

The Lambda Variant in Argentina: Analyzing the Evolution and Spread of SARS-CoV-2 Lineage C.37
 , , , , , , ,
, , , , , , ,  ,
,  , ,
, ,  , ,
, ,  , , add
Show full author list
, , add
Show full author list
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
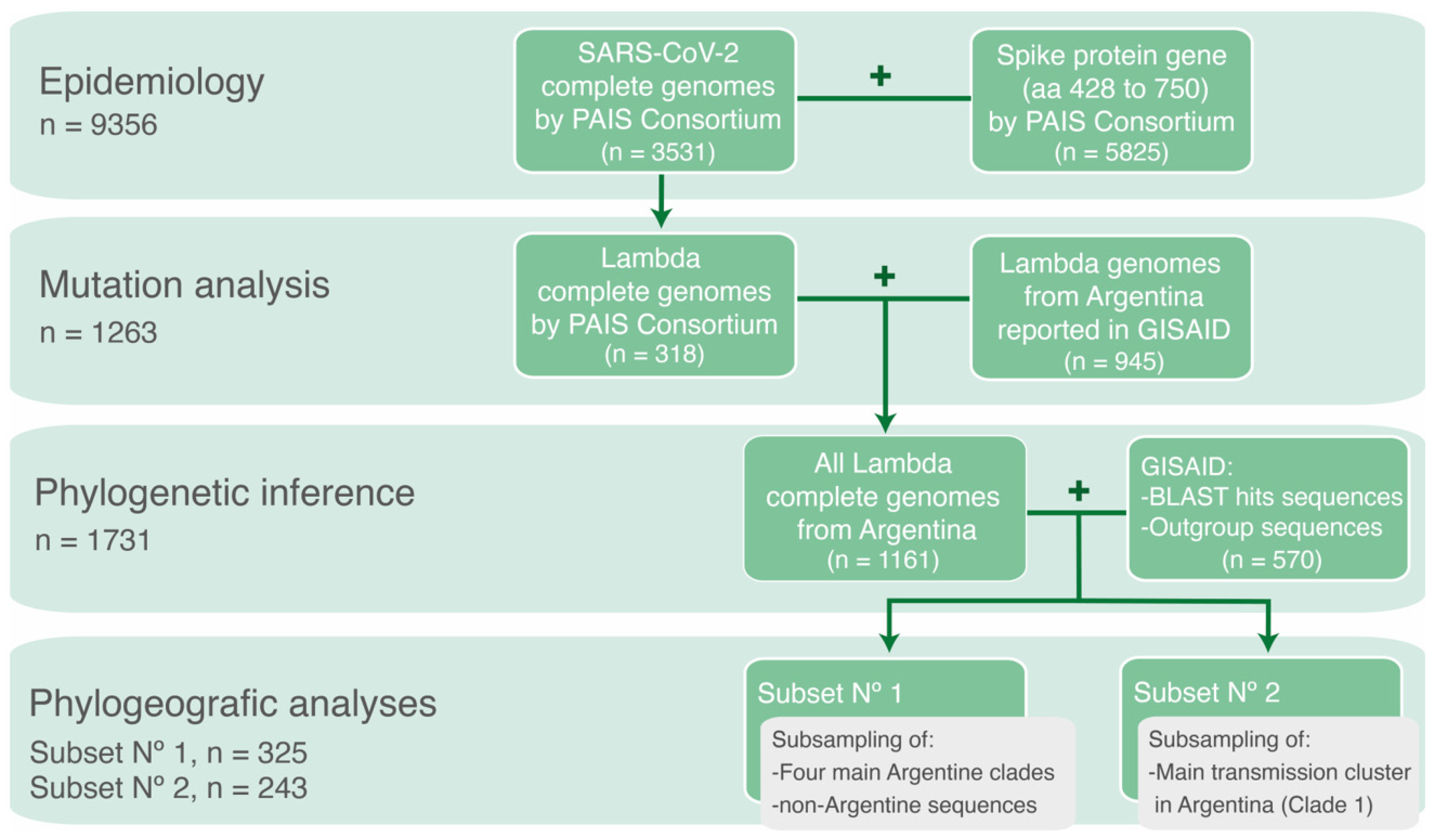
2. Materials and Methods
2.1. SARS-CoV-2 Sample Collection and Sequencing
2.2. SARS-CoV-2 Genomic Datasets
2.3. Phylogenetic Inference
2.4. Phylodynamic Analysis
2.5. Mutation Analysis
2.6. Statistical Analysis
2.7. Data Visualization
3. Results
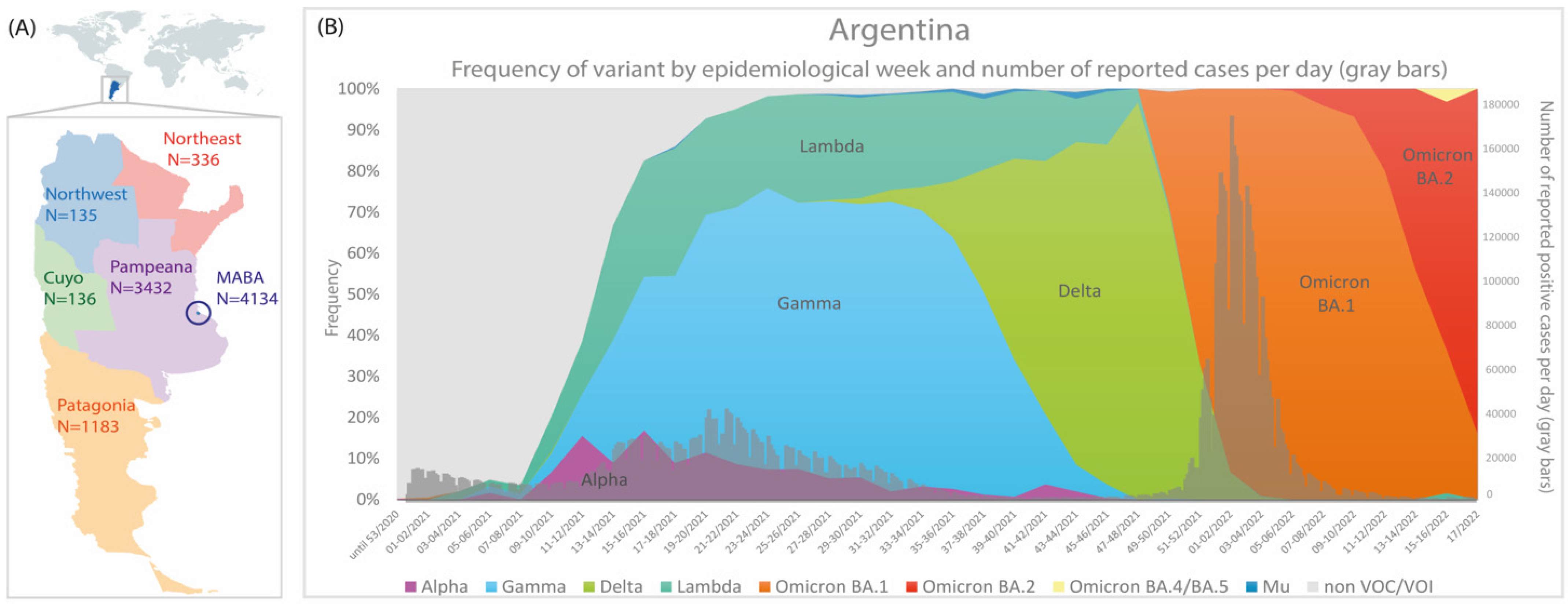
3.1. The Molecular Epidemiology of Lineage C.37 in Argentina
