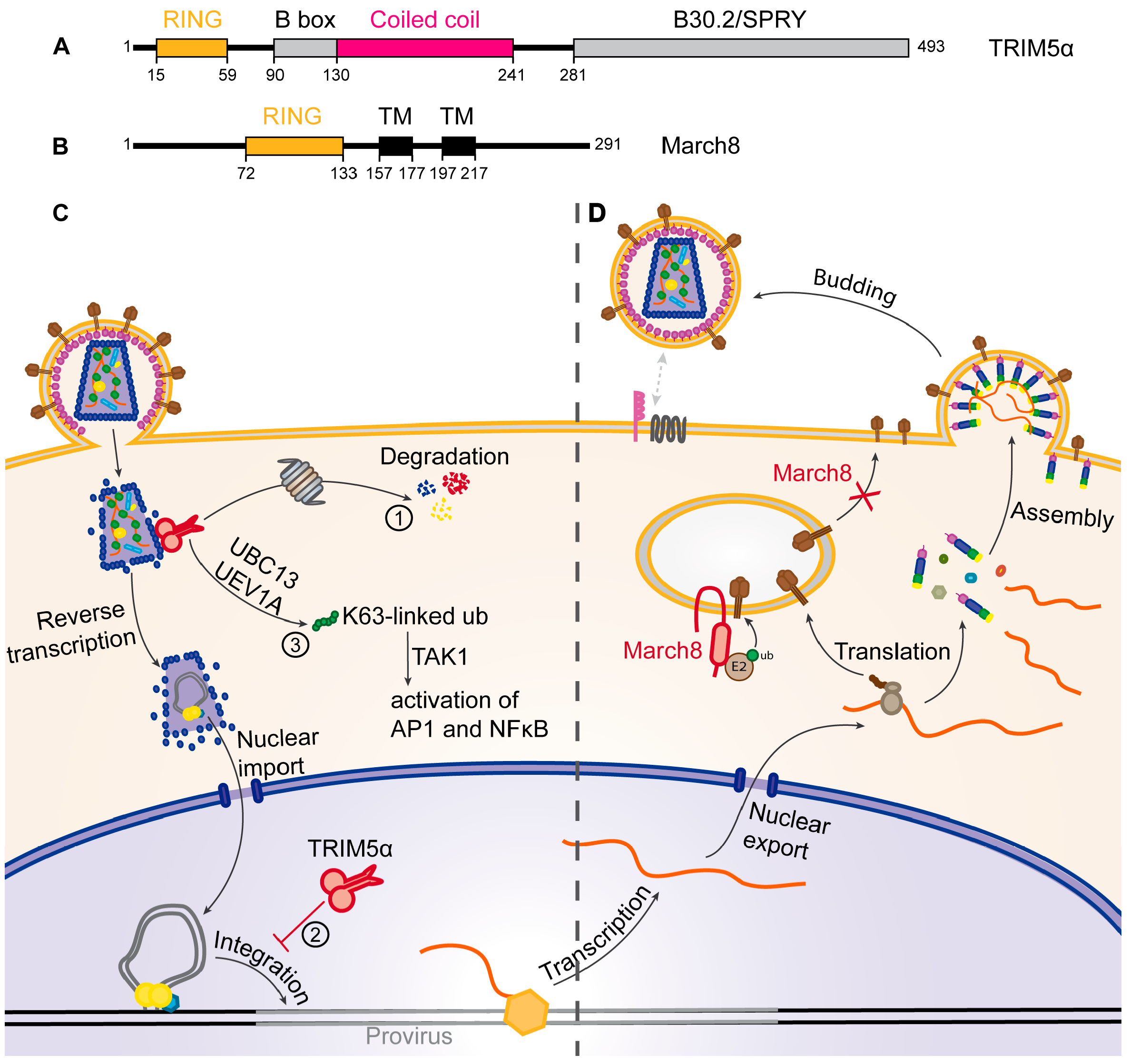
4.1. TRIM5α
4.2. March8
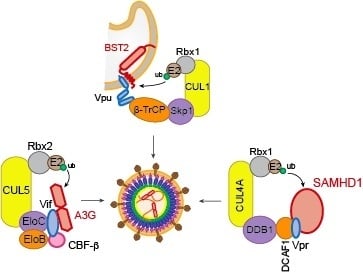
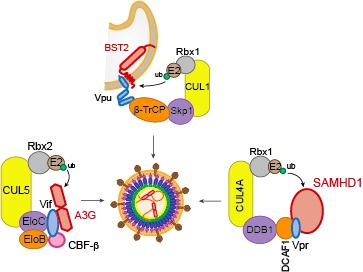
5. Counteraction of Restriction Factors by Viral Auxiliary Proteins Using the UPS
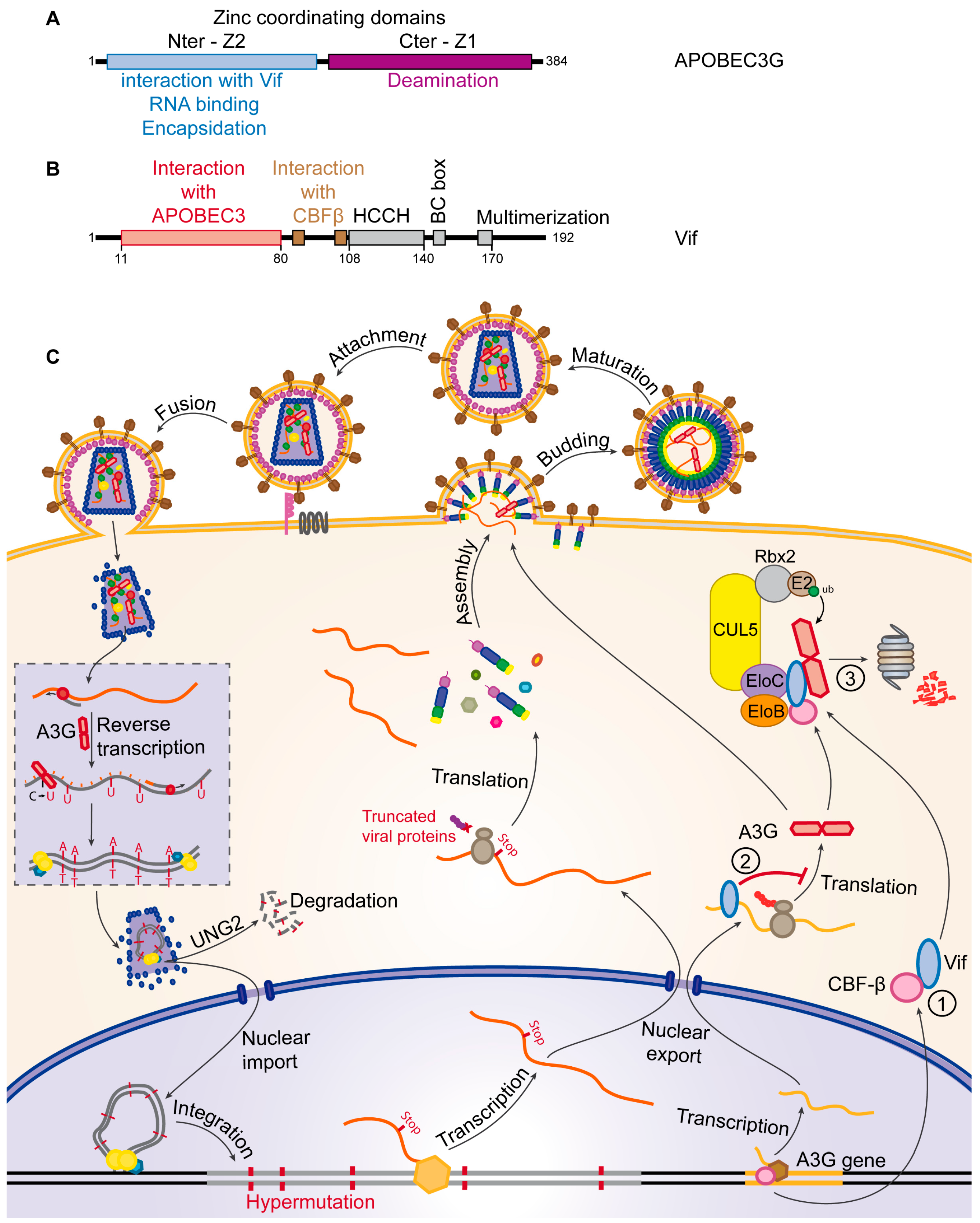
5.1. Vif
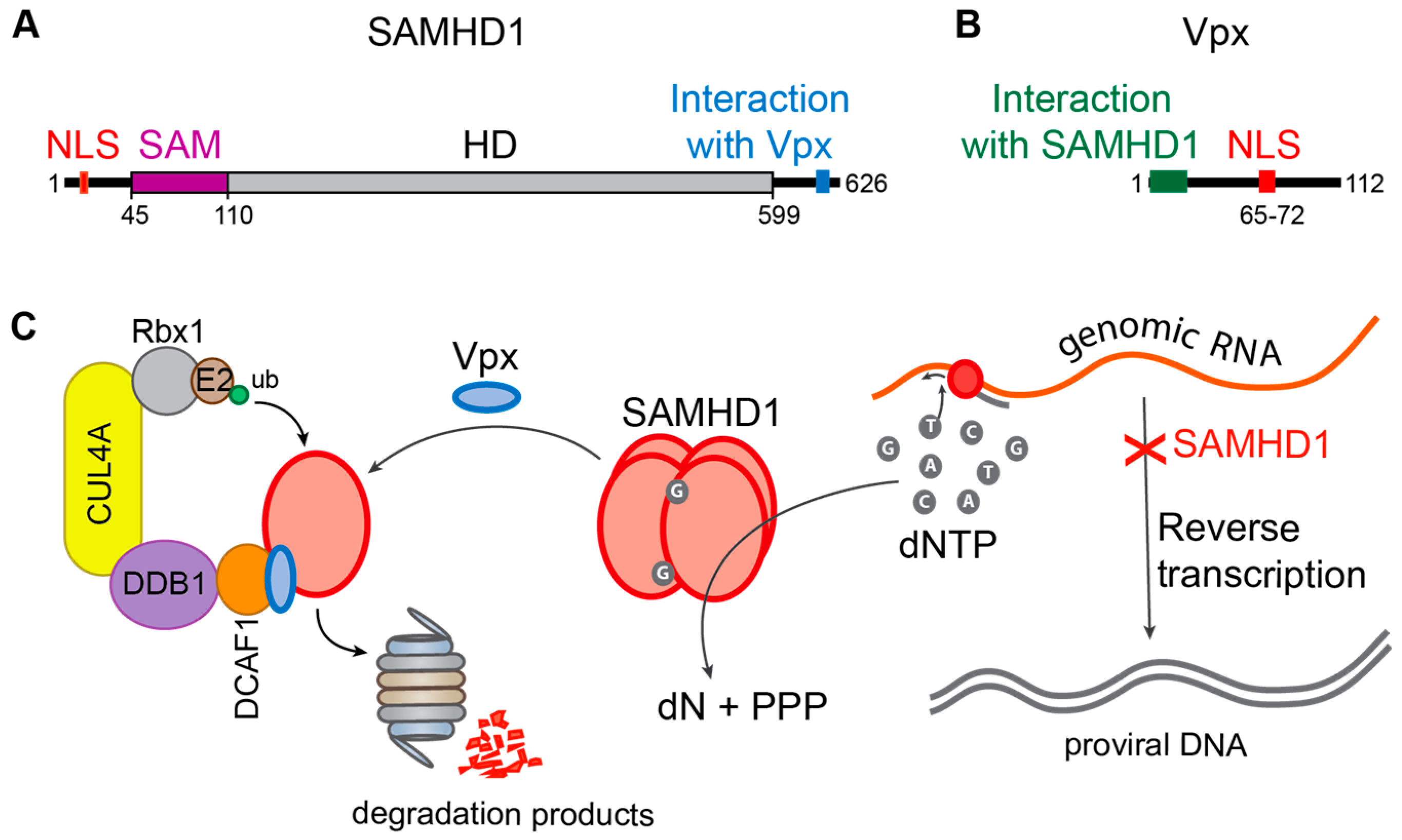
5.2. Vpx
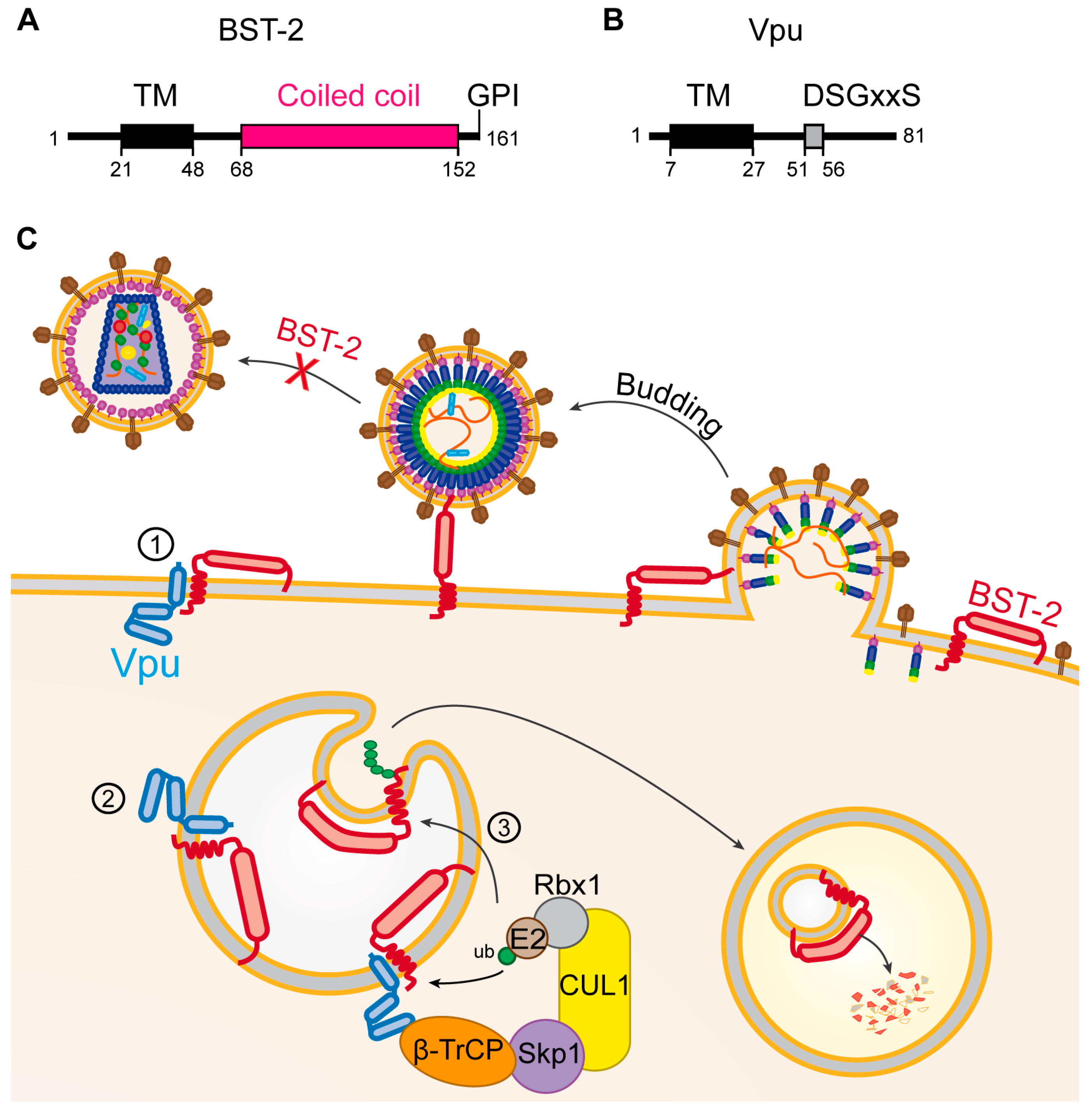
5.3. Vpu
6. Other Cellular Proteins Targeted by the Hijacked UPS
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bailey-Elkin, B.A.; Knaap, R.C.M.; Kikkert, M.; Mark, B.L. Structure and function of viral deubiquitinating enzymes. J. Mol. Biol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Roos-Mattjus, P.; Sistonen, L. The ubiquitin-proteasome pathway. Ann. Med. 2004, 36, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Clague, M.J.; Urbé, S. Ubiquitin: Same molecule, different degradation pathways. Cell 2010, 143, 682–685. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.E.; Gack, M.U. Ubiquitination in the antiviral immune response. Virology 2015, 479–480, 52–65. [Google Scholar] [CrossRef] [PubMed]
- Petroski, M.D.; Deshaies, R.J. Function and regulation of cullin-RING ubiquitin ligases. Nat. Rev. Mol. Cell Biol. 2005, 6, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Hershko, A.; Heller, H.; Elias, S.; Ciechanover, A. Components of ubiquitin-protein ligase system. Resolution, affinity purification, and role in protein breakdown. J. Biol. Chem. 1983, 258, 8206–8214. [Google Scholar] [PubMed]
- Scheffner, M.; Nuber, U.; Huibregtse, J.M. Protein ubiquitination involving an E1-E2-E3 enzyme ubiquitin thioester cascade. Nature 1995, 373, 81–83. [Google Scholar] [CrossRef] [PubMed]
- Gregori, L.; Poosch, M.S.; Cousins, G.; Chau, V. A uniform isopeptide-linked multiubiquitin chain is sufficient to target substrate for degradation in ubiquitin-mediated proteolysis. J. Biol. Chem. 1990, 265, 8354–8357. [Google Scholar] [PubMed]
- Chau, V.; Tobias, J.W.; Bachmair, A.; Marriott, D.; Ecker, D.J.; Gonda, D.K.; Varshavsky, A. A multiubiquitin chain is confined to specific lysine in a targeted short-lived protein. Science 1989, 243, 1576–1583. [Google Scholar] [CrossRef] [PubMed]
- Collins, G.A.; Goldberg, A.L. The logic of the 26S proteasome. Cell 2017, 169, 792–806. [Google Scholar] [CrossRef] [PubMed]
- Matthews, W.; Driscoll, J.; Tanaka, K.; Ichihara, A.; Goldberg, A.L. Involvement of the proteasome in various degradative processes in mammalian cells. Proc. Natl. Acad. Sci. USA 1989, 86, 2597–2601. [Google Scholar] [CrossRef] [PubMed]
- Bashore, C.; Dambacher, C.M.; Goodall, E.A.; Matyskiela, M.E.; Lander, G.C.; Martin, A. Ubp6 deubiquitinase controls conformational dynamics and substrate degradation of the 26S proteasome. Nat. Struct. Mol. Biol. 2015, 22, 712–719. [Google Scholar] [CrossRef] [PubMed]
- Hamazaki, J.; Hirayama, S.; Murata, S. Redundant roles of rpn10 and rpn13 in recognition of ubiquitinated proteins and cellular homeostasis. PLoS Genet. 2015, 11, e1005401. [Google Scholar] [CrossRef] [PubMed]
- Harshbarger, W.; Miller, C.; Diedrich, C.; Sacchettini, J. Crystal structure of the human 20S proteasome in complex with carfilzomib. Structure 2015, 23, 418–424. [Google Scholar] [CrossRef] [PubMed]
- Hurley, J.H.; Emr, S.D. The Escrt complexes: Structure and mechanism of a membrane-trafficking network. Annu. Rev. Biophys. Biomol. Struct. 2006, 35, 277–298. [Google Scholar] [CrossRef] [PubMed]
- Lauwers, E.; Jacob, C.; André, B. K63-linked ubiquitin chains as a specific signal for protein sorting into the multivesicular body pathway. J. Cell Biol. 2009, 185, 493–502. [Google Scholar] [CrossRef] [PubMed]
- Katzmann, D.J.; Babst, M.; Emr, S.D. Ubiquitin-dependent sorting into the multivesicular body pathway requires the function of a conserved endosomal protein sorting complex, ESCRT-I. Cell 2001, 106, 145–155. [Google Scholar] [CrossRef]
- Calistri, A.; Munegato, D.; Carli, I.; Parolin, C.; Palù, G. The ubiquitin-conjugating system: Multiple roles in viral replication and infection. Cells 2014, 3, 386–417. [Google Scholar] [CrossRef] [PubMed]
- Randow, F.; Lehner, P.J. Viral avoidance and exploitation of the ubiquitin system. Nat. Cell Biol. 2009, 11, 527–534. [Google Scholar] [CrossRef] [PubMed]
- Biard-Piechaczyk, M.; Borel, S.; Espert, L.; de Bettignies, G.; Coux, O. HIV-1, ubiquitin and ubiquitin-like proteins: The dialectic interactions of a virus with a sophisticated network of post-translational modifications. Biol. Cell 2012, 104, 165–187. [Google Scholar] [CrossRef] [PubMed]
- Ott, D.E.; Coren, L.V.; Copeland, T.D.; Kane, B.P.; Johnson, D.G.; Sowder, R.C.; Yoshinaka, Y.; Oroszlan, S.; Arthur, L.O.; Henderson, L.E. Ubiquitin is covalently attached to the p6Gag proteins of human immunodeficiency virus type 1 and simian immunodeficiency virus and to the p12Gag protein of moloney murine leukemia virus. J. Virol. 1998, 72, 2962–2968. [Google Scholar] [PubMed]
- Gottwein, E.; Jäger, S.; Habermann, A.; Kräusslich, H.-G. Cumulative mutations of ubiquitin acceptor sites in human immunodeficiency virus type 1 gag cause a late budding defect. J. Virol. 2006, 80, 6267–6275. [Google Scholar] [CrossRef] [PubMed]
- Garrus, J.E.; von Schwedler, U.K.; Pornillos, O.W.; Morham, S.G.; Zavitz, K.H.; Wang, H.E.; Wettstein, D.A.; Stray, K.M.; Côté, M.; Rich, R.L.; et al. Tsg101 and the vacuolar protein sorting pathway are essential for HIV-1 budding. Cell 2001, 107, 55–65. [Google Scholar] [CrossRef]
