Therapeutic Strategies against Epstein-Barr Virus-Associated Cancers Using Proteasome Inhibitors


Abstract
1. Introduction
2. The Ubiquitin-Proteasome System
2.1. Structure and Function of Proteasome
2.2. Proteasomal Degradation Mediated by Ubiquitination
3. Interaction of EBV with Ubiquitin-Proteasome System in Host Cells
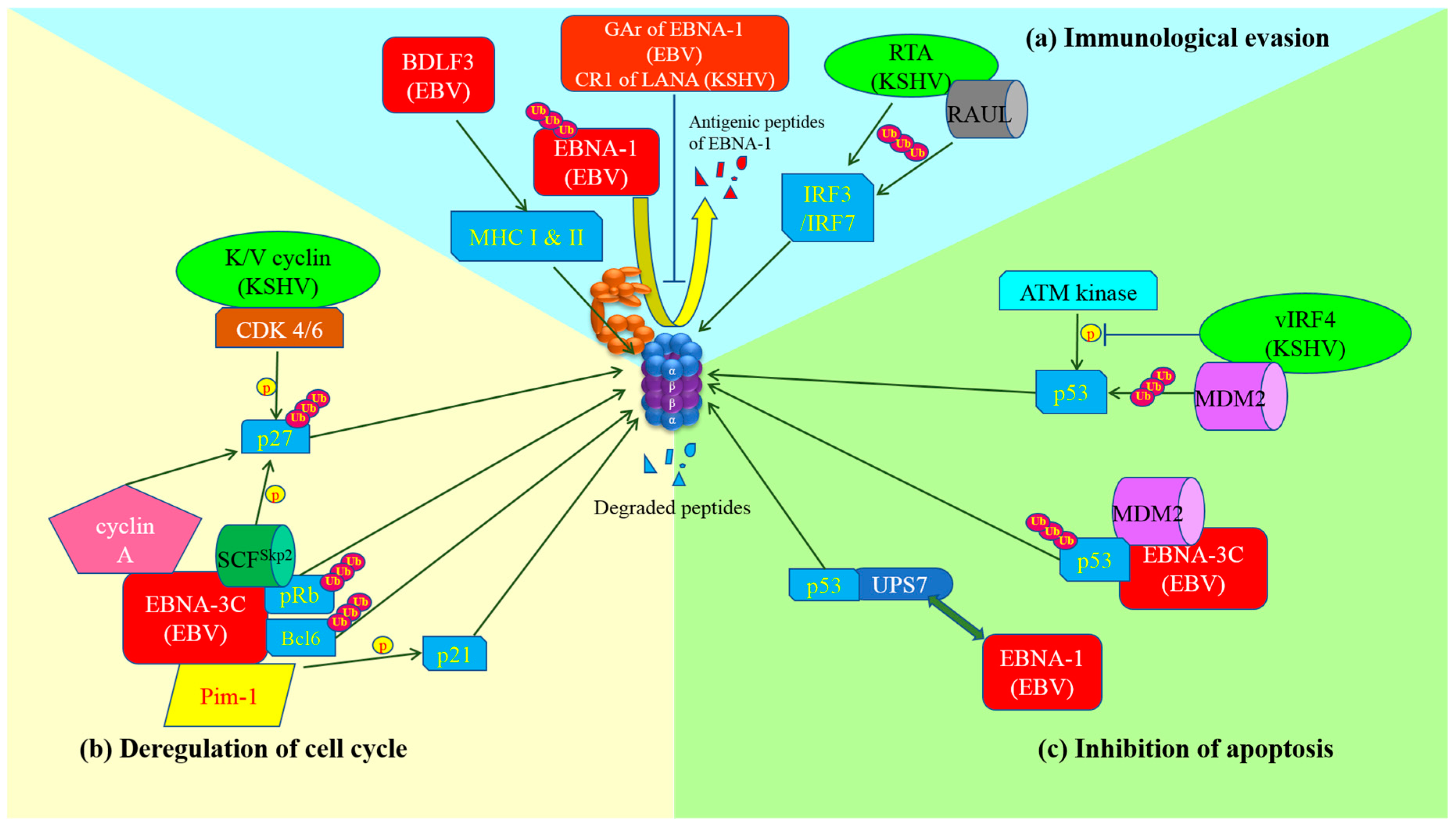
3.1. Immunological Evasion
3.2. Modulation of Cell Cycle Checkpoints
3.3. Inhibition of Apoptosis
4. Rationale of Using Proteasome Inhibitor to Treat EBV-Associated Cancers
4.1. Overview of Proteasome Inhibitors
4.2. Effect of Proteasome Inhibitors on Cell Cycle of EBV-Associated Malignancies
4.3. Effect of Proteasome Inhibitors on Apoptosis of EBV-Associated Malignancies
4.4. Reactivation of Viral Lytic Cycle by Proteasome Inhibitors
4.5. Effect of Proteasome Inhibitors on Immune Evasion
5. Potential Novel Viral-Targeted Strategies against EBV-Associated Cancers by Combination of Proteasome and Histone Deacetylase (HDAC) Inhibitors
5.1. Combination of Proteasome and HDAC Inhibitors on EBV-Associated Epithelial Malignancies
5.2. Combination of Proteasome and HDAC Inhibitors on EBV-Associated Lymphoid Cells
5.3. Pre-Clinical Data of Combination of Proteasome and HDAC Inhibitors on Treatment of EBV-Associated Malignancies
6. Concluding Remarks
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Blanchette, P.; Branton, P.E. Manipulation of the ubiquitin-proteasome pathway by small DNA tumor viruses. Virology 2009, 384, 317–323. [Google Scholar] [CrossRef] [PubMed]
- Ding, F.; Xiao, H.; Wang, M.; Xie, X.; Hu, F. The role of the ubiquitin-proteasome pathway in cancer development and treatment. Front. Biosci. 2014, 19, 886–895. [Google Scholar] [CrossRef]
- Honda, R.; Tanaka, H.; Yasuda, H. Oncoprotein MDM2 is a ubiquitin ligase E3 for tumor suppressor p53. FEBS Lett. 1997, 420, 25–27. [Google Scholar] [CrossRef]
- Knight, J.S.; Sharma, N.; Robertson, E.S. Epstein-Barr virus latent antigen 3C can mediate the degradation of the retinoblastoma protein through an SCF cellular ubiquitin ligase. Proc. Natl. Acad. Sci. USA 2005, 102, 18562–18566. [Google Scholar] [CrossRef] [PubMed]
- Iwahori, S.; Murata, T.; Kudoh, A.; Sato, Y.; Nakayama, S.; Isomura, H.; Kanda, T.; Tsurumi, T. Phosphorylation of P27KIP1 by Epstein-Barr virus protein kinase induces its degradation through SCFSKP2 ubiquitin ligase actions during viral lytic replication. J. Biol. Chem. 2009, 284, 18923–18931. [Google Scholar] [CrossRef] [PubMed]
- Full, F.; Hahn, A.S.; Grosskopf, A.K.; Ensser, A. Gammaherpesviral tegument proteins, PML-nuclear bodies and the ubiquitin-proteasome system. Viruses 2017, 9, 308. [Google Scholar] [CrossRef] [PubMed]
- Groll, M.; Ditzel, L.; Lowe, J.; Stock, D.; Bochtler, M.; Bartunik, H.D.; Huber, R. Structure of 20s proteasome from yeast at 2.4 Å resolution. Nature 1997, 386, 463–471. [Google Scholar] [CrossRef] [PubMed]
- Unno, M.; Mizushima, T.; Morimoto, Y.; Tomisugi, Y.; Tanaka, K.; Yasuoka, N.; Tsukihara, T. The structure of the mammalian 20s proteasome at 2.75 Å resolution. Structure 2002, 10, 609–618. [Google Scholar] [CrossRef]
- Liu, C.W.; Jacobson, A.D. Functions of the 19S complex in proteasomal degradation. Trends Biochem. Sci. 2013, 38, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Hershko, A.; Heller, H.; Elias, S.; Ciechanover, A. Components of ubiquitin-protein ligase system. Resolution, affinity purification and role in protein breakdown. J. Biol. Chem. 1983, 258, 8206–8214. [Google Scholar] [PubMed]
- Ye, Y.; Rape, M. Building ubiquitin chains: E2 enzymes at work. Nat. Rev. Mol. Cell Biol. 2009, 10, 755–764. [Google Scholar] [CrossRef] [PubMed]
- Elsasser, S.; Chandler-Militello, D.; Muller, B.; Hanna, J.; Finley, D. Rad23 and Rpn10 serve as alternative ubiquitin receptors for the proteasome. J. Biol. Chem. 2004, 279, 26817–26822. [Google Scholar] [CrossRef] [PubMed]
- Husnjak, K.; Elsasser, S.; Zhang, N.; Chen, X.; Randles, L.; Shi, Y.; Hofmann, K.; Walters, K.J.; Finley, D.; Dikic, I. Proteasome subunit Rpn13 is a novel ubiquitin receptor. Nature 2008, 453, 481–488. [Google Scholar] [CrossRef] [PubMed]
- Koegl, M.; Hoppe, T.; Schlenker, S.; Ulrich, H.D.; Mayer, T.U.; Jentsch, S. A novel ubiquitination factor, E4, is involved in multiubiquitin chain assembly. Cell 1999, 96, 635–644. [Google Scholar] [CrossRef]
