
Integrating Sequence- and Structure-Based Similarity Metrics for the Demarcation of Multiple Viral Taxonomic Levels
 ,
,  ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Data Sources
2.1.1. RNA Virus Families of the Orthornavirae Kingdom (Riboviria Realm—RNA Viruses)
2.1.2. Microviridae Family (Monodnaviria Realm—ssDNA Viruses, Sangervirae Kingdom)
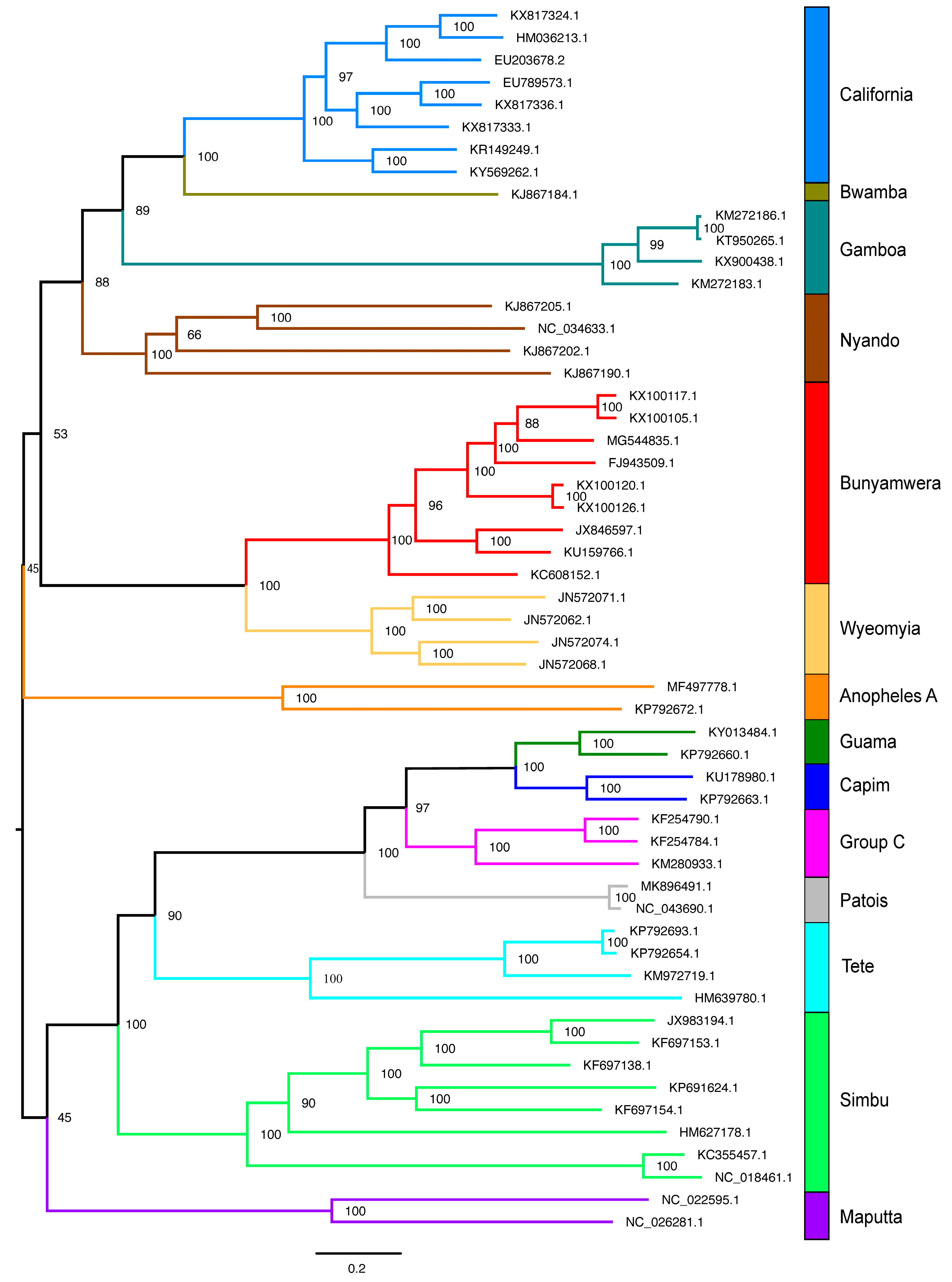
2.1.3. Orthobunyavirus Genus (Negaraviricota Kingdom—Negative-Strand ssRNA Viruses, Riboviria Realm)
2.2. Multiple Sequence Alignment and Phylogenetic Analysis
2.3. Three-Dimensional Protein Structure Prediction
2.4. Implementation of MPACT Program
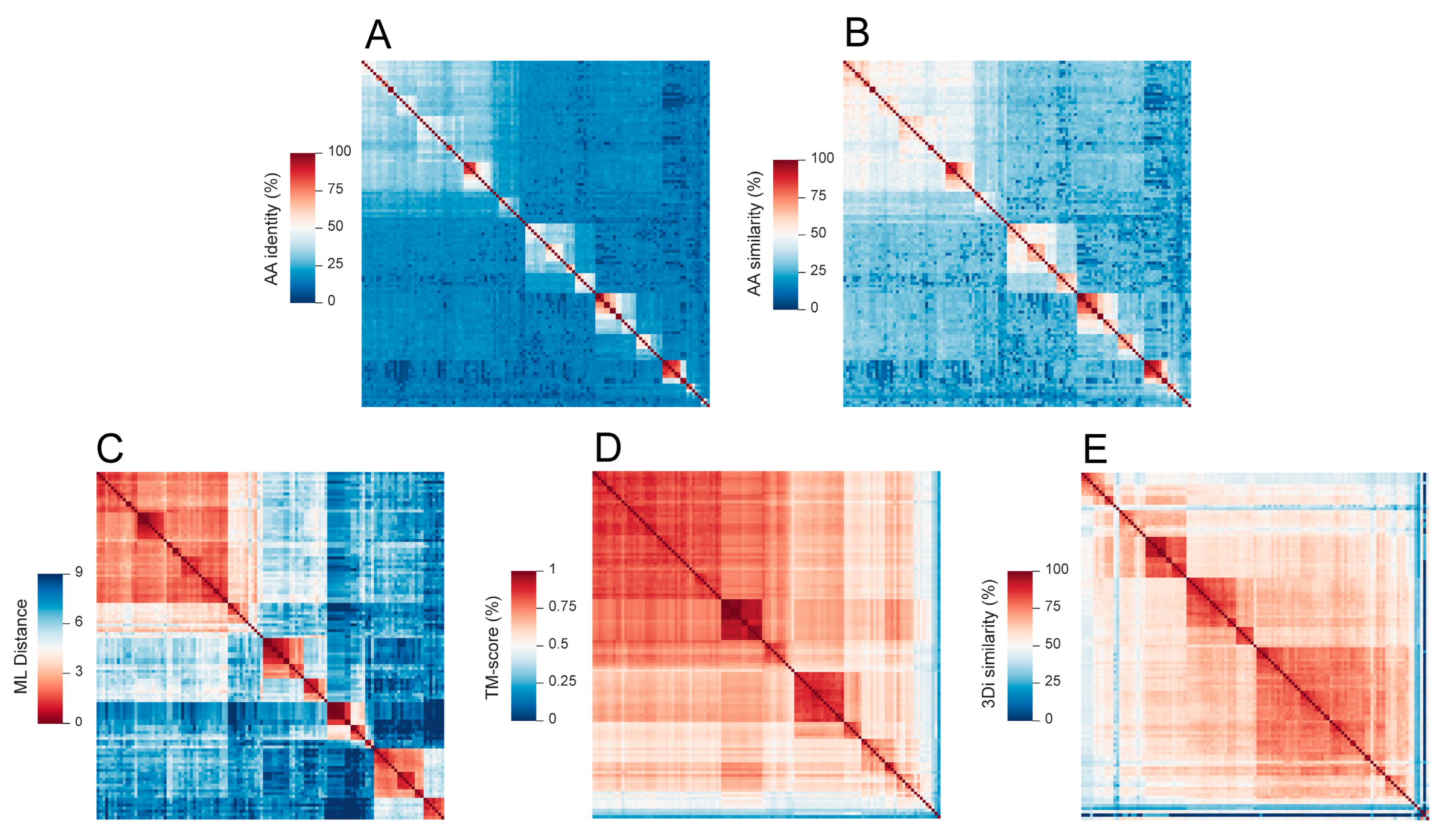
3. Results
3.1. Workflow of the Program
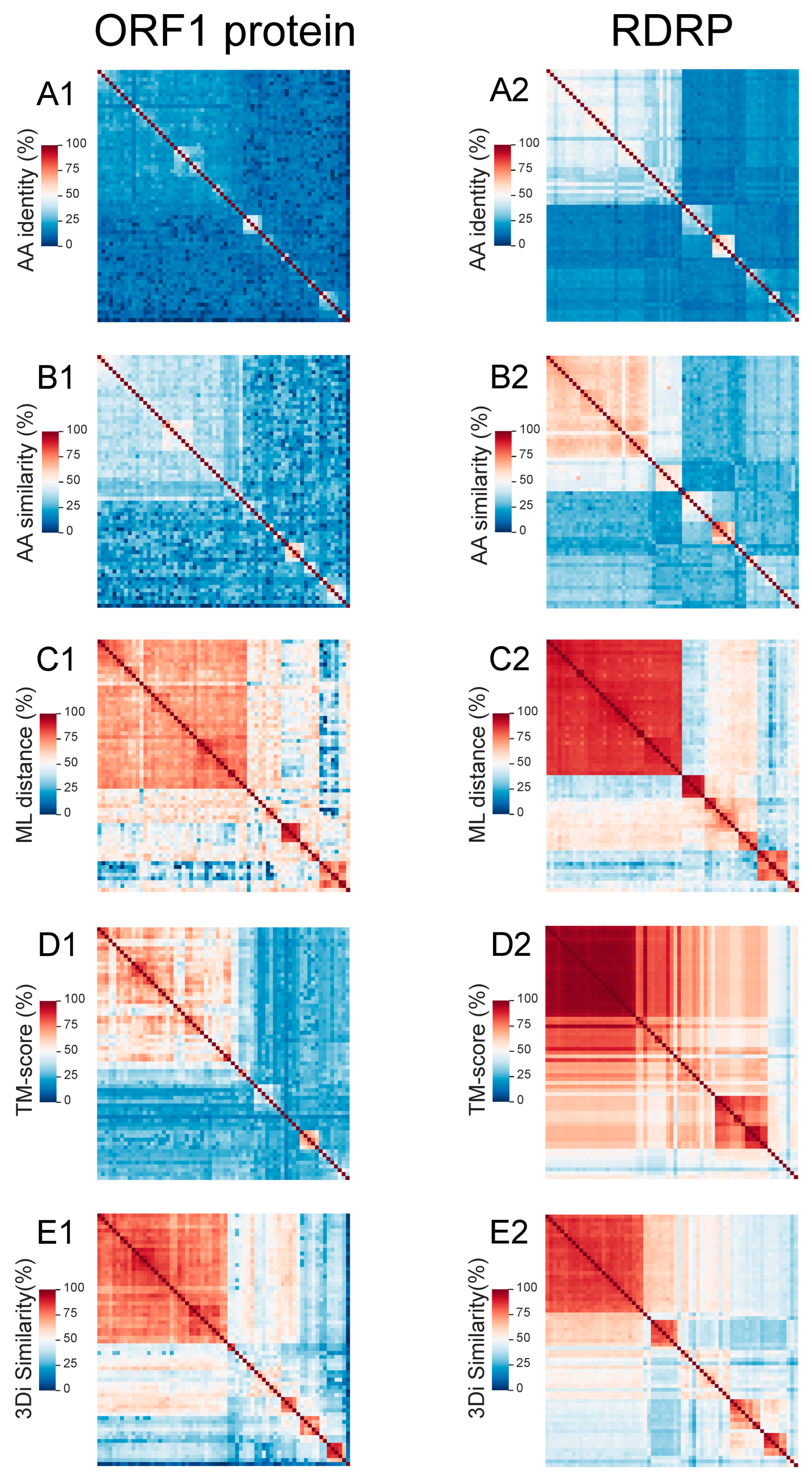
3.2. Using MPACT on Viral Protein Sequence and 3D Structure Datasets
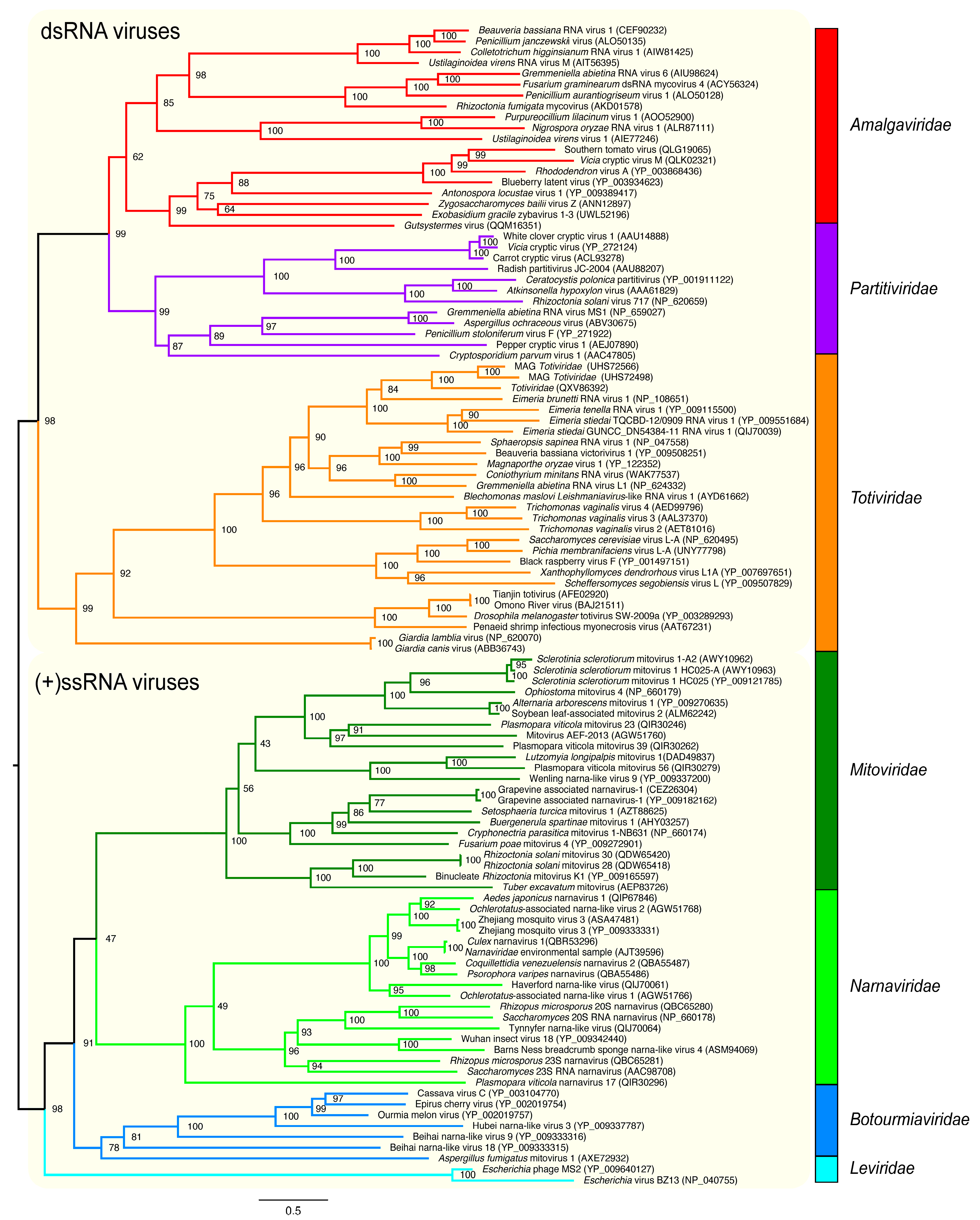
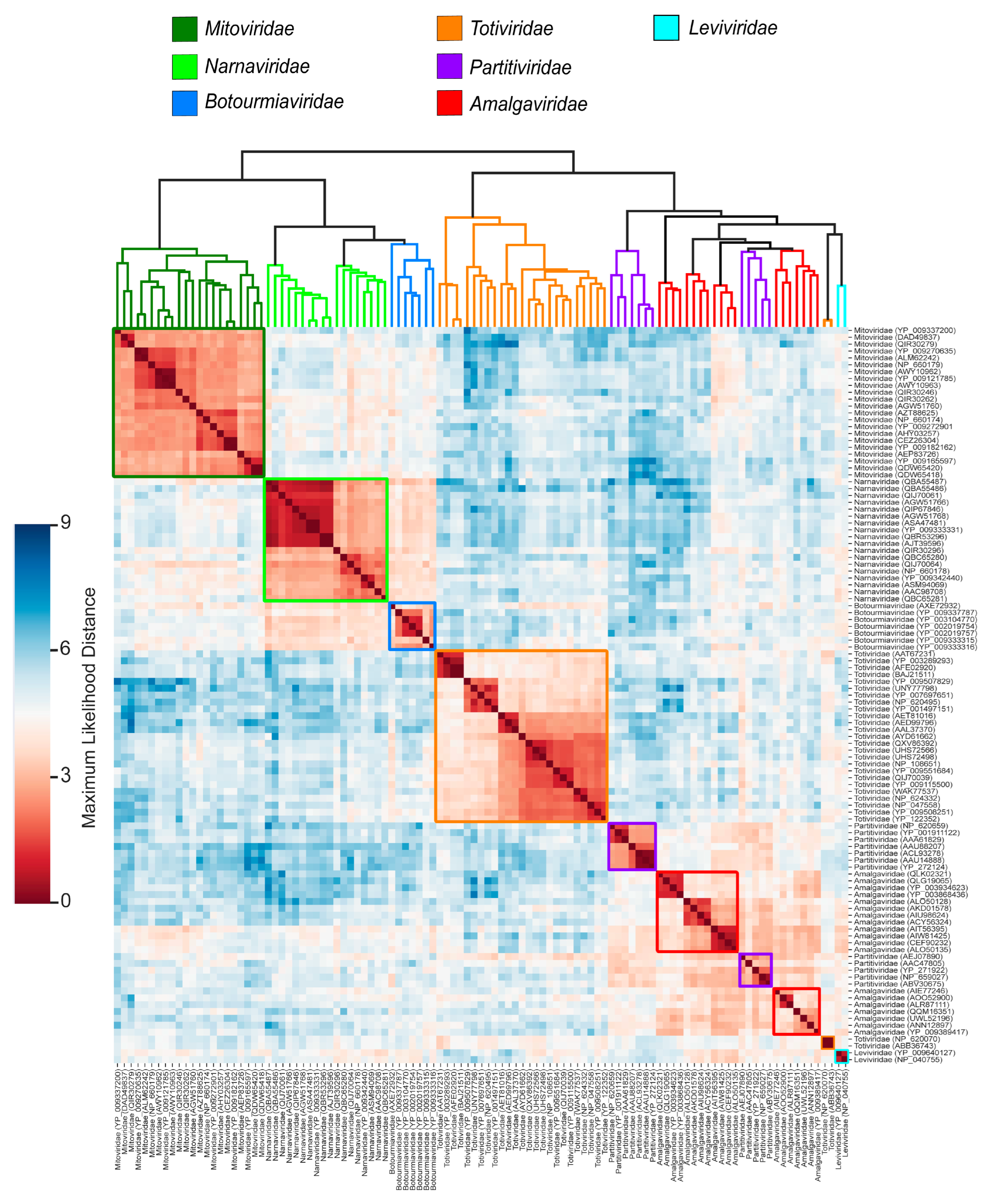
3.2.1. Application on RNA Viral Families of the Orthornavirae Kingdom
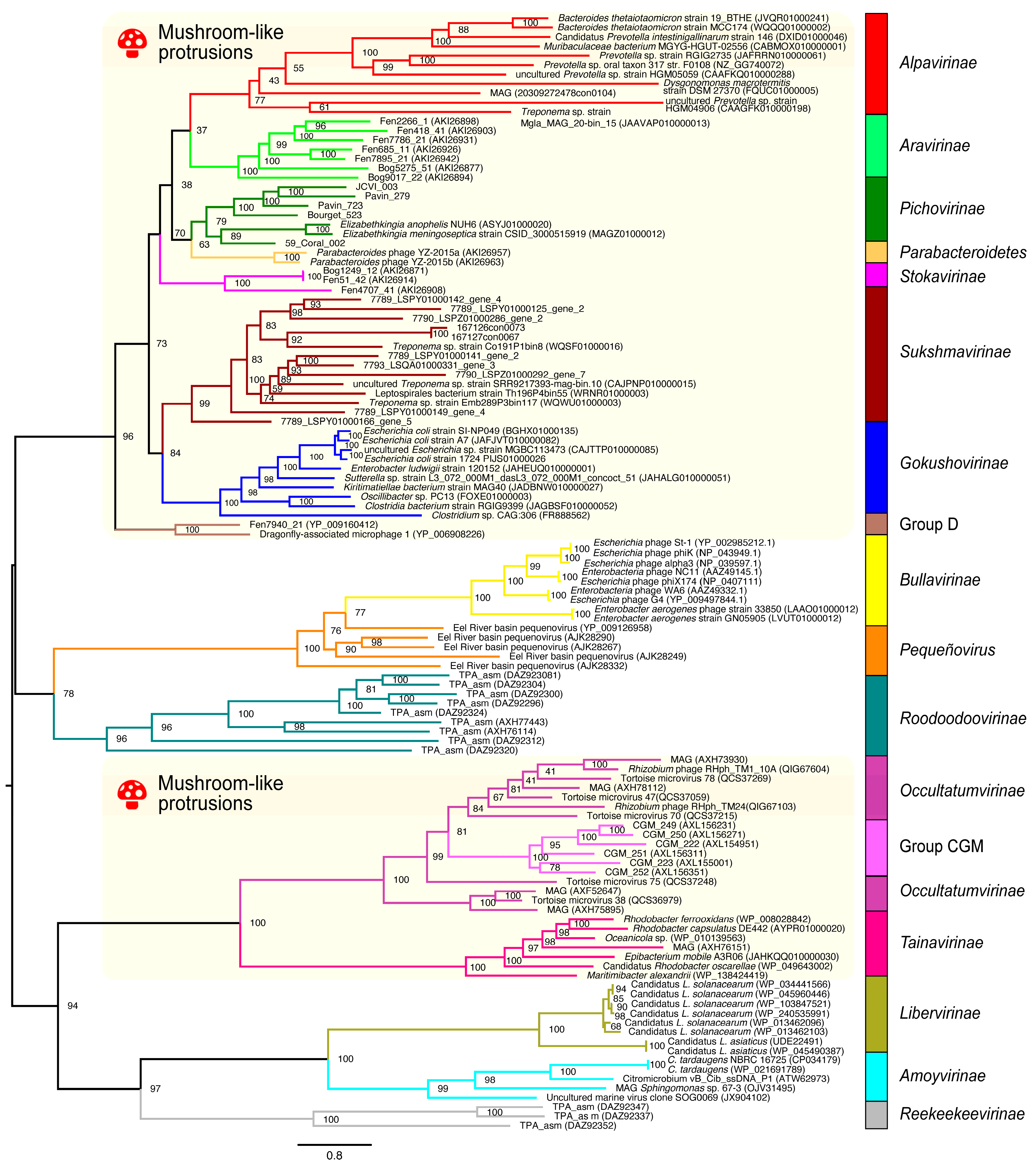
3.2.2. Application on Bacteriophages of the Microviridae Family
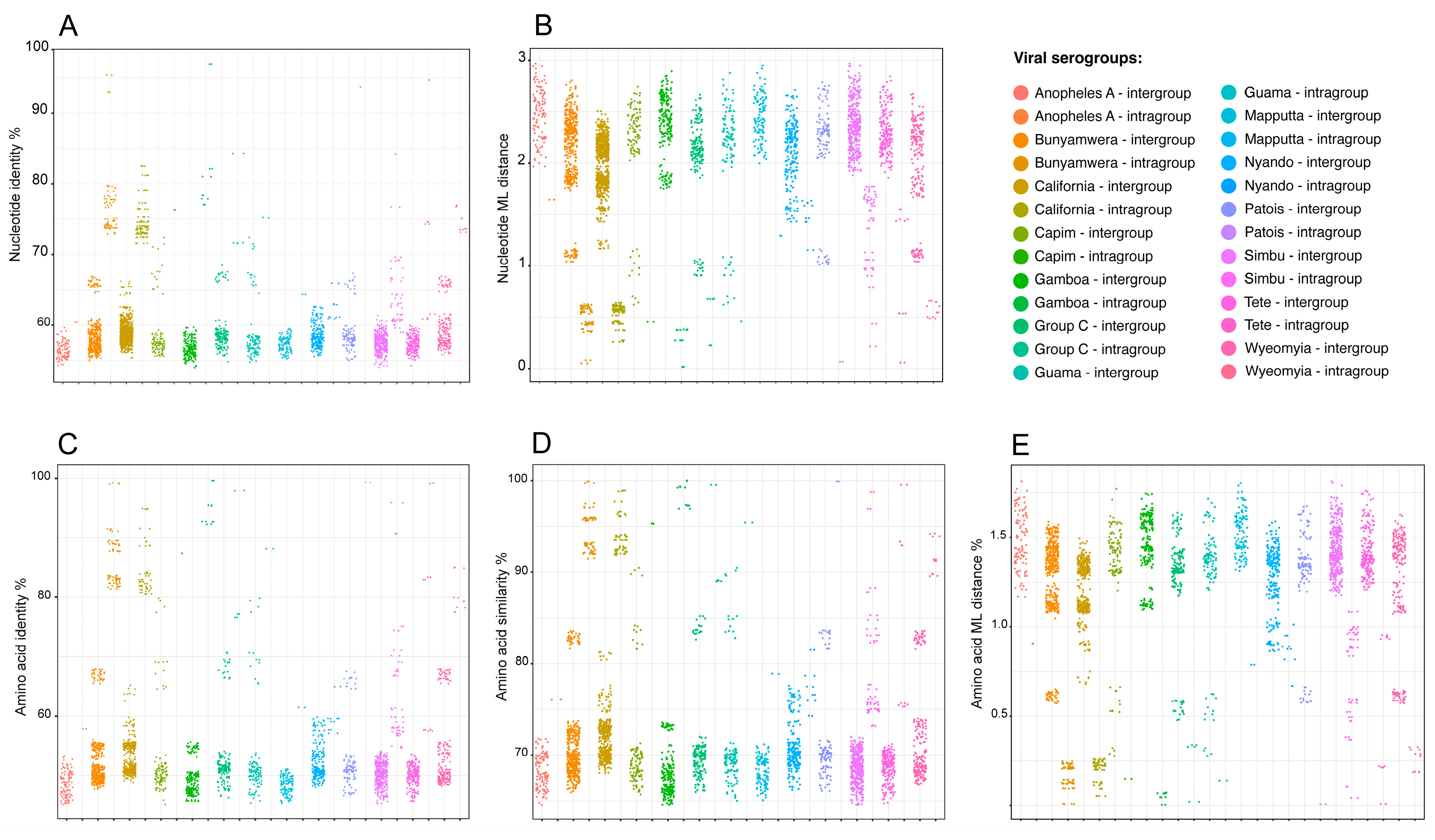
3.3. Viral Group Demarcation
3.4. Data Partitioning
3.5. Comparison of MPACT and Other Similar Tools: SDT and Dali
4. Discussion
4.1. Using MPACT for Virus Comparison and Taxa Demarcation
4.2. Phenotypic and Genotypic Features for Viral Classification
4.3. Single Metrics Versus Molecular Phylogeny
4.4. Protein Structure Information and MSAs
4.5. For Viral Detection and Classification Go Shorter
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 3D | Three-dimensional |
| ICTV | International Committee on Taxonomy of Viruses |
| ML | maximum likelihood |
| NJ | Neighbor-joining |
| ORF | open reading frame |
| pLDDT | predicted local distance difference test |
| MPACT | Multimetric Pairwise Comparison Tool |
| MSA | multiple sequence alignment |
| RDRP | RNA-dependent RNA polymerase |
| NCBI | National Center of Biotechnology Information |
| UPGMA | Unweighted Pair-Group Method using Arithmetic Averages |
| VP1 | major capsid protein |
References
- Simmonds, P.; Aiewsakun, P. Virus Classification—Where Do You Draw the Line? Arch. Virol. 2018, 163, 2037–2046. [Google Scholar] [CrossRef] [PubMed]