- Ott, D.E.; Coren, L.V.; Chertova, E.N.; Gagliardi, T.D.; Schubert, U. Ubiquitination of HIV-1 and MuLV Gag. Virology 2000, 278, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Brès, V.; Kiernan, R.E.; Linares, L.K.; Chable-Bessia, C.; Plechakova, O.; Tréand, C.; Emiliani, S.; Peloponese, J.-M.; Jeang, K.-T.; Coux, O.; et al. A non-proteolytic role for ubiquitin in Tat-mediated transactivation of the HIV-1 promoter. Nat. Cell Biol. 2003, 5, 754–761. [Google Scholar] [CrossRef] [PubMed]
- Faust, T.B.; Li, Y.; Jang, G.M.; Johnson, J.R.; Yang, S.; Weiss, A.; Krogan, N.J.; Frankel, A.D. PJA2 ubiquitinates the HIV-1 Tat protein with atypical chain linkages to activate viral transcription. Sci. Rep. 2017, 7, 45394. [Google Scholar] [CrossRef] [PubMed]
- Fu, B.; Wang, L.; Ding, H.; Schwamborn, J.C.; Li, S.; Dorf, M.E. TRIM32 senses and restricts influenza a virus by ubiquitination of PB1 polymerase. PLoS Pathog. 2015, 11. [Google Scholar] [CrossRef] [PubMed]
- Van Tol, S.; Hage, A.; Giraldo, M.I.; Bharaj, P.; Rajsbaum, R. The TRIMendous role of TRIMs in virus-host interactions. Vaccines 2017, 5. [Google Scholar] [CrossRef] [PubMed]
- Horan, K.A.; Hansen, K.; Jakobsen, M.R.; Holm, C.K.; Søby, S.; Unterholzner, L.; Thompson, M.; West, J.A.; Iversen, M.B.; Rasmussen, S.B.; et al. Proteasomal degradation of herpes simplex virus capsids in macrophages releases DNA to the cytosol for recognition by DNA sensors. J. Immunol. 2013, 190, 2311–2319. [Google Scholar] [CrossRef] [PubMed]
- Reszka, N.; Zhou, C.; Song, B.; Sodroski, J.G.; Knipe, D.M. Simian TRIM5α proteins reduce replication of herpes simplex virus. Virology 2010, 398, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Scheffner, M.; Werness, B.A.; Huibregtse, J.M.; Levine, A.J.; Howley, P.M. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell 1990, 63, 1129–1136. [Google Scholar] [CrossRef]
- Scheffner, M.; Huibregtse, J.M.; Vierstra, R.D.; Howley, P.M. The HPV-16 E6 and E6-AP complex functions as a ubiquitin-protein ligase in the ubiquitination of p53. Cell 1993, 75, 495–505. [Google Scholar] [CrossRef]
- Graff, J.W.; Ettayebi, K.; Hardy, M.E. Rotavirus NSP1 inhibits NFκB activation by inducing proteasome-dependent degradation of β-TrCP: A novel mechanism of IFN antagonism. PLoS Pathog. 2009, 5. [Google Scholar] [CrossRef] [PubMed]
- Morelli, M.; Dennis, A.F.; Patton, J.T. Putative E3 ubiquitin ligase of human rotavirus inhibits NF-κB activation by using molecular mimicry to Target β-TrCP. mBio 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Ishido, S.; Wang, C.; Lee, B.S.; Cohen, G.B.; Jung, J.U. Downregulation of major histocompatibility complex class I molecules by Kaposi’s sarcoma-associated herpesvirus K3 and K5 proteins. J. Virol. 2000, 74, 5300–5309. [Google Scholar] [CrossRef] [PubMed]
- Lorenzo, M.E.; Jung, J.U.; Ploegh, H.L. Kaposi’s sarcoma-associated herpesvirus K3 utilizes the ubiquitin-proteasome system in routing class major histocompatibility complexes to late endocytic compartments. J. Virol. 2002, 76, 5522–5531. [Google Scholar] [CrossRef] [PubMed]
- Chelbi-Alix, M.K.; de Thé, H. Herpes virus induced proteasome-dependent degradation of the nuclear bodies-associated PML and Sp100 proteins. Oncogene 1999, 18, 935–941. [Google Scholar] [CrossRef] [PubMed]
- Everett, R.D.; Rechter, S.; Papior, P.; Tavalai, N.; Stamminger, T.; Orr, A. PML contributes to a cellular mechanism of repression of herpes simplex virus type 1 infection that is inactivated by ICP0. J. Virol. 2006, 80, 7995–8005. [Google Scholar] [CrossRef] [PubMed]
- Setz, C.; Friedrich, M.; Rauch, P.; Fraedrich, K.; Matthaei, A.; Traxdorf, M.; Schubert, U. Inhibitors of deubiquitinating enzymes block HIV-1 replication and augment the presentation of Gag-derived MHC-I epitopes. Viruses 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Schubert, U.; Ott, D.E.; Chertova, E.N.; Welker, R.; Tessmer, U.; Princiotta, M.F.; Bennink, J.R.; Krausslich, H.G.; Yewdell, J.W. Proteasome inhibition interferes with gag polyprotein processing, release, and maturation of HIV-1 and HIV-2. Proc. Natl. Acad. Sci. USA 2000, 97, 13057–13062. [Google Scholar] [CrossRef] [PubMed]
- Ott, D.E.; Coren, L.V.; Sowder, R.C.; Adams, J.; Schubert, U. Retroviruses have differing requirements for proteasome function in the budding process. J. Virol. 2003, 77, 3384–3393. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Mohanram, V.; Simonson, O.E.; Smith, C.I.E.; Spetz, A.-L.; Mohamed, A.J. Proteasome inhibitors block HIV-1 replication by affecting both cellular and viral targets. Biochem. Biophys. Res. Commun. 2009, 385, 100–105. [Google Scholar] [CrossRef] [PubMed]
- Frankel, A.D.; Young, J.A. HIV-1: Fifteen proteins and an RNA. Annu. Rev. Biochem. 1998, 67, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Freed, E.O. HIV-1 replication. Somat. Cell Mol. Genet. 2001, 26, 13–33. [Google Scholar] [CrossRef] [PubMed]
- Azevedo-Pereira, J.M.; Santos-Costa, Q. HIV interaction with human host: HIV-2 as a model of a less virulent infection. AIDS Rev. 2016, 18, 44–53. [Google Scholar] [PubMed]
- Jakobsdottir, G.M.; Iliopoulou, M.; Nolan, R.; Alvarez, L.; Compton, A.A.; Padilla-Parra, S. On the whereabouts of HIV-1 cellular entry and its fusion ports. Trends Mol. Med. 2017. [Google Scholar] [CrossRef] [PubMed]
- Cimarelli, A.; Darlix, J.-L. HIV-1 reverse transcription. Methods Mol. Biol. 2014, 1087, 55–70. [Google Scholar] [CrossRef] [PubMed]
- Craigie, R.; Bushman, F.D. HIV DNA integration. Cold Spring Harb. Perspect. Med. 2012, 2, a006890. [Google Scholar] [CrossRef] [PubMed]
- Sundquist, W.I.; Kräusslich, H.-G. HIV-1 assembly, budding, and maturation. Cold Spring Harb. Perspect. Med. 2012, 2, a006924. [Google Scholar] [CrossRef] [PubMed]
- Usami, Y.; Wu, Y.; Göttlinger, H.G. SERINC3 and SERINC5 restrict HIV-1 infectivity and are counteracted by Nef. Nature 2015, 526, 218–223. [Google Scholar] [CrossRef] [PubMed]