- Michalek, M.T.; Grant, E.P.; Gramm, C.; Goldberg, A.L.; Rock, K.L. A role for the ubiquitin-dependent proteolytic pathway in MHC class I-restricted antigen presentation. Nature 1993, 363, 552–554. [Google Scholar] [CrossRef] [PubMed]
- Levitskaya, J.; Sharipo, A.; Leonchiks, A.; Ciechanover, A.; Masucci, M.G. Inhibition of ubiquitin/proteasome-dependent protein degradation by the Gly-Ala repeat domain of the Epstein-Barr virus nuclear antigen 1. Proc. Natl. Acad. Sci. USA 1997, 94, 12616–12621. [Google Scholar] [CrossRef] [PubMed]
- Kwun, H.J.; da Silva, S.R.; Qin, H.; Ferris, R.L.; Tan, R.; Chang, Y.; Moore, P.S. The central repeat domain 1 of kaposi’s sarcoma-associated herpesvirus (KSHV) latency associated-nuclear antigen 1 (LANA1) prevents cis MHC class I peptide presentation. Virology 2011, 412, 357–365. [Google Scholar] [CrossRef] [PubMed]
- Quinn, L.L.; Williams, L.R.; White, C.; Forrest, C.; Zuo, J.; Rowe, M. The missing link in Epstein-Barr virus immune evasion: The BDLF3 gene induces ubiquitination and downregulation of major histocompatibility complex class I (MHC-I) and MHC-II. J. Virol. 2015, 90, 356–367. [Google Scholar] [CrossRef] [PubMed]
- Coscoy, L.; Ganem, D. Kaposi's sarcoma—Associated herpesvirus encodes two proteins that block cell surface display of MHC class I chains by enhancing their endocytosis. Proc. Natl. Acad. Sci. USA 2000, 97, 8051–8056. [Google Scholar] [CrossRef] [PubMed]
- Honda, K.; Yanai, H.; Negishi, H.; Asagiri, M.; Sato, M.; Mizutani, T.; Shimada, N.; Ohba, Y.; Takaoka, A.; Yoshida, N.; et al. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature 2005, 434, 772–777. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Hayward, G.S. The ubiquitin E3 ligase RAUL negatively regulates type I interferon through ubiquitination of the transcription factors IRF7 and IRF3. Immunity 2010, 33, 863–877. [Google Scholar] [CrossRef] [PubMed]
- Maruo, S.; Zhao, B.; Johannsen, E.; Kieff, E.; Zou, J.; Takada, K. Epstein-Barr virus nuclear antigens 3C and 3A maintain lymphoblastoid cell growth by repressing P16INK4A and p14ARF expression. Proc. Natl. Acad. Sci. USA 2011, 108, 1919–1924. [Google Scholar] [CrossRef] [PubMed]
- Kashuba, E.; Yurchenko, M.; Yenamandra, S.P.; Snopok, B.; Isaguliants, M.; Szekely, L.; Klein, G. Ebv-encoded EBNA-6 binds and targets MRS18–2 to the nucleus, resulting in the disruption of PRB-E2F1 complexes. Proc. Natl. Acad. Sci. USA 2008, 105, 5489–5494. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Lu, J.; Cai, Q.; Sun, Z.; Jha, H.C.; Robertson, E.S. EBNA3C augments PIM-1 mediated phosphorylation and degradation of p21 to promote B-cell proliferation. PLoS Pathog. 2014, 10, e1004304. [Google Scholar] [CrossRef] [PubMed]
- Knight, J.S.; Sharma, N.; Robertson, E.S. SCFSKP2 complex targeted by Epstein-Barr virus essential nuclear antigen. Mol. Cell Biol. 2005, 25, 1749–1763. [Google Scholar] [CrossRef] [PubMed]
- Pei, Y.; Banerjee, S.; Jha, H.C.; Sun, Z.; Robertson, E.S. An essential EBV latent antigen 3C binds Bcl6 for targeted degradation and cell proliferation. PLoS Pathog. 2017, 13, e1006500. [Google Scholar] [CrossRef] [PubMed]
- Vardy, L.; Pesin, J.A.; Orr-Weaver, T.L. Regulation of cyclin a protein in meiosis and early embryogenesis. Proc. Proc. Natl. Acad. Sci. USA 2009, 106, 1838–1843. [Google Scholar] [CrossRef] [PubMed]
- Pagano, M.; Pepperkok, R.; Verde, F.; Ansorge, W.; Draetta, G. Cyclin a is required at two points in the human cell cycle. Embo. J. 1992, 11, 961–971. [Google Scholar] [PubMed]
- Mann, D.J.; Child, E.S.; Swanton, C.; Laman, H.; Jones, N. Modulation of p27(KIP1) levels by the cyclin encoded by kaposi’s sarcoma-associated herpesvirus. EMBO J. 1999, 18, 654–663. [Google Scholar] [CrossRef] [PubMed]
- Ellis, M.; Chew, Y.P.; Fallis, L.; Freddersdorf, S.; Boshoff, C.; Weiss, R.A.; Lu, X.; Mittnacht, S. Degradation of p27(KIP) CDK inhibitor triggered by kaposi’s sarcoma virus cyclin-CDK6 complex. EMBO J. 1999, 18, 644–653. [Google Scholar] [CrossRef] [PubMed]
- Saha, A.; Kaul, R.; Murakami, M.; Robertson, E.S. Tumor viruses and cancer biology: Modulating signaling pathways for therapeutic intervention. Cancer Biol. Ther. 2010, 10, 961–978. [Google Scholar] [CrossRef] [PubMed]
- Eliopoulos, A.G.; Caamano, J.H.; Flavell, J.; Reynolds, G.M.; Murray, P.G.; Poyet, J.L.; Young, L.S. Epstein-Barr virus—Encoded latent infection membrane protein 1 regulates the processing of p100 NF-κB2 to p52 via an IKKγ/nemo-independent signalling pathway. Oncogene 2003, 22, 7557–7569. [Google Scholar] [CrossRef] [PubMed]