- Espino-Vázquez, A.N.; Bermúdez-Barrientos, J.R.; Cabrera-Rangel, J.F.; Córdova-López, G.; Cardoso-Martínez, F.; Martínez-Vázquez, A.; Camarena-Pozos, D.A.; Mondo, S.J.; Pawlowska, T.E.; Abreu-Goodger, C.; et al. Narnaviruses: Novel Players in Fungal–Bacterial Symbioses. ISME J. 2020, 14, 1743–1754. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, P.; Ferreira, F.; da Silva, F.; Oliveira, L.S.; Marques, J.T.; Goes-Neto, A.; Aguiar, E.; Gruber, A. Characterization of a Novel Mitovirus of the Sand Fly Lutzomyia Longipalpis Using Genomic and Virus–Host Interaction Signatures. Viruses 2020, 13, 9. [Google Scholar] [CrossRef]
- Krupovic, M.; Dolja, V.V.; Koonin, E.V. The LUCA and Its Complex Virome. Nat. Rev. Microbiol. 2020, 18, 661–670. [Google Scholar] [CrossRef]
- Forterre, P. The Origin of Viruses and Their Possible Roles in Major Evolutionary Transitions. Virus Res. 2006, 117, 5–16. [Google Scholar] [CrossRef]
- Oliveira, L.S.; Gruber, A. Rational Design of Profile HMMs for Viral Classification and Discovery. In Bioinformatics; Nakaya, H., Ed.; Exon Publications: Brisbane, Australia, 2021; pp. 151–170. ISBN 978-0-645-00171-6. [Google Scholar]
- Reyes, A.; Alves, J.M.P.; Durham, A.M.; Gruber, A. Use of Profile Hidden Markov Models in Viral Discovery: Current Insights. Adv. Genom. Genet. 2017, 7, 29–45. [Google Scholar] [CrossRef]
- Baltimore, D. Expression of Animal Virus Genomes. Bacteriol. Rev. 1971, 35, 235–241. [Google Scholar] [CrossRef]
- Koonin, E.V.; Krupovic, M.; Agol, V.I. The Baltimore Classification of Viruses 50 Years Later: How Does It Stand in the Light of Virus Evolution? Microbiol. Mol. Biol. Rev. 2021, 85, e00053-21. [Google Scholar] [CrossRef]
- Siddell, S.G.; Walker, P.J.; Lefkowitz, E.J.; Mushegian, A.R.; Dutilh, B.E.; Harrach, B.; Harrison, R.L.; Junglen, S.; Knowles, N.J.; Kropinski, A.M.; et al. Binomial Nomenclature for Virus Species: A Consultation. Arch. Virol. 2020, 165, 519–525. [Google Scholar] [CrossRef]
- Zerbini, F.M.; Siddell, S.G.; Mushegian, A.R.; Walker, P.J.; Lefkowitz, E.J.; Adriaenssens, E.M.; Alfenas-Zerbini, P.; Dutilh, B.E.; García, M.L.; Junglen, S.; et al. Differentiating between Viruses and Virus Species by Writing Their Names Correctly. Arch. Virol. 2022, 167, 1231–1234. [Google Scholar] [CrossRef]
- International Committee on Taxonomy of Viruses Executive Committee; Gorbalenya, A.E.; Krupovic, M.; Mushegian, A.; Kropinski, A.M.; Siddell, S.G.; Varsani, A.; Adams, M.J.; Davison, A.J.; Dutilh, B.E.; et al. The New Scope of Virus Taxonomy: Partitioning the Virosphere into 15 Hierarchical Ranks. Nat. Microbiol. 2020, 5, 668–674. [Google Scholar] [CrossRef] [PubMed]