3.2. Phylogenetic Analysis
3.3. Mutational Patterns in Argentine SARS-CoV-2 Whole-Genome Sequences
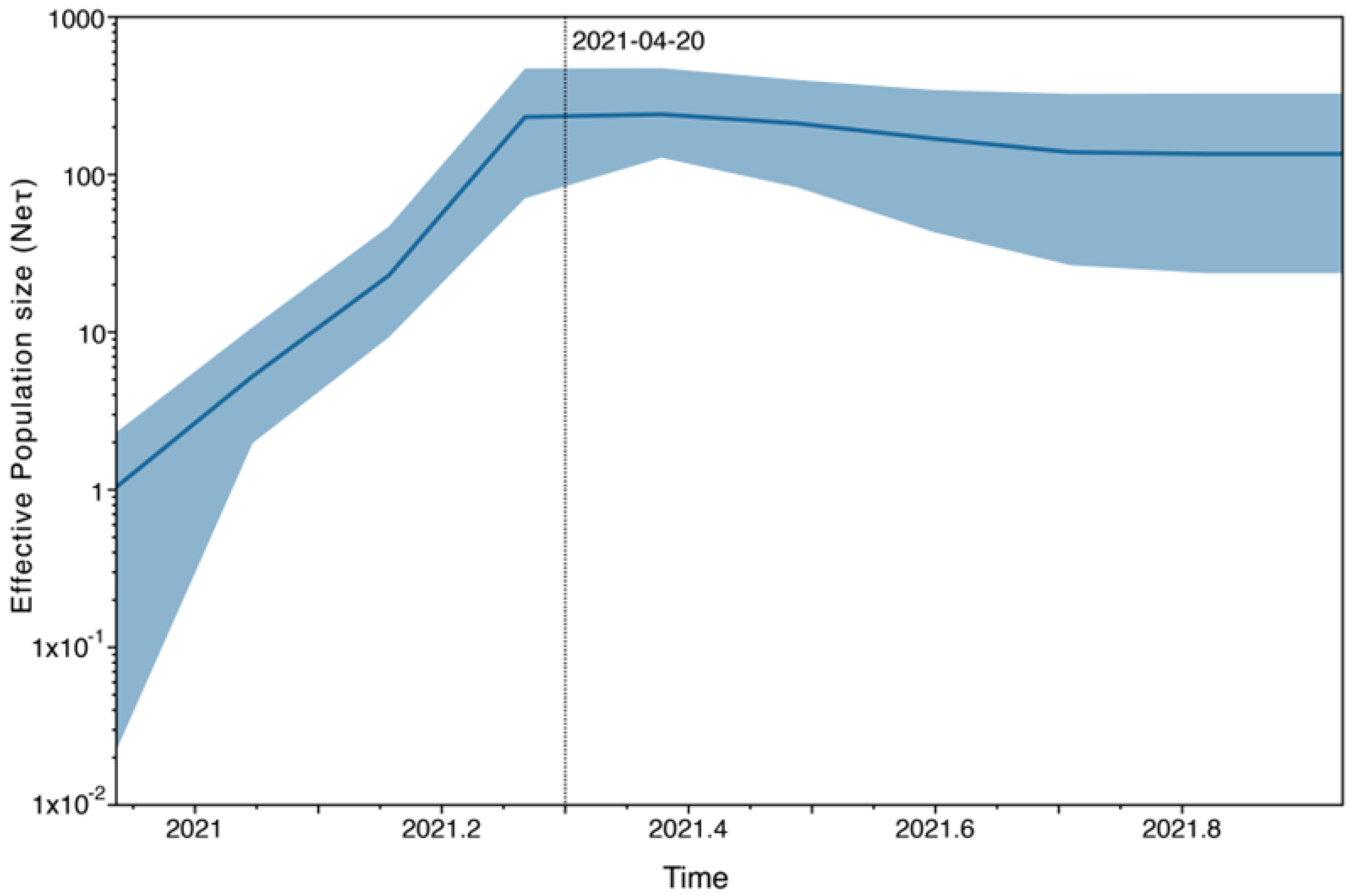
3.4. Phylogeographic Analyses
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Thakur, S.; Sasi, S.; Pillai, S.G.; Nag, A.; Shukla, D.; Singhal, R.; Phalke, S.; Velu, G.S.K. SARS-CoV-2 Mutations and Their Impact on Diagnostics, Therapeutics and Vaccines. Front. Med. 2022, 9, 815389. [Google Scholar] [CrossRef] [PubMed]
- Oude Munnink, B.B.; Worp, N.; Nieuwenhuijse, D.F.; Sikkema, R.S.; Haagmans, B.; Fouchier, R.A.M.; Koopmans, M. The next Phase of SARS-CoV-2 Surveillance: Real-Time Molecular Epidemiology. Nat. Med. 2021, 27, 1518–1524. [Google Scholar] [CrossRef] [PubMed]
- Elbe, S.; Buckland-Merrett, G. Data, Disease and Diplomacy: GISAID’s Innovative Contribution to Global Health: Data, Disease and Diplomacy. Global Chall. 2017, 1, 33–46. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Azman, A.S.; Chen, X.; Zou, J.; Tian, Y.; Sun, R.; Xu, X.; Wu, Y.; Lu, W.; Ge, S.; et al. Global Landscape of SARS-CoV-2 Genomic Surveillance and Data Sharing. Nat. Genet. 2022, 54, 499–507. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Tracking SARS-CoV-2 Variants. 2021. Available online: https://Www.Who.Int/En/Activities/Tracking-SARS-CoV-2-Variants/ (accessed on 25 February 2023).