- Holowaty, M.N.; Zeghouf, M.; Wu, H.; Tellam, J.; Athanasopoulos, V.; Greenblatt, J.; Frappier, L. Protein profiling with Epstein-Barr nuclear antigen-1 reveals an interaction with the herpesvirus-associated ubiquitin-specific protease HAUSP/USP7. J. Biol. Chem. 2003, 278, 29987–29994. [Google Scholar] [CrossRef] [PubMed]
- Saha, A.; Murakami, M.; Kumar, P.; Bajaj, B.; Sims, K.; Robertson, E.S. Epstein-Barr virus nuclear antigen 3C augments MDM2-mediated p53 ubiquitination and degradation by deubiquitinating MDM2. J. Virol 2009, 83, 4652–4669. [Google Scholar] [CrossRef] [PubMed]
- Shin, Y.C.; Nakamura, H.; Liang, X.; Feng, P.; Chang, H.; Kowalik, T.F.; Jung, J.U. Inhibition of the ATM/p53 signal transduction pathway by kaposi’s sarcoma-associated herpesvirus interferon regulatory factor 1. J. Virol. 2006, 80, 2257–2266. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.R.; Toth, Z.; Shin, Y.C.; Lee, J.S.; Chang, H.; Gu, W.; Oh, T.K.; Kim, M.H.; Jung, J.U. Kaposi’s sarcoma-associated herpesvirus viral interferon regulatory factor 4 targets MDM2 to deregulate the p53 tumor suppressor pathway. J. Virol. 2009, 83, 6739–6747. [Google Scholar] [CrossRef] [PubMed]
- Kisselev, A.F.; Goldberg, A.L. Proteasome inhibitors: From research tools to drug candidates. Chem. Biol. 2001, 8, 739–758. [Google Scholar] [CrossRef]
- Manasanch, E.E.; Orlowski, R.Z. Proteasome inhibitors in cancer therapy. Nat. Rev. Clin. Oncol. 2017, 14, 417–433. [Google Scholar] [CrossRef] [PubMed]
- Kane, R.C.; Farrell, A.T.; Sridhara, R.; Pazdur, R. United States food and drug administration approval summary: Bortezomib for the treatment of progressive multiple myeloma after one prior therapy. Clin. Cancer Res. 2006, 12, 2955–2960. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.Y.; Wei, C.C.; Chen, K.C.; Chen, H.J.; Cheng, A.L.; Chen, K.F. Bortezomib enhances radiation-induced apoptosis in solid tumors by inhibiting CIP2A. Cancer Lett. 2012, 317, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Shi, W.Y.; Wang, L.; Xiao, D.; Yao, Y.; Yang, F.; Jiang, X.X.; Leboeuf, C.; Janin, A.; Chen, S.J.; Zhao, W.L. Proteasome inhibitor bortezomib targeted tumor—Endothelial cell interaction in T-cell leukemia/lymphoma. Ann. Hematol. 2011, 90, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Rickinson, A.B.; Kieff, E. Epstein-barr Virus, 5th ed.; Lippincott Williams and Wilkins: Philadelphia, PA, USA, 2007; pp. 2657–2700. [Google Scholar]
- Allday, M.J.; Farrell, P.J. Epstein-Barr virus nuclear antigen EBNA3C/6 expression maintains the level of latent membrane protein 1 in G1-arrested cells. J. Virol. 1994, 68, 3491–3498. [Google Scholar] [PubMed]
- Saha, A.; Halder, S.; Upadhyay, S.K.; Lu, J.; Kumar, P.; Murakami, M.; Cai, Q.; Robertson, E.S. Epstein-Barr virus nuclear antigen 3C facilitates G1-S transition by stabilizing and enhancing the function of cyclin D1. PLoS Pathog. 2011, 7, e1001275. [Google Scholar] [CrossRef] [PubMed]
- Hertle, M.L.; Popp, C.; Petermann, S.; Maier, S.; Kremmer, E.; Lang, R.; Mages, J.; Kempkes, B. Differential gene expression patterns of EBV infected EBNA-3A positive and negative human B lymphocytes. PLoS Pathog. 2009, 5, e1000506. [Google Scholar] [CrossRef] [PubMed]
- Saha, A.; Robertson, E.S. Impact of ebv essential nuclear protein EBNA-3C on B-cell proliferation and apoptosis. Future Microbiol. 2013, 8, 323–352. [Google Scholar] [CrossRef] [PubMed]
- Maruo, S.; Wu, Y.; Ishikawa, S.; Kanda, T.; Iwakiri, D.; Takada, K. Epstein-Barr virus nuclear protein EBNA3C is required for cell cycle progression and growth maintenance of lymphoblastoid cells. Proc. Natl. Acad. Sci. USA 2006, 103, 19500–19505. [Google Scholar] [CrossRef] [PubMed]
- Cai, Q.; Guo, Y.; Xiao, B.; Banerjee, S.; Saha, A.; Lu, J.; Glisovic, T.; Robertson, E.S. Epstein-Barr virus nuclear antigen 3C stabilizes gemin3 to block p53-mediated apoptosis. PLoS Pathog. 2011, 7, e1002418. [Google Scholar] [CrossRef] [PubMed]
- Skalska, L.; White, R.E.; Franz, M.; Ruhmann, M.; Allday, M.J. Epigenetic repression of p16(INK4A) by latent Epstein-Barr virus requires the interaction of EBNA3A and EBNA3C with CTBP. PLoS Pathog. 2010, 6, e1000951. [Google Scholar] [CrossRef] [PubMed]
- Krauer, K.G.; Burgess, A.; Buck, M.; Flanagan, J.; Sculley, T.B.; Gabrielli, B. The EBNA-3 gene family proteins disrupt the G2/M checkpoint. Oncogene 2004, 23, 1342–1353. [Google Scholar] [CrossRef] [PubMed]