- Rosa, A.; Chande, A.; Ziglio, S.; de Sanctis, V.; Bertorelli, R.; Goh, S.L.; McCauley, S.M.; Nowosielska, A.; Antonarakis, S.E.; Luban, J.; et al. HIV-1 Nef promotes infection by excluding SERINC5 from virion incorporation. Nature 2015, 526, 212–217. [Google Scholar] [CrossRef] [PubMed]
- Compton, A.A.; Bruel, T.; Porrot, F.; Mallet, A.; Sachse, M.; Euvrard, M.; Liang, C.; Casartelli, N.; Schwartz, O. IFITM proteins incorporated into HIV-1 virions impair viral fusion and spread. Cell Host Microbe 2014, 16, 736–747. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Pan, Q.; Rong, L.; Liu, S.-L.; Liang, C. The IFITM proteins inhibit HIV-1 infection. J. Virol. 2011, 85, 2126–2137. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Pan, Q.; Ding, S.; Wang, Z.; Yu, J.; Finzi, A.; Liu, S.-L.; Liang, C. The V3 loop of HIV-1 Env determines viral susceptibility to IFITM3 impairment of viral infectivity. J. Virol. 2017, 91, e02441-16. [Google Scholar] [CrossRef] [PubMed]
- Tartour, K.; Appourchaux, R.; Gaillard, J.; Nguyen, X.-N.; Durand, S.; Turpin, J.; Beaumont, E.; Roch, E.; Berger, G.; Mahieux, R.; et al. IFITM proteins are incorporated onto HIV-1 virion particles and negatively imprint their infectivity. Retrovirology 2014, 11, 103. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Li, M.; Wilkins, J.; Ding, S.; Swartz, T.H.; Esposito, A.M.; Zheng, Y.-M.; Freed, E.O.; Liang, C.; Chen, B.K.; et al. IFITM proteins restrict HIV-1 infection by antagonizing the envelope glycoprotein. Cell Rep. 2015, 13, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Foster, T.L.; Wilson, H.; Iyer, S.S.; Coss, K.; Doores, K.; Smith, S.; Kellam, P.; Finzi, A.; Borrow, P.; Hahn, B.H.; et al. Resistance of transmitted founder HIV-1 to IFITM-mediated restriction. Cell Host Microbe 2016, 20, 429–442. [Google Scholar] [CrossRef] [PubMed]
- Roa, A.; Hayashi, F.; Yang, Y.; Lienlaf, M.; Zhou, J.; Shi, J.; Watanabe, S.; Kigawa, T.; Yokoyama, S.; Aiken, C.; et al. RING domain mutations uncouple TRIM5α restriction of HIV-1 from inhibition of reverse transcription and acceleration of uncoating. J. Virol. 2012, 86, 1717–1727. [Google Scholar] [CrossRef] [PubMed]
- Stremlau, M.; Perron, M.; Lee, M.; Li, Y.; Song, B.; Javanbakht, H.; Diaz-Griffero, F.; Anderson, D.J.; Sundquist, W.I.; Sodroski, J. Specific recognition and accelerated uncoating of retroviral capsids by the TRIM5α restriction factor. Proc. Natl. Acad. Sci. USA 2006, 103, 5514–5519. [Google Scholar] [CrossRef] [PubMed]
- Stremlau, M.; Owens, C.M.; Perron, M.J.; Kiessling, M.; Autissier, P.; Sodroski, J. The cytoplasmic body component TRIM5α restricts HIV-1 infection in old world monkeys. Nature 2004, 427, 848–853. [Google Scholar] [CrossRef] [PubMed]
- Kane, M.; Yadav, S.S.; Bitzegeio, J.; Kutluay, S.B.; Zang, T.; Wilson, S.J.; Schoggins, J.W.; Rice, C.M.; Yamashita, M.; Hatziioannou, T.; et al. MX2 is an interferon-induced inhibitor of HIV-1 infection. Nature 2013, 502, 563–566. [Google Scholar] [CrossRef] [PubMed]
- Goujon, C.; Moncorgé, O.; Bauby, H.; Doyle, T.; Ward, C.C.; Schaller, T.; Hué, S.; Barclay, W.S.; Schulz, R.; Malim, M.H. Human MX2 is an interferon-induced post-entry inhibitor of HIV-1 infection. Nature 2013, 502, 559–562. [Google Scholar] [CrossRef] [PubMed]
- Schulte, B.; Buffone, C.; Opp, S.; di Nunzio, F.; de Souza Aranha Vieira, D.A.; Brandariz-Nuñez, A.; Diaz-Griffero, F. Restriction of HIV-1 requires the N-terminal region of MxB as a capsid-binding motif but not as a nuclear localization signal. J. Virol. 2015, 89, 8599–8610. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Pan, Q.; Liang, Z.; Qiao, W.; Cen, S.; Liang, C. The highly polymorphic cyclophilin A-binding loop in HIV-1 capsid modulates viral resistance to MxB. Retrovirology 2015, 12, 1. [Google Scholar] [CrossRef] [PubMed]
- Busnadiego, I.; Kane, M.; Rihn, S.J.; Preugschas, H.F.; Hughes, J.; Blanco-Melo, D.; Strouvelle, V.P.; Zang, T.M.; Willett, B.J.; Boutell, C.; et al. Host and viral determinants of Mx2 antiretroviral activity. J. Virol. 2014, 88, 7738–7752. [Google Scholar] [CrossRef] [PubMed]
- Matreyek, K.A.; Wang, W.; Serrao, E.; Singh, P.K.; Levin, H.L.; Engelman, A. Host and viral determinants for MxB restriction of HIV-1 infection. Retrovirology 2014, 11, 90. [Google Scholar] [CrossRef] [PubMed]
- Laguette, N.; Sobhian, B.; Casartelli, N.; Ringeard, M.; Chable-Bessia, C.; Ségéral, E.; Yatim, A.; Emiliani, S.; Schwartz, O.; Benkirane, M. SAMHD1 is the dendritic- and myeloid-cell-specific HIV-1 restriction factor counteracted by Vpx. Nature 2011, 474, 654–657. [Google Scholar] [CrossRef] [PubMed]
- Lahouassa, H.; Daddacha, W.; Hofmann, H.; Ayinde, D.; Logue, E.C.; Dragin, L.; Bloch, N.; Maudet, C.; Bertrand, M.; Gramberg, T.; et al. SAMHD1 restricts the replication of human immunodeficiency virus type 1 by depleting the intracellular pool of deoxynucleoside triphosphates. Nat. Immunol. 2012, 13, 223–228. [Google Scholar] [CrossRef] [PubMed]
- Sheehy, A.M.; Gaddis, N.C.; Choi, J.D.; Malim, M.H. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature 2002, 418, 646–650. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Yang, B.; Pomerantz, R.J.; Zhang, C.; Arunachalam, S.C.; Gao, L. The cytidine deaminase CEM15 induces hypermutation in newly synthesized HIV-1 DNA. Nature 2003, 424, 94–98. [Google Scholar] [CrossRef] [PubMed]
- Mangeat, B.; Turelli, P.; Caron, G.; Friedli, M.; Perrin, L.; Trono, D. Broad antiretroviral defence by human APOBEC3G through lethal editing of nascent reverse transcripts. Nature 2003, 424, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Kao, E.; Gao, X.; Sandig, H.; Limmer, K.; Pavon-Eternod, M.; Jones, T.E.; Landry, S.; Pan, T.; Weitzman, M.D.; et al. Codon-usage-based inhibition of HIV protein synthesis by human schlafen 11. Nature 2012, 491, 125–128. [Google Scholar] [CrossRef] [PubMed]