- Sabino, E.C.; Buss, L.F.; Carvalho, M.P.S.; Prete, C.A.; Crispim, M.A.E.; Fraiji, N.A.; Pereira, R.H.M.; Parag, K.V.; Da Silva Peixoto, P.; Kraemer, M.U.G.; et al. Resurgence of COVID-19 in Manaus, Brazil, despite High Seroprevalence. Lancet 2021, 397, 452–455. [Google Scholar] [CrossRef] [PubMed]
- Zahradník, J.; Nunvar, J.; Schreiber, G. Perspectives: SARS-CoV-2 Spike Convergent Evolution as a Guide to Explore Adaptive Advantage. Front. Cell. Infect. Microbiol. 2022, 12, 748948. [Google Scholar] [CrossRef] [PubMed]
- Consorcio Argentino de Genómica de SARS-CoV-2 Proyecto Argentino Interinstitucional de Genómica de SARS-CoV-2. Available online: http://Pais.Qb.Fcen.Uba.Ar/ (accessed on 25 February 2023).
- Ministerio de Salud de la Republica Argentina Monitor Público de Vacunación. Available online: https://Www.Argentina.Gob.Ar/Coronavirus/Vacuna/Aplicadas (accessed on 25 February 2023).
- Ministerio de Justicia y Derechos Humanos. Presidencia de la Republica Argentina Decreto 67/2021. Distanciamiento Social, Preventivo y Obligatorio y Aislamiento Social, Preventivo y Obligatorio. Available online: https://www.argentina.gob.ar/normativa/nacional/decreto-67-2021-346580 (accessed on 25 February 2023).
- Torres, C.; Mojsiejczuk, L.; Acuña, D.; Alexay, S.; Amadio, A.; Aulicino, P.; Debat, H.; Fay, F.; Fernández, F.; Giri, A.A.; et al. Cost-Effective Method to Perform SARS-CoV-2 Variant Surveillance: Detection of Alpha, Gamma, Lambda, Delta, Epsilon, and Zeta in Argentina. Front. Med. 2021, 8, 755463. [Google Scholar] [CrossRef]
- Zambrana Montaño, R.; Culasso, A.C.A.; Fernández, F.; Marquez, N.; Debat, H.; Salmerón, M.; Zamora, A.M.; Ruíz de Huidobro, G.; Costas, D.; Alabarse, G.; et al. Evolution of SARS-CoV-2 during the First Year of the COVID-19 Pandemic in Northwestern Argentina. Virus Res. 2023, 323, 198936. [Google Scholar] [CrossRef]
- World Health Organization (WHO). COVID-19 Weekly Epidemiological Update, 44th ed.; World Health Organization: Geneva, Switzerland, 2021. Available online: https://apps.who.int/iris/handle/10665/341904 (accessed on 25 February 2023).
- Josh Quick 2020. NCoV-2019 Sequencing Protocol. Protocols.Io. Available online: https://dx.doi.org/10.17504/protocols.io.bbmuik6w (accessed on 25 February 2023).
- Freed, N.E.; Vlková, M.; Faisal, M.B.; Silander, O.K. Rapid and Inexpensive Whole-Genome Sequencing of SARS-CoV-2 Using 1200 Bp Tiled Amplicons and Oxford Nanopore Rapid Barcoding. Biol. Methods Protoc. 2020, 5, bpaa014. [Google Scholar] [CrossRef]
- Schmieder, R.; Edwards, R. Fast Identification and Removal of Sequence Contamination from Genomic and Metagenomic Datasets. PLoS ONE 2011, 6, e17288. [Google Scholar] [CrossRef]
- Bushnell, B. BBMap—Sourceforge.Net/Projects/Bbmap/. Available online: https://sourceforge.net/projects/bbmap/ (accessed on 25 February 2023).
- Andrews, S. FastQC: A Quality Control Tool for High throughput Sequence Data. 2010. Available online: http://www.Bioinformatics.Babraham.Ac.Uk/Projects/Fastqc/ (accessed on 25 February 2023).