- Choudhuri, T.; Verma, S.C.; Lan, K.; Murakami, M.; Robertson, E.S. The ATM/ATR signaling effector CHK2 is targeted by Epstein-Barr virus nuclear antigen 3C to release the G2/M cell cycle block. J. Virol. 2007, 81, 6718–6730. [Google Scholar] [CrossRef] [PubMed]
- Parker, G.A.; Touitou, R.; Allday, M.J. Epstein-Barr virus EBNA3C can disrupt multiple cell cycle checkpoints and induce nuclear division divorced from cytokinesis. Oncogene 2000, 19, 700–709. [Google Scholar] [CrossRef] [PubMed]
- Wade, M.; Allday, M.J. Epstein-Barr virus suppresses a G(2)/M checkpoint activated by genotoxins. Mol. Cell Biol. 2000, 20, 1344–1360. [Google Scholar] [CrossRef] [PubMed]
- White, R.E.; Groves, I.J.; Turro, E.; Yee, J.; Kremmer, E.; Allday, M.J. Extensive co-operation between the Epstein-Barr virus EBNA3 proteins in the manipulation of host gene expression and epigenetic chromatin modification. PLoS ONE 2010, 5, e13979. [Google Scholar] [CrossRef] [PubMed]
- Rapino, F.; Naumann, I.; Fulda, S. Bortezomib antagonizes microtubule-interfering drug-induced apoptosis by inhibiting G2/M transition and MCL-1 degradation. Cell Death Dis. 2013, 4, e925. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.S.; Hong, S.W.; Kim, S.M.; Jin, D.H.; Shin, J.S.; Yoon, D.H.; Kim, K.P.; Lee, J.L.; Heo, D.S.; Lee, J.S.; et al. Bortezomib induces G2-M arrest in human colon cancer cells through ROS-inducible phosphorylation of ATM-CHK1. Int. J. Oncol. 2012, 41, 76–82. [Google Scholar] [PubMed]
- Bonvini, P.; Zorzi, E.; Basso, G.; Rosolen, A. Bortezomib-mediated 26S proteasome inhibition causes cell-cycle arrest and induces apoptosis in CD30+ anaplastic large cell lymphoma. Leukemia 2007, 21, 838–842. [Google Scholar] [PubMed]
- Hui, K.F.; Leung, Y.Y.; Yeung, P.L.; Middeldorp, J.M.; Chiang, A.K. Combination of SAHA and bortezomib up-regulates CDKN2A and CDKN1A and induces apoptosis of Epstein-Barr virus-positive WP-restricted burkitt lymphoma and lymphoblastoid cell lines. Br. J. Haematol. 2014, 167, 639–650. [Google Scholar] [CrossRef] [PubMed]
- Vaysberg, M.; Hatton, O.; Lambert, S.L.; Snow, A.L.; Wong, B.; Krams, S.M.; Martinez, O.M. Tumor-derived variants of Epstein-Barr virus latent membrane protein 1 induce sustained ERK activation and C-FOS. J. Biol. Chem. 2008, 283, 36573–36585. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.A.; Dawson, C.W.; Young, L.S. Role of the Epstein-Barr virus-encoded latent membrane protein-1, LMP1, in the pathogenesis of nasopharyngeal carcinoma. Future Oncol. 2009, 5, 811–825. [Google Scholar] [CrossRef] [PubMed]
- Hui, K.F.; Lam, B.H.; Ho, D.N.; Tsao, S.W.; Chiang, A.K. Bortezomib and SAHA synergistically induce ROS-driven caspase-dependent apoptosis of nasopharyngeal carcinoma and block replication of Epstein-Barr virus. Mol. Cancer Ther. 2013, 12, 747–758. [Google Scholar] [CrossRef] [PubMed]
- Zou, P.; Kawada, J.; Pesnicak, L.; Cohen, J.I. Bortezomib induces apoptosis of Epstein-Barr virus (EBV)-transformed B cells and prolongs survival of mice inoculated with ebv-transformed B cells. J. Virol. 2007, 81, 10029–10036. [Google Scholar] [CrossRef] [PubMed]
- Leao, M.; Anderton, E.; Wade, M.; Meekings, K.; Allday, M.J. Epstein-Barr virus-induced resistance to drugs that activate the mitotic spindle assembly checkpoint in burkitt’s lymphoma cells. J. Virol. 2007, 81, 248–260. [Google Scholar] [CrossRef] [PubMed]
- Kelly, G.L.; Milner, A.E.; Tierney, R.J.; Croom-Carter, D.S.; Altmann, M.; Hammerschmidt, W.; Bell, A.I.; Rickinson, A.B. Epstein-Barr virus nuclear antigen 2 (EBNA2) gene deletion is consistently linked with EBNA3A, -3B and -3C expression in burkitt’s lymphoma cells and with increased resistance to apoptosis. J. Virol. 2005, 79, 10709–10717. [Google Scholar] [CrossRef] [PubMed]
- Vereide, D.T.; Sugden, B. Lymphomas differ in their dependence on Epstein-Barr virus. Blood 2011, 117, 1977–1985. [Google Scholar] [CrossRef] [PubMed]
- Picksley, S.M.; Vojtesek, B.; Sparks, A.; Lane, D.P. Immunochemical analysis of the interaction of p53 with MDM2—Fine mapping of the MDM2 binding site on p53 using synthetic peptides. Oncogene 1994, 9, 2523–2529. [Google Scholar] [PubMed]
- Haupt, Y.; Maya, R.; Kazaz, A.; Oren, M. MDM2 promotes the rapid degradation of p53. Nature 1997, 387, 296–299. [Google Scholar] [CrossRef] [PubMed]