- Simmonds, P.; Adriaenssens, E.M.; Zerbini, F.M.; Abrescia, N.G.A.; Aiewsakun, P.; Alfenas-Zerbini, P.; Bao, Y.; Barylski, J.; Drosten, C.; Duffy, S.; et al. Four Principles to Establish a Universal Virus Taxonomy. PLoS Biol. 2023, 21, e3001922. [Google Scholar] [CrossRef] [PubMed]
- Gorbalenya, A.E.; Lauber, C. Bioinformatics of Virus Taxonomy: Foundations and Tools for Developing Sequence-Based Hierarchical Classification. Curr. Opin. Virol. 2022, 52, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Evseev, P.; Gutnik, D.; Shneider, M.; Miroshnikov, K. Use of an Integrated Approach Involving AlphaFold Predictions for the Evolutionary Taxonomy of Duplodnaviria Viruses. Biomolecules 2023, 13, 110. [Google Scholar] [CrossRef]
- Aiewsakun, P.; Simmonds, P. The Genomic Underpinnings of Eukaryotic Virus Taxonomy: Creating a Sequence-Based Framework for Family-Level Virus Classification. Microbiome 2018, 6, 38. [Google Scholar] [CrossRef]
- Aiewsakun, P.; Adriaenssens, E.M.; Lavigne, R.; Kropinski, A.M.; Simmonds, P. Evaluation of the Genomic Diversity of Viruses Infecting Bacteria, Archaea and Eukaryotes Using a Common Bioinformatic Platform: Steps towards a Unified Taxonomy. J. Gen. Virol. 2018, 99, 1331–1343. [Google Scholar] [CrossRef]
- Bin Jang, H.; Bolduc, B.; Zablocki, O.; Kuhn, J.H.; Roux, S.; Adriaenssens, E.M.; Brister, J.R.; Kropinski, A.M.; Krupovic, M.; Lavigne, R.; et al. Taxonomic Assignment of Uncultivated Prokaryotic Virus Genomes Is Enabled by Gene-Sharing Networks. Nat. Biotechnol. 2019, 37, 632–639. [Google Scholar] [CrossRef]
- Meier-Kolthoff, J.P.; Göker, M. VICTOR: Genome-Based Phylogeny and Classification of Prokaryotic Viruses. Bioinformatics 2017, 33, 3396–3404. [Google Scholar] [CrossRef]
- Bao, Y.; Chetvernin, V.; Tatusova, T. PAirwise Sequence Comparison (PASC) and Its Application in the Classification of Filoviruses. Viruses 2012, 4, 1318–1327. [Google Scholar] [CrossRef]
- Lauber, C.; Gorbalenya, A.E. Partitioning the Genetic Diversity of a Virus Family: Approach and Evaluation through a Case Study of Picornaviruses. J. Virol. 2012, 86, 3890–3904. [Google Scholar] [CrossRef]
- Muhire, B.M.; Varsani, A.; Martin, D.P. SDT: A Virus Classification Tool Based on Pairwise Sequence Alignment and Identity Calculation. PLoS ONE 2014, 9, e108277. [Google Scholar] [CrossRef] [PubMed]