- Li, H.; Durbin, R. Fast and Accurate Long-Read Alignment with Burrows–Wheeler Transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve Years of SAMtools and BCFtools. GigaScience 2021, 10, giab008. [Google Scholar] [CrossRef] [PubMed]
- Benton, M. 2021 Nanopore Guppy GPU Basecalling on Windows Using WSL2. 2022. Available online: https://medium.com/analytics-vidhya/explained-output-of-nvidia-smi-utility-fc4fbee3b124 (accessed on 3 August 2022).
- ARTIC SARS-CoV-2 Workflow. EPI2ME. Oxford Nanopore Technologies Ltd. Available online: https://github.com/epi2me-labs/wf-artic (accessed on 25 February 2023).
- Medaka. Oxford Nanopore Technologies Ltd. Available online: https://github.com/nanoporetech/medaka (accessed on 25 February 2023).
- O’Toole, Á.; Scher, E.; Underwood, A.; Jackson, B.; Hill, V.; McCrone, J.T.; Colquhoun, R.; Ruis, C.; Abu-Dahab, K.; Taylor, B.; et al. Assignment of Epidemiological Lineages in an Emerging Pandemic Using the Pangolin Tool. Virus Evol. 2021, 7, veab064. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; Von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast Model Selection for Accurate Phylogenetic Estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Nguyen, L.-T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Hoang, D.T.; Chernomor, O.; Von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the Ultrafast Bootstrap Approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef]
- Guindon, S.; Dufayard, J.-F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing the Performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef]
- Pango Designation. Available online: https://github.com/cov-lineages/pango-designation (accessed on 10 June 2022).
- Rambaut, A.; Lam, T.T.; Max Carvalho, L.; Pybus, O.G. Exploring the Temporal Structure of Heterochronous Sequences Using TempEst (Formerly Path-O-Gen). Virus Evol. 2016, 2, vew007. [Google Scholar] [CrossRef]
- Drummond, A.J.; Ho, S.Y.W.; Phillips, M.J.; Rambaut, A. Relaxed Phylogenetics and Dating with Confidence. PLoS Biol. 2006, 4, e88. [Google Scholar] [CrossRef]
- Drummond, A.J. Bayesian Coalescent Inference of Past Population Dynamics from Molecular Sequences. Mol. Biol. Evol. 2005, 22, 1185–1192. [Google Scholar] [CrossRef] [PubMed]
- Suchard, M.A.; Lemey, P.; Baele, G.; Ayres, D.L.; Drummond, A.J.; Rambaut, A. Bayesian Phylogenetic and Phylodynamic Data Integration Using BEAST 1.10. Virus Evol. 2018, 4. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior Summarization in Bayesian Phylogenetics Using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [PubMed]
- FigTree v1.4.4. Available online: https://github.com/rambaut/figtree/releases (accessed on 1 March 2019).
- Bielejec, F.; Baele, G.; Vrancken, B.; Suchard, M.A.; Rambaut, A.; Lemey, P. SpreaD3: Interactive Visualization of Spatiotemporal History and Trait Evolutionary Processes. Mol. Biol. Evol. 2016, 33, 2167–2169. [Google Scholar] [CrossRef] [PubMed]
- Tzou, P.L.; Tao, K.; Pond, S.L.K.; Shafer, R.W. Coronavirus Resistance Database (CoV-RDB): SARS-CoV-2 Susceptibility to Monoclonal Antibodies, Convalescent Plasma, and Plasma from Vaccinated Persons. PLoS ONE 2022, 17, e0261045. [Google Scholar] [CrossRef]
- Ministerio de Hacienda Resolución 426-E/2017. Available online: https://www.argentina.gob.ar/normativa/nacional/resoluci%C3%B3n-426-2017-279545 (accessed on 10 June 2022).
- Organización Panamericana de la Salud Situación de COVID-19 En Argentina. Distribución Espacio-Temporal de Casos y Muertes. Available online: https://paho-covid19-response-who.hub.arcgis.com/pages/paho-argentina-covid-19-response (accessed on 15 November 2022).