- Shirley, C.M.; Chen, J.; Shamay, M.; Li, H.; Zahnow, C.A.; Hayward, S.D.; Ambinder, R.F. Bortezomib induction of C/EBPβ mediates Epstein-Barr virus lytic activation in burkitt lymphoma. Blood 2011, 117, 6297–6303. [Google Scholar] [CrossRef] [PubMed]
- Fu, D.X.; Tanhehco, Y.; Chen, J.; Foss, C.A.; Fox, J.J.; Chong, J.M.; Hobbs, R.F.; Fukayama, M.; Sgouros, G.; Kowalski, J.; et al. Bortezomib-induced enzyme-targeted radiation therapy in herpesvirus-associated tumors. Nat. Med. 2008, 14, 1118–1122. [Google Scholar] [CrossRef] [PubMed]
- Sarosiek, K.A.; Cavallin, L.E.; Bhatt, S.; Toomey, N.L.; Natkunam, Y.; Blasini, W.; Gentles, A.J.; Ramos, J.C.; Mesri, E.A.; Lossos, I.S. Efficacy of bortezomib in a direct xenograft model of primary effusion lymphoma. Proc. Natl. Acad. Sci. USA 2010, 107, 13069–13074. [Google Scholar] [CrossRef] [PubMed]
- Granato, M.; Romeo, M.A.; Tiano, M.S.; Santarelli, R.; Gonnella, R.; Gilardini Montani, M.S.; Faggioni, A.; Cirone, M. Bortezomib promotes KHSV and EBV lytic cycle by activating JNK and autophagy. Sci. Rep. 2017, 7, 13052. [Google Scholar] [CrossRef] [PubMed]
- Hui, K.F.; Yeung, P.L.; Chiang, A.K. Induction of MAPK- and ROS-dependent autophagy and apoptosis in gastric carcinoma by combination of romidepsin and bortezomib. Oncotarget 2015, 7, 4454. [Google Scholar]
- Hui, K.F.; Chiang, A.K. Suberoylanilide hydroxamic acid induces viral lytic cycle in Epstein-Barr virus-positive epithelial malignancies and mediates enhanced cell death. Int. J. Cancer 2010, 126, 2479–2489. [Google Scholar] [CrossRef] [PubMed]
- Hui, K.F.; Ho, D.N.; Tsang, C.M.; Middeldorp, J.M.; Tsao, G.S.; Chiang, A.K. Activation of lytic cycle of Epstein-Barr virus by suberoylanilide hydroxamic acid leads to apoptosis and tumor growth suppression of nasopharyngeal carcinoma. Int. J. Cancer 2012, 131, 1930–1940. [Google Scholar] [CrossRef] [PubMed]
- Choi, C.K.; Ho, D.N.; Hui, K.F.; Kao, R.Y.; Chiang, A.K. Identification of novel small organic compounds with diverse structures for the induction of Epstein-Barr virus (EBV) lytic cycle in EBV-positive epithelial malignancies. PLoS ONE 2015, 10, e0145994. [Google Scholar] [CrossRef] [PubMed]
- Hui, K.F.; Cheung, A.K.; Choi, C.K.; Yeung, P.L.; Middeldorp, J.M.; Lung, M.L.; Tsao, S.W.; Chiang, A.K. Inhibition of class I histone deacetylases by romidepsin potently induces Epstein-Barr virus lytic cycle and mediates enhanced cell death with ganciclovir. Int. J. Cancer 2016, 138, 125–136. [Google Scholar] [CrossRef] [PubMed]
- Pellom, S.T., Jr.; Dudimah, D.F.; Thounaojam, M.C.; Uzhachenko, R.V.; Singhal, A.; Richmond, A.; Shanker, A. Bortezomib augments lymphocyte stimulatory cytokine signaling in the tumor microenvironment to sustain CD8+ T cell antitumor function. Oncotarget 2017, 8, 8604–8621. [Google Scholar] [CrossRef] [PubMed]
- Jiang, G.M.; Wang, H.S.; Du, J.; Ma, W.F.; Wang, H.; Qiu, Y.; Zhang, Q.G.; Xu, W.; Liu, H.F.; Liang, J.P. Bortezomib relieves immune tolerance in nasopharyngeal carcinoma via STAT1 suppression and indoleamine 2,3-dioxygenase downregulation. Cancer Immunol. Res. 2017, 5, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Pei, X.Y.; Dai, Y.; Grant, S. Synergistic induction of oxidative injury and apoptosis in human multiple myeloma cells by the proteasome inhibitor bortezomib and histone deacetylase inhibitors. Clin. Cancer Res. 2004, 10, 3839–3852. [Google Scholar] [CrossRef] [PubMed]
- Heider, U.; von Metzler, I.; Kaiser, M.; Rosche, M.; Sterz, J.; Rotzer, S.; Rademacher, J.; Jakob, C.; Fleissner, C.; Kuckelkorn, U.; et al. Synergistic interaction of the histone deacetylase inhibitor SAHA with the proteasome inhibitor bortezomib in mantle cell lymphoma. Eur. J. Haematol. 2008, 80, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Heider, U.; Rademacher, J.; Lamottke, B.; Mieth, M.; Moebs, M.; von Metzler, I.; Assaf, C.; Sezer, O. Synergistic interaction of the histone deacetylase inhibitor SAHA with the proteasome inhibitor bortezomib in cutaneous T cell lymphoma. Eur. J. Haematol. 2009, 82, 440–449. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Rahmani, M.; Conrad, D.; Subler, M.; Dent, P.; Grant, S. The proteasome inhibitor bortezomib interacts synergistically with histone deacetylase inhibitors to induce apoptosis in BCR/ABL+ cells sensitive and resistant to STI571. Blood 2003, 102, 3765–3774. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.L.; Wang, L.; Zhang, Y.W.; Jiang, X.X.; Yang, F.; Wu, W.L.; Janin, A.; Chen, Z.; Shen, Z.X.; Chen, S.J.; et al. The proteasome inhibitor bortezomib interacts synergistically with the histone deacetylase inhibitor suberoylanilide hydroxamic acid to induce T-leukemia/lymphoma cells apoptosis. Leukemia 2009, 23, 1507–1514. [Google Scholar] [CrossRef] [PubMed]