- Brenner, S.E.; Chothia, C.; Hubbard, T.J.P. Assessing Sequence Comparison Methods with Reliable Structurally Identified Distant Evolutionary Relationships. Proc. Natl. Acad. Sci. USA 1998, 95, 6073–6078. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Karplus, K.; Barrett, C.; Hughey, R.; Haussler, D.; Hubbard, T.; Chothia, C. Sequence Comparisons Using Multiple Sequences Detect Three Times as Many Remote Homologues as Pairwise Methods. J. Mol. Biol. 1998, 284, 1201–1210. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, L.S.; Reyes, A.; Dutilh, B.E.; Gruber, A. Rational Design of Profile HMMs for Sensitive and Specific Sequence Detection with Case Studies Applied to Viruses, Bacteriophages, and Casposons. Viruses 2023, 15, 519. [Google Scholar] [CrossRef]
- Caetano-Anollés, G.; Nasir, A. Benefits of Using Molecular Structure and Abundance in Phylogenomic Analysis. Front. Genet. 2012, 3, 172. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Varadi, M.; Velankar, S. The Impact of AlphaFold Protein Structure Database on the Fields of Life Sciences. Proteomics 2023, 23, 2200128. [Google Scholar] [CrossRef]
- Lin, Z.; Akin, H.; Rao, R.; Hie, B.; Zhu, Z.; Lu, W.; Smetanin, N.; Verkuil, R.; Kabeli, O.; Shmueli, Y.; et al. Evolutionary-Scale Prediction of Atomic-Level Protein Structure with a Language Model. Science 2023, 379, 1123–1130. [Google Scholar] [CrossRef]
- Holm, L. Dali Server: Structural Unification of Protein Families. Nucleic Acids Res. 2022, 50, W210–W215. [Google Scholar] [CrossRef]
- Zhang, Y.; Skolnick, J. TM-Align: A Protein Structure Alignment Algorithm Based on the TM-Score. Nucleic Acids Res. 2005, 33, 2302–2309. [Google Scholar] [CrossRef]
- Van Kempen, M.; Kim, S.S.; Tumescheit, C.; Mirdita, M.; Lee, J.; Gilchrist, C.L.M.; Söding, J.; Steinegger, M. Fast and Accurate Protein Structure Search with Foldseek. Nat. Biotechnol. 2023, 42, 243–246. [Google Scholar] [CrossRef] [PubMed]
- Steinegger, M.; Söding, J. MMseqs2 Enables Sensitive Protein Sequence Searching for the Analysis of Massive Data Sets. Nat. Biotechnol. 2017, 35, 1026–1028. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, J.H.; Abe, J.; Adkins, S.; Alkhovsky, S.V.; Avšič-Županc, T.; Ayllón, M.A.; Bahl, J.; Balkema-Buschmann, A.; Ballinger, M.J.; Kumar Baranwal, V.; et al. Annual (2023) Taxonomic Update of RNA-Directed RNA Polymerase-Encoding Negative-Sense RNA Viruses (Realm Riboviria: Kingdom Orthornavirae: Phylum Negarnaviricota). J. Gen. Virol. 2023, 104, 001864. [Google Scholar] [CrossRef] [PubMed]
- Martin, R.R.; Zhou, J.; Tzanetakis, I.E. Blueberry Latent Virus: An Amalgam of the Partitiviridae and Totiviridae. Virus Res. 2011, 155, 175–180. [Google Scholar] [CrossRef]