- Stabell, A.C.; Hawkins, J.; Li, M.; Gao, X.; David, M.; Press, W.H.; Sawyer, S.L. Non-human primate schlafen11 inhibits production of both host and viral proteins. PLoS Pathog. 2016, 12, e1006066. [Google Scholar] [CrossRef] [PubMed]
- Van Weringh, A.; Ragonnet-Cronin, M.; Pranckeviciene, E.; Pavon-Eternod, M.; Kleiman, L.; Xia, X. HIV-1 modulates the tRNA pool to improve translation efficiency. Mol. Biol. Evol. 2011, 28, 1827–1834. [Google Scholar] [CrossRef] [PubMed]
- Neil, S.J.D.; Eastman, S.W.; Jouvenet, N.; Bieniasz, P.D. HIV-1 Vpu promotes release and prevents endocytosis of nascent retrovirus particles from the plasma membrane. PLoS Pathog. 2006, 2, e39. [Google Scholar] [CrossRef] [PubMed]
- Neil, S.J.D.; Zang, T.; Bieniasz, P.D. Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature 2008, 451, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Van Damme, N.; Goff, D.; Katsura, C.; Jorgenson, R.L.; Mitchell, R.; Johnson, M.C.; Stephens, E.B.; Guatelli, J. The interferon-induced protein BST-2 restricts HIV-1 release and is downregulated from the cell surface by the viral Vpu protein. Cell Host Microbe 2008, 3, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Tada, T.; Zhang, Y.; Koyama, T.; Tobiume, M.; Tsunetsugu-Yokota, Y.; Yamaoka, S.; Fujita, H.; Tokunaga, K. MARCH8 inhibits HIV-1 infection by reducing virion incorporation of envelope glycoproteins. Nat. Med. 2015, 21, 1502–1507. [Google Scholar] [CrossRef] [PubMed]
- Trautz, B.; Pierini, V.; Wombacher, R.; Stolp, B.; Chase, A.J.; Pizzato, M.; Fackler, O.T. The antagonism of HIV-1 Nef to SERINC5 particle infectivity restriction involves the counteraction of virion-associated pools of the restriction factor. J. Virol. 2016, 90, 10915–10927. [Google Scholar] [CrossRef] [PubMed]
- Sheehy, A.M.; Gaddis, N.C.; Malim, M.H. The antiretroviral enzyme APOBEC3G is degraded by the proteasome in response to HIV-1 Vif. Nat. Med. 2003, 9, 1404–1407. [Google Scholar] [CrossRef] [PubMed]
- Anderson, B.D.; Harris, R.S. Transcriptional regulation of APOBEC3 antiviral immunity through the CBF-β/RUNX axis. Sci. Adv. 2015, 1, e1500296. [Google Scholar] [CrossRef] [PubMed]
- Stopak, K.; de Noronha, C.; Yonemoto, W.; Greene, W.C. HIV-1 Vif blocks the antiviral activity of APOBEC3G by impairing both its translation and intracellular stability. Mol. Cell 2003, 12, 591–601. [Google Scholar] [CrossRef]
- Hrecka, K.; Hao, C.; Gierszewska, M.; Swanson, S.K.; Kesik-Brodacka, M.; Srivastava, S.; Florens, L.; Washburn, M.P.; Skowronski, J. Vpx relieves inhibition of HIV-1 infection of macrophages mediated by the SAMHD1 protein. Nature 2011, 474, 658–661. [Google Scholar] [CrossRef] [PubMed]
- Lim, E.S.; Fregoso, O.I.; McCoy, C.O.; Matsen, F.A.; Malik, H.S.; Emerman, M. The ability of primate lentiviruses to degrade the monocyte restriction factor SAMHD1 preceded the birth of the viral accessory protein Vpx. Cell Host Microbe 2012, 11, 194–204. [Google Scholar] [CrossRef] [PubMed]
- McNatt, M.W.; Zang, T.; Bieniasz, P.D. Vpu binds directly to tetherin and displaces it from nascent virions. PLoS Pathog. 2013, 9, e1003299. [Google Scholar] [CrossRef] [PubMed]
- Shibata, R.; Sakai, H.; Kawamura, M.; Tokunaga, K.; Adachi, A. Early replication block of human immunodeficiency virus type 1 in monkey cells. J. Gen. Virol. 1995, 76 Pt 11, 2723–2730. [Google Scholar] [CrossRef] [PubMed]
- Himathongkham, S.; Luciw, P.A. Restriction of HIV-1 (subtype B) replication at the entry step in rhesus macaque cells. Virology 1996, 219, 485–488. [Google Scholar] [CrossRef] [PubMed]
- Danielson, C.M.; Cianci, G.C.; Hope, T.J. Recruitment and dynamics of proteasome association with rhTRIM5α cytoplasmic complexes during HIV-1 infection. Traffic 2012, 13, 1206–1217. [Google Scholar] [CrossRef] [PubMed]
- Lukic, Z.; Hausmann, S.; Sebastian, S.; Rucci, J.; Sastri, J.; Robia, S.L.; Luban, J.; Campbell, E.M. TRIM5α associates with proteasomal subunits in cells while in complex with HIV-1 virions. Retrovirology 2011, 8, 93. [Google Scholar] [CrossRef] [PubMed]
- Rold, C.J.; Aiken, C. Proteasomal degradation of TRIM5α during retrovirus restriction. PLoS Pathog. 2008, 4, e1000074. [Google Scholar] [CrossRef] [PubMed]
- Campbell, E.M.; Weingart, J.; Sette, P.; Opp, S.; Sastri, J.; O’Connor, S.K.; Talley, S.; Diaz-Griffero, F.; Hirsch, V.; Bouamr, F. TRIM5α-mediated ubiquitin chain conjugation is required for inhibition of HIV-1 reverse transcription and capsid destabilization. J. Virol. 2015, 90, 1849–1857. [Google Scholar] [CrossRef] [PubMed]
- Kutluay, S.B.; Perez-Caballero, D.; Bieniasz, P.D. Fates of retroviral core components during unrestricted and TRIM5-restricted infection. PLoS Pathog. 2013, 9, e1003214. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.L.; Campbell, E.M.; Wu, X.; Vandegraaff, N.; Engelman, A.; Hope, T.J. Proteasome inhibition reveals that a functional preintegration complex intermediate can be generated during restriction by diverse TRIM5 proteins. J. Virol. 2006, 80, 9754–9760. [Google Scholar] [CrossRef] [PubMed]
- Pertel, T.; Hausmann, S.; Morger, D.; Züger, S.; Guerra, J.; Lascano, J.; Reinhard, C.; Santoni, F.A.; Uchil, P.D.; Chatel, L.; et al. TRIM5 is an innate immune sensor for the retrovirus capsid lattice. Nature 2011, 472, 361–365. [Google Scholar] [CrossRef] [PubMed]
- Yudina, Z.; Roa, A.; Johnson, R.; Biris, N.; de Souza Aranha Vieira, D.A.; Tsiperson, V.; Reszka, N.; Taylor, A.B.; Hart, P.J.; Demeler, B.; et al. RING dimerization links higher-order assembly of TRIM5α to synthesis of K63-linked polyubiquitin. Cell Rep. 2015, 12, 788–797. [Google Scholar] [CrossRef] [PubMed]