- Emanuele, S.; Lauricella, M.; Carlisi, D.; Vassallo, B.; D’Anneo, A.; di Fazio, P.; Vento, R.; Tesoriere, G. SAHA induces apoptosis in hepatoma cells and synergistically interacts with the proteasome inhibitor bortezomib. Apoptosis 2007, 12, 1327–1338. [Google Scholar] [CrossRef] [PubMed]
- Denlinger, C.E.; Rundall, B.K.; Jones, D.R. Proteasome inhibition sensitizes non-small cell lung cancer to histone deacetylase inhibitor-induced apoptosis through the generation of reactive oxygen species. J. Thorac. Cardiovasc. Surg. 2004, 128, 740–748. [Google Scholar] [CrossRef] [PubMed]
- Sato, A.; Asano, T.; Ito, K.; Sumitomo, M. Suberoylanilide hydroxamic acid (SAHA) combined with bortezomib inhibits renal cancer growth by enhancing histone acetylation and protein ubiquitination synergistically. BJU Int. 2012, 109, 1258–1268. [Google Scholar] [CrossRef] [PubMed]
- Miller, C.P.; Rudra, S.; Keating, M.J.; Wierda, W.G.; Palladino, M.; Chandra, J. Caspase-8 dependent histone acetylation by a novel proteasome inhibitor, NPI-0052: A mechanism for synergy in leukemia cells. Blood 2009, 113, 4289–4299. [Google Scholar] [CrossRef] [PubMed]
- Nawrocki, S.T.; Carew, J.S.; Pino, M.S.; Highshaw, R.A.; Andtbacka, R.H.; Dunner, K., Jr.; Pal, A.; Bornmann, W.G.; Chiao, P.J.; Huang, P.; et al. Aggresome disruption: A novel strategy to enhance bortezomib-induced apoptosis in pancreatic cancer cells. Cancer Res. 2006, 66, 3773–3781. [Google Scholar] [CrossRef] [PubMed]
- Marks, P.A. The mechanism of the anti-tumor activity of the histone deacetylase inhibitor, suberoylanilide hydroxamic acid (SAHA). Cell Cycle 2004, 3, 534–535. [Google Scholar] [CrossRef] [PubMed]
- Trachootham, D.; Alexandre, J.; Huang, P. Targeting cancer cells by ROS-mediated mechanisms: A radical therapeutic approach? Nat. Rev. Drug Discov. 2009, 8, 579–591. [Google Scholar] [CrossRef] [PubMed]
- Hui, K.F.; Chiang, A.K. Combination of proteasome and class I HDAC inhibitors induces apoptosis of NPC cells through an HDAC6-independent ER stress-induced mechanism. Int. J. Cancer 2014, 135, 2950–2961. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Zhao, S.; Zhang, Y.; Wu, J.; Peng, H.; Fan, J.; Liao, J. Reactive oxygen species-mediated endoplasmic reticulum stress and mitochondrial dysfunction contribute to polydatin-induced apoptosis in human nasopharyngeal carcinoma CNE cells. J. Cell Biochem. 2011, 112, 3695–3703. [Google Scholar] [CrossRef] [PubMed]
- Quan, Z.; Gu, J.; Dong, P.; Lu, J.; Wu, X.; Wu, W.; Fei, X.; Li, S.; Wang, Y.; Wang, J.; et al. Reactive oxygen species-mediated endoplasmic reticulum stress and mitochondrial dysfunction contribute to cirsimaritin-induced apoptosis in human gallbladder carcinoma GBC-SD cells. Cancer Lett. 2010, 295, 252–259. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.S.; Kim, A.K. Platycodin D induces reactive oxygen species-mediated apoptosis signal-regulating kinase 1 activation and endoplasmic reticulum stress response in human breast cancer cells. J. Med. Food 2012, 15, 691–699. [Google Scholar] [CrossRef] [PubMed]
- Nawrocki, S.T.; Carew, J.S.; Maclean, K.H.; Courage, J.F.; Huang, P.; Houghton, J.A.; Cleveland, J.L.; Giles, F.J.; McConkey, D.J. Myc regulates aggresome formation, the induction of NOXA and apoptosis in response to the combination of bortezomib and SAHA. Blood 2008, 112, 2917–2926. [Google Scholar] [CrossRef] [PubMed]
- Brunk, U.T.; Dalen, H.; Roberg, K.; Hellquist, H.B. Photo-oxidative disruption of lysosomal membranes causes apoptosis of cultured human fibroblasts. Free Radic. Biol. Med. 1997, 23, 616–626. [Google Scholar] [CrossRef]