- Vainio, E.J.; Chiba, S.; Ghabrial, S.A.; Maiss, E.; Roossinck, M.; Sabanadzovic, S.; Suzuki, N.; Xie, J.; Nibert, M.; ICTV Report Consortium. ICTV Virus Taxonomy Profile: Partitiviridae. J. Gen. Virol. 2018, 99, 17–18. [Google Scholar] [CrossRef]
- Nibert, M.L.; Pyle, J.D.; Firth, A.E. A +1 Ribosomal Frameshifting Motif Prevalent among Plant Amalgaviruses. Virology 2016, 498, 201–208. [Google Scholar] [CrossRef]
- Depierreux, D.; Vong, M.; Nibert, M.L. Nucleotide Sequence of Zygosaccharomyces Bailii Virus Z: Evidence for +1 Programmed Ribosomal Frameshifting and for Assignment to Family Amalgaviridae. Virus Res. 2016, 217, 115–124. [Google Scholar] [CrossRef]
- Roux, S.; Krupovic, M.; Poulet, A.; Debroas, D.; Enault, F. Evolution and Diversity of the Microviridae Viral Family through a Collection of 81 New Complete Genomes Assembled from Virome Reads. PLoS ONE 2012, 7, e40418. [Google Scholar] [CrossRef]
- Brentlinger, K.L.; Hafenstein, S.; Novak, C.R.; Fane, B.A.; Borgon, R.; McKenna, R.; Agbandje-McKenna, M. Microviridae, a Family Divided: Isolation, Characterization, and Genome Sequence of phiMH2K, a Bacteriophage of the Obligate Intracellular Parasitic Bacterium Bdellovibrio bacteriovorus. J. Bacteriol. 2002, 184, 1089–1094. [Google Scholar] [CrossRef]
- Carstens, E.B. Ratification Vote on Taxonomic Proposals to the International Committee on Taxonomy of Viruses (2009). Arch. Virol. 2010, 155, 133–146. [Google Scholar] [CrossRef]
- Tikhe, C.V.; Husseneder, C. Metavirome Sequencing of the Termite Gut Reveals the Presence of an Unexplored Bacteriophage Community. Front. Microbiol. 2018, 8, 2548. [Google Scholar] [CrossRef] [PubMed]
- Rosario, K.; Dayaram, A.; Marinov, M.; Ware, J.; Kraberger, S.; Stainton, D.; Breitbart, M.; Varsani, A. Diverse Circular ssDNA Viruses Discovered in Dragonflies (Odonata: Epiprocta). J. Gen. Virol. 2012, 93, 2668–2681. [Google Scholar] [CrossRef] [PubMed]
- Quaiser, A.; Dufresne, A.; Ballaud, F.; Roux, S.; Zivanovic, Y.; Colombet, J.; Sime-Ngando, T.; Francez, A.-J. Diversity and Comparative Genomics of Microviridae in Sphagnum—Dominated Peatlands. Front. Microbiol. 2015, 6, 375. [Google Scholar] [CrossRef]
- Zhang, L.; Li, Z.; Bao, M.; Li, T.; Fang, F.; Zheng, Y.; Liu, Y.; Xu, M.; Chen, J.; Deng, X.; et al. A Novel Microviridae Phage (CLasMV1) From “Candidatus Liberibacter Asiaticus”. Front. Microbiol. 2021, 12, 754245. [Google Scholar] [CrossRef]
- Zheng, Q.; Chen, Q.; Xu, Y.; Suttle, C.A.; Jiao, N. A Virus Infecting Marine Photoheterotrophic Alphaproteobacteria (Citromicrobium Spp.) Defines a New Lineage of ssDNA Viruses. Front. Microbiol. 2018, 9, 1418. [Google Scholar] [CrossRef]