- Ohmura-Hoshino, M.; Matsuki, Y.; Aoki, M.; Goto, E.; Mito, M.; Uematsu, M.; Kakiuchi, T.; Hotta, H.; Ishido, S. Inhibition of MHC class II expression and immune responses by c-MIR. J. Immunol. 2006, 177, 341–354. [Google Scholar] [CrossRef] [PubMed]
- Van de Kooij, B.; Verbrugge, I.; de Vries, E.; Gijsen, M.; Montserrat, V.; Maas, C.; Neefjes, J.; Borst, J. Ubiquitination by the membrane-associated RING-CH-8 (MARCH-8) ligase controls steady-state cell surface expression of tumor necrosis factor-related apoptosis inducing ligand (TRAIL) receptor 1. J. Biol. Chem. 2013, 288, 6617–6628. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Li, M.; Zhang, Y.; Zhou, Q.; Shu, H.-B. The E3 ubiquitin ligase MARCH8 negatively regulates IL-1β-induced NF-κB activation by targeting the IL1RAP coreceptor for ubiquitination and degradation. Proc. Natl. Acad. Sci. USA 2012, 109, 14128–14133. [Google Scholar] [CrossRef] [PubMed]
- Eyster, C.A.; Cole, N.B.; Petersen, S.; Viswanathan, K.; Früh, K.; Donaldson, J.G. MARCH ubiquitin ligases alter the itinerary of clathrin-independent cargo from recycling to degradation. Mol. Biol. Cell 2011, 22, 3218–3230. [Google Scholar] [CrossRef] [PubMed]
- Ito, F.; Fu, Y.; Kao, S.-C.A.; Yang, H.; Chen, X.S. Family-wide comparative analysis of cytidine and methylcytidine deamination by eleven human APOBEC proteins. J. Mol. Biol. 2017, 429, 1787–1799. [Google Scholar] [CrossRef] [PubMed]
- Liddament, M.T.; Brown, W.L.; Schumacher, A.J.; Harris, R.S. APOBEC3F properties and hypermutation preferences indicate activity against HIV-1 In Vivo. Curr. Biol. 2004, 14, 1385–1391. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.S.; Bishop, K.N.; Sheehy, A.M.; Craig, H.M.; Petersen-Mahrt, S.K.; Watt, I.N.; Neuberger, M.S.; Malim, M.H. DNA deamination mediates innate immunity to retroviral infection. Cell 2003, 113, 803–809. [Google Scholar] [CrossRef]
- Yang, B.; Chen, K.; Zhang, C.; Huang, S.; Zhang, H. Virion-associated uracil DNA glycosylase-2 and apurinic/apyrimidinic endonuclease are involved in the degradation of APOBEC3G-edited nascent HIV-1 DNA. J. Biol. Chem. 2007, 282, 11667–11675. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Yu, Y.; Liu, B.; Luo, K.; Kong, W.; Mao, P.; Yu, X.-F. Induction of APOBEC3G ubiquitination and degradation by an HIV-1 Vif-Cul5-SCF complex. Science 2003, 302, 1056–1060. [Google Scholar] [CrossRef] [PubMed]
- Mehle, A.; Thomas, E.R.; Rajendran, K.S.; Gabuzda, D. A zinc-binding region in Vif binds Cul5 and determines cullin selection. J. Biol. Chem. 2006, 281, 17259–17265. [Google Scholar] [CrossRef] [PubMed]
- Mehle, A.; Goncalves, J.; Santa-Marta, M.; McPike, M.; Gabuzda, D. Phosphorylation of a novel SOCS-box regulates assembly of the HIV-1 Vif-Cul5 complex that promotes APOBEC3G degradation. Genes Dev. 2004, 18, 2861–2866. [Google Scholar] [CrossRef] [PubMed]
- Valera, M.-S.; de Armas-Rillo, L.; Barroso-González, J.; Ziglio, S.; Batisse, J.; Dubois, N.; Marrero-Hernández, S.; Borel, S.; García-Expósito, L.; Biard-Piechaczyk, M.; et al. The HDAC6/APOBEC3G complex regulates HIV-1 infectiveness by inducing Vif autophagic degradation. Retrovirology 2015, 12. [Google Scholar] [CrossRef] [PubMed]
- Izumi, T.; Takaori-Kondo, A.; Shirakawa, K.; Higashitsuji, H.; Itoh, K.; Io, K.; Matsui, M.; Iwai, K.; Kondoh, H.; Sato, T.; et al. MDM2 is a novel E3 ligase for HIV-1 Vif. Retrovirology 2009, 6, 1. [Google Scholar] [CrossRef] [PubMed]
- Matsui, Y.; Shindo, K.; Nagata, K.; Yoshinaga, N.; Shirakawa, K.; Kobayashi, M.; Takaori-Kondo, A. Core binding factor β protects HIV, type 1 accessory protein viral infectivity factor from MDM2-mediated degradation. J. Biol. Chem. 2016, 291, 24892–24899. [Google Scholar] [CrossRef] [PubMed]
- Jäger, S.; Kim, D.Y.; Hultquist, J.F.; Shindo, K.; LaRue, R.S.; Kwon, E.; Li, M.; Anderson, B.D.; Yen, L.; Stanley, D.; et al. Vif hijacks CBF-β to degrade APOBEC3G and promote HIV-1 infection. Nature 2011, 481, 371–375. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Du, J.; Evans, S.L.; Yu, Y.; Yu, X.-F. T-cell differentiation factor CBF-β regulates HIV-1 Vif-mediated evasion of host restriction. Nature 2011, 481, 376–379. [Google Scholar] [CrossRef] [PubMed]
- Mercenne, G.; Bernacchi, S.; Richer, D.; Bec, G.; Henriet, S.; Paillart, J.-C.; Marquet, R. HIV-1 Vif binds to APOBEC3G mRNA and inhibits its translation. Nucleic Acids Res. 2010, 38, 633–646. [Google Scholar] [CrossRef] [PubMed]
- Guerrero, S.; Libre, C.; Batisse, J.; Mercenne, G.; Richer, D.; Laumond, G.; Decoville, T.; Moog, C.; Marquet, R.; Paillart, J.-C. Translational regulation of APOBEC3G mRNA by Vif requires its 5’UTR and contributes to restoring HIV-1 infectivity. Sci. Rep. 2016, 6, 39507. [Google Scholar] [CrossRef] [PubMed]
- Hultquist, J.F.; Lengyel, J.A.; Refsland, E.W.; LaRue, R.S.; Lackey, L.; Brown, W.L.; Harris, R.S. Human and rhesus APOBEC3D, APOBEC3F, APOBEC3G, and APOBEC3H demonstrate a conserved capacity to restrict Vif-deficient HIV-1. J. Virol. 2011, 85, 11220–11234. [Google Scholar] [CrossRef] [PubMed]
- Binka, M.; Ooms, M.; Steward, M.; Simon, V. The activity spectrum of Vif from multiple HIV-1 subtypes against APOBEC3G, APOBEC3F, and APOBEC3H. J. Virol. 2012, 86, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Baig, T.T.; Love, R.P.; Chelico, L. Suppression of APOBEC3-mediated restriction of HIV-1 by Vif. Front. Microbiol. 2014, 5, 450. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Sarkis, P.T.N.; Luo, K.; Yu, Y.; Yu, X.-F. Regulation of Apobec3F and human immunodeficiency virus type 1 Vif by Vif-Cul5-ElonB/C E3 ubiquitin ligase. J. Virol. 2005, 79, 9579–9587. [Google Scholar] [CrossRef] [PubMed]