- Chen, Y.; McMillan-Ward, E.; Kong, J.; Israels, S.J.; Gibson, S.B. Oxidative stress induces autophagic cell death independent of apoptosis in transformed and cancer cells. Cell Death Differ. 2008, 15, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Kiffin, R.; Bandyopadhyay, U.; Cuervo, A.M. Oxidative stress and autophagy. Antioxid. Redox. Signal 2006, 8, 152–162. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, E.; Richardson, D.R.; Jansson, P.J. The anticancer agent Di-2-pyridylketone 4,4-dimethyl-3-thiosemicarbazone (DP44MT) overcomes prosurvival autophagy by two mechanisms: Persistent induction of autophagosome synthesis and impairment of lysosomal integrity. J. Biol. Chem. 2014, 289, 33568–33589. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Kamranvar, S.A.; Masucci, M.G. Oxidative stress enables Epstein-Barr virus-induced B-cell transformation by posttranscriptional regulation of viral and cellular growth-promoting factors. Oncogene 2016, 35, 3807–3816. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.Y.; Fang, C.Y.; Wu, C.C.; Tsai, C.H.; Lin, S.F.; Chen, J.Y. Reactive oxygen species mediate Epstein-Barr virus reactivation by N-methyl-N′-nitro-N-nitrosoguanidine. PLoS ONE 2013, 8, e84919. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.Y.; Mansouri, S.; Frappier, L. Changes in the nasopharyngeal carcinoma nuclear proteome induced by the EBNA1 protein of Epstein-Barr virus reveal potential roles for ebna1 in metastasis and oxidative stress responses. J. Virol. 2012, 86, 382–394. [Google Scholar] [CrossRef] [PubMed]
- Cooper, A.; Johannsen, E.; Maruo, S.; Cahir-McFarland, E.; Illanes, D.; Davidson, D.; Kieff, E. EBNA3A association with RBP-Jκ down-regulates c-myc and Epstein-Barr virus-transformed lymphoblast growth. J. Virol. 2003, 77, 999–1010. [Google Scholar] [CrossRef] [PubMed]
- Saha, A.; Bamidele, A.; Murakami, M.; Robertson, E.S. EBNA3C attenuates the function of p53 through interaction with inhibitor of growth family proteins 4 and 5. J. Virol. 2011, 85, 2079–2088. [Google Scholar] [CrossRef] [PubMed]
- Anderton, E.; Yee, J.; Smith, P.; Crook, T.; White, R.E.; Allday, M.J. Two Epstein-Barr virus (EBV) oncoproteins cooperate to repress expression of the proapoptotic tumour-suppressor BIM: Clues to the pathogenesis of burkitt’s lymphoma. Oncogene 2008, 27, 421–433. [Google Scholar] [CrossRef] [PubMed]
- Paschos, K.; Smith, P.; Anderton, E.; Middeldorp, J.M.; White, R.E.; Allday, M.J. Epstein-Barr virus latency in B cells leads to epigenetic repression and CPG methylation of the tumour suppressor gene BIM. PLoS Pathog. 2009, 5, e1000492. [Google Scholar] [CrossRef] [PubMed]
- Chinnadurai, G. CTBP, an unconventional transcriptional corepressor in development and oncogenesis. Mol. Cell 2002, 9, 213–224. [Google Scholar] [CrossRef]
- Radkov, S.A.; Touitou, R.; Brehm, A.; Rowe, M.; West, M.; Kouzarides, T.; Allday, M.J. Epstein-Barr virus nuclear antigen 3C interacts with histone deacetylase to repress transcription. J. Virol. 1999, 73, 5688–5697. [Google Scholar] [PubMed]
- Knight, J.S.; Lan, K.; Subramanian, C.; Robertson, E.S. Epstein-Barr virus nuclear antigen 3C recruits histone deacetylase activity and associates with the corepressors MSIN3A and NCOR in human B-cell lines. J. Virol. 2003, 77, 4261–4272. [Google Scholar] [CrossRef] [PubMed]
- Boutros, R.; Lobjois, V.; Ducommun, B. CDC25 phosphatases in cancer cells: Key players? Good targets? Nat. Rev. Cancer 2007, 7, 495–507. [Google Scholar] [CrossRef] [PubMed]
- Hermeking, H.; Benzinger, A. 14–3-3 proteins in cell cycle regulation. Semin. Cancer Biol. 2006, 16, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Harrison, S.J.; Bishton, M.; Bates, S.E.; Grant, S.; Piekarz, R.L.; Johnstone, R.W.; Dai, Y.; Lee, B.; Araujo, M.E.; Prince, H.M. A focus on the preclinical development and clinical status of the histone deacetylase inhibitor, romidepsin (depsipeptide, istodax®). Epigenomics 2012, 4, 571–589. [Google Scholar] [CrossRef] [PubMed]
- Moreau, P.; Richardson, P.G.; Cavo, M.; Orlowski, R.Z.; San Miguel, J.F.; Palumbo, A.; Harousseau, J.L. Proteasome inhibitors in multiple myeloma: 10 years later. Blood 2012, 120, 947–959. [Google Scholar] [CrossRef] [PubMed]
- Piekarz, R.L.; Frye, R.; Prince, H.M.; Kirschbaum, M.H.; Zain, J.; Allen, S.L.; Jaffe, E.S.; Ling, A.; Turner, M.; Peer, C.J.; et al. Phase 2 trial of romidepsin in patients with peripheral T-cell lymphoma. Blood 2011, 117, 5827–5834. [Google Scholar] [CrossRef] [PubMed]
- Bross, P.F.; Kane, R.; Farrell, A.T.; Abraham, S.; Benson, K.; Brower, M.E.; Bradley, S.; Gobburu, J.V.; Goheer, A.; Lee, S.L.; et al. Approval summary for bortezomib for injection in the treatment of multiple myeloma. Clin. Cancer Res. 2004, 10, 3954–3964. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Fang, J. Implication of heme oxygenase-1 in the sensitivity of nasopharyngeal carcinomas to radiotherapy. J. Exp. Clin. Cancer Res. 2008, 27, 13. [Google Scholar] [CrossRef] [PubMed]