- Krupovic, M.; Dutilh, B.E.; Adriaenssens, E.M.; Wittmann, J.; Vogensen, F.K.; Sullivan, M.B.; Rumnieks, J.; Prangishvili, D.; Lavigne, R.; Kropinski, A.M.; et al. Taxonomy of Prokaryotic Viruses: Update from the ICTV Bacterial and Archaeal Viruses Subcommittee. Arch. Virol. 2016, 161, 1095–1099. [Google Scholar] [CrossRef]
- Bryson, S.J.; Thurber, A.R.; Correa, A.M.S.; Orphan, V.J.; Vega Thurber, R. A Novel Sister Clade to the Enterobacteria Microviruses (Family Microviridae) Identified in Methane Seep Sediments: DNA Phages Associated with Methane Seeps. Environ. Microbiol. 2015, 17, 3708–3721. [Google Scholar] [CrossRef]
- Creasy, A.; Rosario, K.; Leigh, B.; Dishaw, L.; Breitbart, M. Unprecedented Diversity of ssDNA Phages from the Family Microviridae Detected within the Gut of a Protochordate Model Organism (Ciona robusta). Viruses 2018, 10, 404. [Google Scholar] [CrossRef]
- Zucker, F.; Bischoff, V.; Olo Ndela, E.; Heyerhoff, B.; Poehlein, A.; Freese, H.M.; Roux, S.; Simon, M.; Enault, F.; Moraru, C. New Microviridae Isolated from Sulfitobacter Reveals Two Cosmopolitan Subfamilies of Single-Stranded DNA Phages Infecting Marine and Terrestrial Alphaproteobacteria. Virus Evol. 2022, 8, veac070. [Google Scholar] [CrossRef]
- Olo Ndela, E.; Roux, S.; Henke, C.; Sczyrba, A.; Sime Ngando, T.; Varsani, A.; Enault, F. Reekeekee- and Roodoodooviruses, Two Different Microviridae Clades Constituted by the Smallest DNA Phages. Virus Evol. 2023, 9, veac123. [Google Scholar] [CrossRef]
- De Souza, W.M.; Calisher, C.H.; Carrera, J.P.; Hughes, H.R.; Nunes, M.R.T.; Russell, B.; Tilson-Lunel, N.L.; Venter, M.; Xia, H. ICTV Virus Taxonomy Profile: Peribunyaviridae 2024: This Article Is Part of the ICTV Virus Taxonomy Profiles Collection. J. Gen. Virol. 2024, 105, 002034. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast Model Selection for Accurate Phylogenetic Estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Hoang, D.T.; Chernomor, O.; von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the Ultrafast Bootstrap Approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef]
- Mirdita, M.; Schütze, K.; Moriwaki, Y.; Heo, L.; Ovchinnikov, S.; Steinegger, M. ColabFold: Making Protein Folding Accessible to All. Nat. Methods 2022, 19, 679–682. [Google Scholar] [CrossRef]
- Mariani, V.; Biasini, M.; Barbato, A.; Schwede, T. lDDT: A Local Superposition-Free Score for Comparing Protein Structures and Models Using Distance Difference Tests. Bioinformatics 2013, 29, 2722–2728. [Google Scholar] [CrossRef]