- Goldstone, D.C.; Ennis-Adeniran, V.; Hedden, J.J.; Groom, H.C.T.; Rice, G.I.; Christodoulou, E.; Walker, P.A.; Kelly, G.; Haire, L.F.; Yap, M.W.; et al. HIV-1 restriction factor SAMHD1 is a deoxynucleoside triphosphate triphosphohydrolase. Nature 2011, 480, 379–382. [Google Scholar] [CrossRef] [PubMed]
- Powell, R.D.; Holland, P.J.; Hollis, T.; Perrino, F.W. Aicardi-goutieres syndrome gene and HIV-1 restriction factor SAMHD1 is a dGTP-regulated deoxynucleotide triphosphohydrolase. J. Biol. Chem. 2011, 286, 43596–43600. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.; Nguyen, L.A.; Daddacha, W.; Hollenbaugh, J.A. Tight interplay among SAMHD1 protein level, cellular dNTP levels, and HIV-1 proviral DNA synthesis kinetics in human primary monocyte-derived macrophages. J. Biol. Chem. 2012, 287, 21570–21574. [Google Scholar] [CrossRef] [PubMed]
- Baldauf, H.-M.; Pan, X.; Erikson, E.; Schmidt, S.; Daddacha, W.; Burggraf, M.; Schenkova, K.; Ambiel, I.; Wabnitz, G.; Gramberg, T.; et al. SAMHD1 restricts HIV-1 infection in resting CD4(+) T cells. Nat. Med. 2012, 18, 1682–1687. [Google Scholar] [CrossRef] [PubMed]
- Bergamaschi, A.; Ayinde, D.; David, A.; le Rouzic, E.; Morel, M.; Collin, G.; Descamps, D.; Damond, F.; Brun-Vezinet, F.; Nisole, S.; et al. The human immunodeficiency virus type 2 Vpx protein usurps the CUL4A-DDB1 DCAF1 ubiquitin ligase to overcome a postentry block in macrophage infection. J. Virol. 2009, 83, 4854–4860. [Google Scholar] [CrossRef] [PubMed]
- Brandariz-Nuñez, A.; Valle-Casuso, J.C.; White, T.E.; Laguette, N.; Benkirane, M.; Brojatsch, J.; Diaz-Griffero, F. Role of SAMHD1 nuclear localization in restriction of HIV-1 and SIVmac. Retrovirology 2012, 9, 49. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, H.; Logue, E.C.; Bloch, N.; Daddacha, W.; Polsky, S.B.; Schultz, M.L.; Kim, B.; Landau, N.R. The Vpx lentiviral accessory protein targets SAMHD1 for degradation in the nucleus. J. Virol. 2012, 86, 12552–12560. [Google Scholar] [CrossRef] [PubMed]
- Tan, L.; Ehrlich, E.; Yu, X.-F. DDB1 and Cul4A are required for human immunodeficiency virus type 1 Vpr-induced G2 arrest. J. Virol. 2007, 81, 10822–10830. [Google Scholar] [CrossRef] [PubMed]
- Belzile, J.-P.; Duisit, G.; Rougeau, N.; Mercier, J.; Finzi, A.; Cohen, E.A. HIV-1 Vpr-mediated G2 arrest involves the DDB1-CUL4AVPRBP E3 ubiquitin ligase. PLoS Pathog. 2007, 3, e85. [Google Scholar] [CrossRef] [PubMed]
- Laguette, N.; Brégnard, C.; Hue, P.; Basbous, J.; Yatim, A.; Larroque, M.; Kirchhoff, F.; Constantinou, A.; Sobhian, B.; Benkirane, M. Premature activation of the SLX4 complex by Vpr promotes G2/M arrest and escape from innate immune sensing. Cell 2014, 156, 134–145. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; DeLucia, M.; Ahn, J. SLX4-SLX1 protein-independent down-regulation of MUS81-EME1 protein by HIV-1 viral protein R (Vpr). J. Biol. Chem. 2016, 291, 16936–16947. [Google Scholar] [CrossRef] [PubMed]
- Tristem, M.; Purvis, A.; Quicke, D.L.J. Complex evolutionary history of primate lentiviralvprgenes. Virology 1998, 240, 232–237. [Google Scholar] [CrossRef] [PubMed]
- Fregoso, O.I.; Ahn, J.; Wang, C.; Mehrens, J.; Skowronski, J.; Emerman, M. Evolutionary toggling of Vpx/Vpr specificity results in divergent recognition of the restriction factor SAMHD1. PLoS Pathog. 2013, 9, e1003496. [Google Scholar] [CrossRef] [PubMed]
- Pauls, E.; Ruiz, A.; Badia, R.; Permanyer, M.; Gubern, A.; Riveira-Muñoz, E.; Torres-Torronteras, J.; Alvarez, M.; Mothe, B.; Brander, C.; et al. Cell cycle control and HIV-1 susceptibility are linked by CDK6-dependent CDK2 phosphorylation of SAMHD1 in myeloid and lymphoid cells. J. Immunol. 2014, 193, 1988–1997. [Google Scholar] [CrossRef] [PubMed]
- Rocha-Perugini, V.; Suárez, H.; Álvarez, S.; López-Martín, S.; Lenzi, G.M.; Vences-Catalán, F.; Levy, S.; Kim, B.; Muñoz-Fernández, M.A.; Sánchez-Madrid, F.; et al. CD81 association with SAMHD1 enhances HIV-1 reverse transcription by increasing dNTP levels. Nat. Microbiol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Kyei, G.B.; Cheng, X.; Ramani, R.; Ratner, L. Cyclin L2 is a critical HIV dependency factor in macrophages that controls SAMHD1 abundance. Cell Host Microbe 2015, 17, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Hinz, A.; Miguet, N.; Natrajan, G.; Usami, Y.; Yamanaka, H.; Renesto, P.; Hartlieb, B.; McCarthy, A.A.; Simorre, J.-P.; Göttlinger, H.; et al. Structural basis of HIV-1 tethering to membranes by the BST-2/tetherin ectodomain. Cell Host Microbe 2010, 7, 314–323. [Google Scholar] [CrossRef] [PubMed]
- Venkatesh, S.; Bieniasz, P.D. Mechanism of HIV-1 virion entrapment by tetherin. PLoS Pathog. 2013, 9, e1003483. [Google Scholar] [CrossRef] [PubMed]
- Skasko, M.; Wang, Y.; Tian, Y.; Tokarev, A.; Munguia, J.; Ruiz, A.; Stephens, E.B.; Opella, S.J.; Guatelli, J. HIV-1 Vpu protein antagonizes innate restriction factor BST-2 via lipid-embedded helix-helix interactions. J. Biol. Chem. 2012, 287, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, R.S.; Katsura, C.; Skasko, M.A.; Fitzpatrick, K.; Lau, D.; Ruiz, A.; Stephens, E.B.; Margottin-Goguet, F.; Benarous, R.; Guatelli, J.C. Vpu antagonizes BST-2-mediated restriction of HIV-1 release via β-TrCP and endo-lysosomal trafficking. PLoS Pathog. 2009, 5, e1000450. [Google Scholar] [CrossRef] [PubMed]
- Dubé, M.; Roy, B.B.; Guiot-Guillain, P.; Binette, J.; Mercier, J.; Chiasson, A.; Cohen, E.A. Antagonism of tetherin restriction of HIV-1 release by Vpu involves binding and sequestration of the restriction factor in a perinuclear compartment. PLoS Pathog. 2010, 6, e1000856. [Google Scholar] [CrossRef] [PubMed]