- Herndon, T.M.; Deisseroth, A.; Kaminskas, E.; Kane, R.C.; Koti, K.M.; Rothmann, M.D.; Habtemariam, B.; Bullock, J.; Bray, J.D.; Hawes, J.; et al. Food and drug administration approval: Carfilzomib for the treatment of multiple myeloma. Clin. Cancer Res. 2013, 19, 4559–4563. [Google Scholar] [CrossRef] [PubMed]
- Shirley, M. Ixazomib: First global approval. Drugs 2016, 76, 405–411. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Viruses | Oncogenic Proteins/Molecules Involved | Proteins Processed by UPS | Mechanisms | Cell Function Affected | References |
|---|---|---|---|---|---|
| EBV | BDLF3 | MHC I & II | Postulated E3 ligase for degradation of MHC molecules is not identified yet | Immune evasion | [18] |
| EBNA-1 | EBNA-1 | Inhibits proteasomal processing of EBNA-1 antigenic peptides | Immune evasion | [16] | |
| p53 | Interacts with USP7, leading to ubiquitination and proteasomal degradation | Apoptosis inhibition | [33] | ||
| EBNA-3C | pRb | Stabilizes cyclin D1/CDK6 and recruits SCFSkp2 E3-ubiquitin ligase to facilitate the proteasomal degradation of pRb | Cell cycle deregulation (Bypass sub-G1 arrest) | [22,23] | |
| p21WAF1 | Physically interacts with Pim-1 which in turn phosphorylates p21 and enhances poly-ubiquitination of p21 for degradation | Cell cycle deregulation (Bypass G1 arrest) Apoptosis inhibition | [24] | ||
| p27KIP1 | Enhances the phosphorylation and proteasomal degradation of p27KIP1 through SCFSkp2 E3-ubiquitin ligase | Cell cycle deregulation (Bypass G1 & G2/M arrest) | [25] | ||
| Bcl-6 | Interacts with Bcl-6 and promotes its ubiquitination and proteasomal degradation | Cell cycle deregulation (Bypass G1 arrest) Apoptosis inhibition (release of Bcl-2) | [26] | ||
| p53 | Recruits and stabilizes MDM2 E3 ligase for proteasomal degradation of p53 | Apoptosis inhibition | [34,66,67] | ||
| LMP-1 | p100 | Induces proteolysis of p100 to p52 through proteasome and activates non-canonical NF-κB pathway | Apoptosis inhibition | [32] | |
| KSHV | K/V cyclin | p27KIP1 | Interacts with CDK6 and phosphorylates p27KIP1 for proteasomal degradation | Cell cycle deregulation (Bypass G1 & G2/M arrest) | [29,30] |
| LANA (CR1 repeat) | N/A | Inhibits proteasomal processing of LANA antigenic peptides | Immune evasion | [17] | |
| RTA | IRF3 & IRF7 | Promotes proteasomal degradation of IRF3 & IRF7 directly or through stabilization of RAUL | Immune evasion | [21] | |
| vIRF4 | p53 | Inhibits phosphorylation of p53 by ATM and interacts with MDM2 to facilitate proteasomal degradation of p53 | Apoptosis inhibition | [35,36] |
| Proteasome Inhibitor | Type | Viral Protein Affected | Lytic Reactivation | Clinical Development | Structure |
|---|---|---|---|---|---|
| Bortezomib | Boronate | EBNA-3C (combination with SAHA or romidepsin) | EBV KSHV HSV-1 | FDA-approved for MM, MCL and RRMM [39] |  |
| Carfilzomib | Epoxyketone | N.D. | EBV | FDA-approved for RRMM [117] |  |
| Ixazomib | Boronate | N.D. | N.D. | FDA-approved for RRMM Phase I clinical trials in AML, follicular lymphoma and peripheral T-cell lymphoma [118] |  |
| Marizomib | β-lactone | N.D. | N.D. | Phase I clinical trials in RRMM, solid tumors and lymphoma |  |
| CEP-18770 | Boronate | N.D. | N.D. | Phase I–II clinical trials in RRMM |  |
| ONX-0912 | Epoxyketone | N.D. | N.D. | Phase I clinical trials in haematological solid malignancies |  |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hui, K.F.; Tam, K.P.; Chiang, A.K.S. Therapeutic Strategies against Epstein-Barr Virus-Associated Cancers Using Proteasome Inhibitors. Viruses 2017, 9, 352. https://doi.org/10.3390/v9110352
Hui KF, Tam KP, Chiang AKS. Therapeutic Strategies against Epstein-Barr Virus-Associated Cancers Using Proteasome Inhibitors. Viruses. 2017; 9(11):352. https://doi.org/10.3390/v9110352
Chicago/Turabian StyleHui, Kwai Fung, Kam Pui Tam, and Alan Kwok Shing Chiang. 2017. "Therapeutic Strategies against Epstein-Barr Virus-Associated Cancers Using Proteasome Inhibitors" Viruses 9, no. 11: 352. https://doi.org/10.3390/v9110352
APA StyleHui, K. F., Tam, K. P., & Chiang, A. K. S. (2017). Therapeutic Strategies against Epstein-Barr Virus-Associated Cancers Using Proteasome Inhibitors. Viruses, 9(11), 352. https://doi.org/10.3390/v9110352