- Rice, P.; Longden, I.; Bleasby, A. EMBOSS: The European Molecular Biology Open Software Suite. Trends Genet. 2000, 16, 276–277. [Google Scholar] [CrossRef]
- Sabanadzovic, S.; Valverde, R.A.; Brown, J.K.; Martin, R.R.; Tzanetakis, I.E. Southern Tomato Virus: The Link between the Families Totiviridae and Partitiviridae. Virus Res. 2009, 140, 130–137. [Google Scholar] [CrossRef]
- Ghabrial, S.A.; Nibert, M.L. Victorivirus, a New Genus of Fungal Viruses in the Family Totiviridae. Arch. Virol. 2009, 154, 373–379. [Google Scholar] [CrossRef]
- Isogai, M.; Nakamura, T.; Ishii, K.; Watanabe, M.; Yamagishi, N.; Yoshikawa, N. Histochemical Detection of Blueberry Latent Virus in Highbush Blueberry Plant. J. Gen. Plant Pathol. 2011, 77, 304–306. [Google Scholar] [CrossRef]
- Kirchberger, P.C.; Ochman, H. Microviruses: A World Beyond phiX174. Annu. Rev. Virol. 2023, 10, 99–118. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Baxter, A.J.; Bator, C.M.; Fane, B.A.; Hafenstein, S.L. Cryo-EM Structure of Gokushovirus ΦEC6098 Reveals a Novel Capsid Architecture for a Single-Scaffolding Protein, Microvirus Assembly System. J. Virol. 2022, 96, e0099022. [Google Scholar] [CrossRef]
- Gago, S.; Elena, S.F.; Flores, R.; Sanjuan, R. Extremely High Mutation Rate of a Hammerhead Viroid. Science 2009, 323, 1308. [Google Scholar] [CrossRef]
- Sanjuán, R.; Nebot, M.R.; Chirico, N.; Mansky, L.M.; Belshaw, R. Viral Mutation Rates. J. Virol. 2010, 84, 9733–9748. [Google Scholar] [CrossRef]
- Holland, J.; Spindler, K.; Horodyski, F.; Grabau, E.; Nichol, S.; VandePol, S. Rapid Evolution of RNA Genomes. Science 1982, 215, 1577–1585. [Google Scholar] [CrossRef]
- Drake, J.W. Rates of Spontaneous Mutation among RNA Viruses. Proc. Natl. Acad. Sci. USA 1993, 90, 4171–4175. [Google Scholar] [CrossRef]
- Peck, K.M.; Lauring, A.S. Complexities of Viral Mutation Rates. J. Virol. 2018, 92, e01031-17. [Google Scholar] [CrossRef]
- Dias, H.G.; Dos Santos, F.B.; Pauvolid-Corrêa, A. An Overview of Neglected Orthobunyaviruses in Brazil. Viruses 2022, 14, 987. [Google Scholar] [CrossRef]
- Briese, T.; Calisher, C.H.; Higgs, S. Viruses of the Family Bunyaviridae: Are All Available Isolates Reassortants? Virology 2013, 446, 207–216. [Google Scholar] [CrossRef]
- Calisher, C.H. History, Classification, and Taxonomy of Viruses in the Family Bunyaviridae. In The Bunyaviridae; Elliott, R.M., Ed.; Springer: Boston, MA, USA, 1996; pp. 1–17. ISBN 978-1-4899-1366-1. [Google Scholar]