- Roy, N.; Pacini, G.; Berlioz-Torrent, C.; Janvier, K. Characterization of E3 ligases involved in lysosomal sorting of the HIV-1 restriction factor BST2. J. Cell Sci. 2017, 130, 1596–1611. [Google Scholar] [CrossRef] [PubMed]
- Margottin, F.; Bour, S.P.; Durand, H.; Selig, L.; Benichou, S.; Richard, V.; Thomas, D.; Strebel, K.; Benarous, R. A novel human WD protein, h-β TrCp, that interacts with HIV-1 Vpu connects CD4 to the ER degradation pathway through an F-box motif. Mol. Cell 1998, 1, 565–574. [Google Scholar] [CrossRef]
- Iwabu, Y.; Fujita, H.; Kinomoto, M.; Kaneko, K.; Ishizaka, Y.; Tanaka, Y.; Sata, T.; Tokunaga, K. HIV-1 accessory protein Vpu internalizes cell-surface BST-2/tetherin through transmembrane interactions leading to lysosomes. J. Biol. Chem. 2009, 284, 35060–35072. [Google Scholar] [CrossRef] [PubMed]
- Douglas, J.L.; Viswanathan, K.; McCarroll, M.N.; Gustin, J.K.; Früh, K.; Moses, A.V. Vpu directs the degradation of the human immunodeficiency virus restriction factor BST-2/Tetherin via a {β}TrCP-dependent mechanism. J. Virol. 2009, 83, 7931–7947. [Google Scholar] [CrossRef] [PubMed]
- Mangeat, B.; Gers-Huber, G.; Lehmann, M.; Zufferey, M.; Luban, J.; Piguet, V. HIV-1 Vpu neutralizes the antiviral factor Tetherin/BST-2 by binding it and directing its β-TrCP2-dependent degradation. PLoS Pathog. 2009, 5, e1000574. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, P.W.; DePaula-Silva, A.B.; Szaniawski, M.; Barker, E.; Bosque, A.; Planelles, V. HIV-1 Vpu utilizes both cullin-RING ligase (CRL) dependent and independent mechanisms to downmodulate host proteins. Retrovirology 2015, 12, 65. [Google Scholar] [CrossRef] [PubMed]
- Kueck, T.; Foster, T.L.; Weinelt, J.; Sumner, J.C.; Pickering, S.; Neil, S.J.D. Serine phosphorylation of HIV-1 Vpu and its binding to tetherin regulates interaction with clathrin adaptors. PLoS Pathog. 2015, 11, e1005141. [Google Scholar] [CrossRef] [PubMed]
- Tervo, H.-M.; Homann, S.; Ambiel, I.; Fritz, J.V.; Fackler, O.T.; Keppler, O.T. β-TrCP is dispensable for Vpu’s ability to overcome the CD317/Tetherin-imposed restriction to HIV-1 release. Retrovirology 2011, 8, 9. [Google Scholar] [CrossRef] [PubMed]
- Madjo, U.; Leymarie, O.; Frémont, S.; Kuster, A.; Nehlich, M.; Gallois-Montbrun, S.; Janvier, K.; Berlioz-Torrent, C. LC3C contributes to Vpu-mediated antagonism of BST2/Tetherin restriction on HIV-1 release through a non-canonical autophagy pathway. Cell Rep. 2016, 17, 2221–2233. [Google Scholar] [CrossRef] [PubMed]
- Pujol, F.M.; Laketa, V.; Schmidt, F.; Mukenhirn, M.; Müller, B.; Boulant, S.; Grimm, D.; Keppler, O.T.; Fackler, O.T. HIV-1 Vpu antagonizes CD317/Tetherin by adaptor protein-1-mediated exclusion from virus assembly sites. J. Virol. 2016, 90, 6709–6723. [Google Scholar] [CrossRef] [PubMed]
- Janvier, K.; Pelchen-Matthews, A.; Renaud, J.-B.; Caillet, M.; Marsh, M.; Berlioz-Torrent, C. The ESCRT-0 component HRS is required for HIV-1 Vpu-mediated BST-2/tetherin down-regulation. PLoS Pathog. 2011, 7, e1001265. [Google Scholar] [CrossRef] [PubMed]
- Krapp, C.; Hotter, D.; Gawanbacht, A.; McLaren, P.J.; Kluge, S.F.; Stürzel, C.M.; Mack, K.; Reith, E.; Engelhart, S.; Ciuffi, A.; et al. Guanylate binding protein (GBP) 5 is an interferon-inducible inhibitor of HIV-1 infectivity. Cell Host Microbe 2016, 19, 504–514. [Google Scholar] [CrossRef] [PubMed]
- McLaren, P.J.; Gawanbacht, A.; Pyndiah, N.; Krapp, C.; Hotter, D.; Kluge, S.F.; Götz, N.; Heilmann, J.; Mack, K.; Sauter, D.; et al. Identification of potential HIV restriction factors by combining evolutionary genomic signatures with functional analyses. Retrovirology 2015, 12, 41. [Google Scholar] [CrossRef] [PubMed]
- Schubert, U.; Bour, S.; Willey, R.L.; Strebel, K. Regulation of virus release by the macrophage-tropic human immunodeficiency virus type 1 AD8 isolate is redundant and can be controlled by either Vpu or Env. J. Virol. 1999, 73, 887–896. [Google Scholar] [PubMed]
- Bour, S.; Schubert, U.; Peden, K.; Strebel, K. The envelope glycoprotein of human immunodeficiency virus type 2 enhances viral particle release: A Vpu-like factor? J. Virol. 1996, 70, 820–829. [Google Scholar] [PubMed]
- Bour, S.; Strebel, K. The human immunodeficiency virus (HIV) type 2 envelope protein is a functional complement to HIV type 1 Vpu that enhances particle release of heterologous retroviruses. J. Virol. 1996, 70, 8285–8300. [Google Scholar] [PubMed]
- Zhang, F.; Wilson, S.J.; Landford, W.C.; Virgen, B.; Gregory, D.; Johnson, M.C.; Munch, J.; Kirchhoff, F.; Bieniasz, P.D.; Hatziioannou, T. Nef proteins from simian immunodeficiency viruses are tetherin antagonists. Cell Host Microbe 2009, 6, 54–67. [Google Scholar] [CrossRef] [PubMed]
- Jia, B.; Serra-Moreno, R.; Neidermyer, W.; Rahmberg, A.; Mackey, J.; Fofana, I.B.; Johnson, W.E.; Westmoreland, S.; Evans, D.T. Species-specific activity of SIV Nef and HIV-1 Vpu in overcoming restriction by tetherin/BST2. PLoS Pathog. 2009, 5, e1000429. [Google Scholar] [CrossRef] [PubMed]
- Sauter, D.; Schindler, M.; Specht, A.; Landford, W.N.; Münch, J.; Kim, K.-A.; Votteler, J.; Schubert, U.; Bibollet-Ruche, F.; Keele, B.F.; et al. The evolution of pandemic and non-pandemic HIV-1 strains has been driven by Tetherin antagonism. Cell Host Microbe 2009, 6, 409–421. [Google Scholar] [CrossRef] [PubMed]
- Willey, R.L.; Maldarelli, F.; Martin, M.A.; Strebel, K. Human immunodeficiency virus type 1 Vpu protein induces rapid degradation of CD4. J. Virol. 1992, 66, 7193–7200. [Google Scholar] [PubMed]
- Schubert, U.; Antón, L.C.; Bacík, I.; Cox, J.H.; Bour, S.; Bennink, J.R.; Orlowski, M.; Strebel, K.; Yewdell, J.W. CD4 glycoprotein degradation induced by human immunodeficiency virus type 1 Vpu protein requires the function of proteasomes and the ubiquitin-conjugating pathway. J. Virol. 1998, 72, 2280–2288. [Google Scholar] [PubMed]
- Binette, J.; Dubé, M.; Mercier, J.; Halawani, D.; Latterich, M.; Cohen, E.A. Requirements for the selective degradation of CD4 receptor molecules by the human immunodeficiency virus type 1 Vpu protein in the endoplasmic reticulum. Retrovirology 2007, 4, 75. [Google Scholar] [CrossRef] [PubMed]
- Magadán, J.G.; Pérez-Victoria, F.J.; Sougrat, R.; Ye, Y.; Strebel, K.; Bonifacino, J.S. Multilayered mechanism of CD4 downregulation by HIV-1 Vpu involving distinct ER retention and ERAD targeting steps. PLoS Pathog. 2010, 6, e1000869. [Google Scholar] [CrossRef] [PubMed]
- Magadán, J.G.; Bonifacino, J.S. Transmembrane domain determinants of CD4 downregulation by HIV-1 Vpu. J. Virol. 2012, 86, 757–772. [Google Scholar] [CrossRef] [PubMed]
- Petris, G.; Casini, A.; Sasset, L.; Cesaratto, F.; Bestagno, M.; Cereseto, A.; Burrone, O.R. CD4 and BST-2/tetherin proteins retro-translocate from endoplasmic reticulum to cytosol as partially folded and multimeric molecules. J. Biol. Chem. 2014, 289, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Sugden, S.M.; Pham, T.N. Q.; Cohen, É.A. HIV-1 Vpu downmodulates ICAM-1 expression, resulting in decreased killing of infected CD4(+) T cells by NK cells. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed]
- Matheson, N.J.; Sumner, J.; Wals, K.; Rapiteanu, R.; Weekes, M.P.; Vigan, R.; Weinelt, J.; Schindler, M.; Antrobus, R.; Costa, A.S.H.; et al. Cell surface proteomic map of HIV infection reveals antagonism of amino acid metabolism by Vpu and Nef. Cell Host Microbe 2015, 18, 409–423. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.; Vu, T.; Novince, Z.; Guerrero-Santoro, J.; Rapic-Otrin, V.; Gronenborn, A.M. HIV-1 Vpr loads uracil DNA glycosylase-2 onto DCAF1, a substrate recognition subunit of a cullin 4A-ring E3 ubiquitin ligase for proteasome-dependent degradation. J. Biol. Chem. 2010, 285, 37333–37341. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Zhou, X.; Barnes, C.O.; DeLucia, M.; Cohen, A.E.; Gronenborn, A.M.; Ahn, J.; Calero, G. The DDB1-DCAF1-Vpr-UNG2 crystal structure reveals how HIV-1 Vpr steers human UNG2 toward destruction. Nat. Struct. Mol. Biol. 2016, 23, 933–940. [Google Scholar] [CrossRef] [PubMed]
- Schröfelbauer, B.; Yu, Q.; Zeitlin, S.G.; Landau, N.R. Human immunodeficiency virus type 1 Vpr induces the degradation of the UNG and SMUG uracil-DNA glycosylases. J. Virol. 2005, 79, 10978–10987. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Wang, Y.; Tokunaga, K.; Huang, F.; Sun, B.; Yang, R. The HIV-1 accessory protein Vpr induces the degradation of the anti-HIV-1 agent APOBEC3G through a VprBP-mediated proteasomal pathway. Virus Res. 2015, 195, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Casey Klockow, L.; Sharifi, H.J.; Wen, X.; Flagg, M.; Furuya, A.K.M.; Nekorchuk, M.; de Noronha, C.M.C. The HIV-1 protein Vpr targets the endoribonuclease Dicer for proteasomal degradation to boost macrophage infection. Virology 2013, 444, 191–202. [Google Scholar] [CrossRef] [PubMed]
- Lahouassa, H.; Blondot, M.-L.; Chauveau, L.; Chougui, G.; Morel, M.; Leduc, M.; Guillonneau, F.; Ramirez, B.C.; Schwartz, O.; Margottin-Goguet, F. HIV-1 Vpr degrades the HLTF DNA translocase in T cells and macrophages. Proc. Natl. Acad. Sci. USA 2016, 113, 5311–5316. [Google Scholar] [CrossRef] [PubMed]
- Romani, B.; Shaykh Baygloo, N.; Aghasadeghi, M.R.; Allahbakhshi, E. HIV-1 Vpr protein Enhances proteasomal degradation of MCM10 DNA replication factor through the Cul4-DDB1[VprBP] E3 ubiquitin ligase to induce G2/M cell cycle arrest. J. Biol. Chem. 2015, 290, 17380–17389. [Google Scholar] [CrossRef] [PubMed]
- Maudet, C.; Sourisce, A.; Dragin, L.; Lahouassa, H.; Rain, J.-C.; Bouaziz, S.; Ramirez, B.C.; Margottin-Goguet, F. HIV-1 Vpr induces the degradation of ZIP and sZIP, adaptors of the NuRD chromatin remodeling complex, by hijacking DCAF1/VprBP. PLoS ONE 2013, 8, e77320. [Google Scholar] [CrossRef] [PubMed]
- Romani, B.; Baygloo, N.S.; Hamidi-Fard, M.; Aghasadeghi, M.R.; Allahbakhshi, E. HIV-1 Vpr protein induces proteasomal degradation of chromatin-associated class I HDACs to overcome latent infection of macrophages. J. Biol. Chem. 2016, 291, 2696–2711. [Google Scholar] [CrossRef] [PubMed]
- Klinger, P.P.; Schubert, U. The ubiquitin-proteasome system in HIV replication: Potential targets for antiretroviral therapy. Expert Rev. Anti-Infect. Ther. 2005, 3, 61–79. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Guo, H.; Su, J.; Rui, Y.; Zheng, W.; Gao, W.; Zhang, W.; Li, Z.; Liu, G.; Markham, R.B.; et al. Inhibition of Vpx-mediated SAMHD1 and Vpr-mediated host helicase transcription factor degradation by selective disruption of viral CRL4 (DCAF1) E3 ubiquitin ligase assembly. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed]






© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seissler, T.; Marquet, R.; Paillart, J.-C. Hijacking of the Ubiquitin/Proteasome Pathway by the HIV Auxiliary Proteins. Viruses 2017, 9, 322. https://doi.org/10.3390/v9110322
Seissler T, Marquet R, Paillart J-C. Hijacking of the Ubiquitin/Proteasome Pathway by the HIV Auxiliary Proteins. Viruses. 2017; 9(11):322. https://doi.org/10.3390/v9110322
Chicago/Turabian StyleSeissler, Tanja, Roland Marquet, and Jean-Christophe Paillart. 2017. "Hijacking of the Ubiquitin/Proteasome Pathway by the HIV Auxiliary Proteins" Viruses 9, no. 11: 322. https://doi.org/10.3390/v9110322
APA StyleSeissler, T., Marquet, R., & Paillart, J.-C. (2017). Hijacking of the Ubiquitin/Proteasome Pathway by the HIV Auxiliary Proteins. Viruses, 9(11), 322. https://doi.org/10.3390/v9110322