- Caetano-Anollés, G.; Claverie, J.-M.; Nasir, A. A Critical Analysis of the Current State of Virus Taxonomy. Front. Microbiol. 2023, 14, 1240993. [Google Scholar] [CrossRef] [PubMed]
- Caetano-Anollés, G. Are Viruses Taxonomic Units? A Protein Domain and Loop-Centric Phylogenomic Assessment. Viruses 2024, 16, 1061. [Google Scholar] [CrossRef] [PubMed]
- Gilchrist, C.L.M.; Mirdita, M.; Steinegger, M. Multiple Protein Structure Alignment at Scale with FoldMason. bioRxiv 2024. [Google Scholar] [CrossRef]
- Edgar, R.C. Protein Structure Alignment by Reseek Improves Sensitivity to Remote Homologs. Bioinformatics 2024, 40, btae687. [Google Scholar] [CrossRef]
- Edgar, R.C. Muscle5: High-Accuracy Alignment Ensembles Enable Unbiased Assessments of Sequence Homology and Phylogeny. Nat. Commun. 2022, 13, 6968. [Google Scholar] [CrossRef]
- Edgar, R.C.; Tolstoy, I. Muscle-3D: Scalable Multiple Protein Structure Alignment. bioRxiv 2024. [Google Scholar] [CrossRef]
- Alves, J.M.P.; de Oliveira, A.L.; Sandberg, T.O.M.; Moreno-Gallego, J.L.; de Toledo, M.A.F.; de Moura, E.M.M.; Oliveira, L.S.; Durham, A.M.; Mehnert, D.U.; Zanotto, P.M.d.A.; et al. GenSeed-HMM: A Tool for Progressive Assembly Using Profile HMMs as Seeds and Its Application in Alpavirinae Viral Discovery from Metagenomic Data. Front. Microbiol. 2016, 7, 269. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
dos Santos, I.C.; de Souza, R.d.S.; Tolstoy, I.; Oliveira, L.S.; Gruber, A. Integrating Sequence- and Structure-Based Similarity Metrics for the Demarcation of Multiple Viral Taxonomic Levels. Viruses 2025, 17, 642. https://doi.org/10.3390/v17050642
dos Santos IC, de Souza RdS, Tolstoy I, Oliveira LS, Gruber A. Integrating Sequence- and Structure-Based Similarity Metrics for the Demarcation of Multiple Viral Taxonomic Levels. Viruses. 2025; 17(5):642. https://doi.org/10.3390/v17050642
Chicago/Turabian Styledos Santos, Igor C., Rebecca di Stephano de Souza, Igor Tolstoy, Liliane S. Oliveira, and Arthur Gruber. 2025. "Integrating Sequence- and Structure-Based Similarity Metrics for the Demarcation of Multiple Viral Taxonomic Levels" Viruses 17, no. 5: 642. https://doi.org/10.3390/v17050642
APA Styledos Santos, I. C., de Souza, R. d. S., Tolstoy, I., Oliveira, L. S., & Gruber, A. (2025). Integrating Sequence- and Structure-Based Similarity Metrics for the Demarcation of Multiple Viral Taxonomic Levels. Viruses, 17(5), 642. https://doi.org/10.3390/v17050642