- Torres, C.; Nabaes Jodar, M.; Acuña, D.; Montaño, R.M.Z.; Culasso, A.C.A.; Amadio, A.F.; Aulicino, P.; Ceballos, S.; Cacciabue, M.; Debat, H.; et al. Omicron Waves in Argentina: Dynamics of SARS-CoV-2 Lineages BA.1, BA.2 and the Emerging BA.2.12.1 and BA.4/BA.5. Viruses 2023, 15, 312. [Google Scholar] [CrossRef]
- C.37 Lineage Defining Mutations. Available online: https://cov-lineages.org/constellations.html (accessed on 25 January 2022).
- Romero, P.E.; Dávila-Barclay, A.; Gonzáles, L.; Salvatierra, G.; Cuicapuza, D.; Solis, L.; Marcos, P.; Huancachoque, J.; Carhuaricra, D.; Rosadio, R.; et al. Novel Sublineage within B.1.1.1 Currently Expanding in Peru and Chile, with a Convergent Deletion in the ORF1a Gene (Δ3675-3677) and a Novel Deletion in the Spike Gene (Δ246-252, G75V, T76I, L452Q, F490S, T859N). Available online: https://virological.org/t/novel-sublineage-within-b-1-1-1-currently-expanding-in-peru-and-chile-with-a-convergent-deletion-in-the-orf1a-gene-3675-3677-and-a-novel-deletion-in-the-spike-gene-246-252-g75v-t76i-l452q-f490s-t859n/685/1 (accessed on 1 June 2021).
- Padilla-Rojas, C.; Jimenez-Vasquez, V.; Hurtado, V.; Mestanza, O.; Molina, I.S.; Barcena, L.; Morales Ruiz, S.; Acedo, S.; Lizarraga, W.; Bailon, H.; et al. Genomic Analysis Reveals a Rapid Spread and Predominance of Lambda (C.37) SARS-CoV-2 Lineage in Peru despite Circulation of Variants of Concern. J. Med. Virol. 2021, 93, 6845–6849. [Google Scholar] [CrossRef] [PubMed]
- Vargas-Herrera, N.; Araujo-Castillo, R.V.; Mestanza, O.; Galarza, M.; Rojas-Serrano, N.; Solari-Zerpa, L. SARS-CoV-2 Lambda and Gamma Variants Competition in Peru, a Country with High Seroprevalence. Lancet Reg. Health—Am. 2022, 6, 100112. [Google Scholar] [CrossRef] [PubMed]
- Quispe-Ricalde, M.A.; Castelán-Sánchez, H.G.; Meza-Rodríguez, P.M.; Dávila-Ramos, S.; Sierra, J.L.; Batista-Garcia, R.; Concha-Velasco, F.; Lucana, S.F.; De Santa Cruz, J.; Zea, V.; et al. Evidence of Natural Selection and Dominance of SARS-CoV-2 Variant Lambda (C.37) over Variants of Concern in Cusco, Peru. Arch. Virol. 2023, 168, 88. [Google Scholar] [CrossRef]
- Kashima, S.; Slavov, S.N.; Giovanetti, M.; Rodrigues, E.S.; Patané, J.S.L.; Viala, V.L.; Santos, E.V.; Evaristo, M.; Lima, L.P.O.; Martins, A.J.; et al. Introduction of SARS-CoV-2 C.37 (WHO VOI Lambda) in the Sao Paulo State, Southeast Brazil. J. Med. Virol. 2022, 94, 1206–1211. [Google Scholar] [CrossRef]
- Molina-Mora, J.A.; Reales-González, J.; Camacho, E.; Duarte-Martínez, F.; Tsukayama, P.; Soto-Garita, C.; Brenes, H.; Cordero-Laurent, E.; Ribeiro Dos Santos, A.; Guedes Salgado, C.; et al. Overview of the SARS-CoV-2 Genotypes Circulating in Latin America during 2021. Front. Public Health 2023, 11, 1095202. [Google Scholar] [CrossRef] [PubMed]
- Stadtmüller, M.; Laubner, A.; Rost, F.; Winkler, S.; Patrasová, E.; Šimůnková, L.; Reinhardt, S.; Beil, J.; Dalpke, A.H.; Yi, B. Emergence and Spread of a Sub-Lineage of SARS-CoV-2 Alpha Variant, B.1.1.7 in Europe, and with Further Evolution of Spike Mutation Accumulations Shared with the Beta and Gamma Variants. Virus Evol. 2022, 8, veac010. [Google Scholar] [CrossRef] [PubMed]
- Presidencia de la Nacion Argentina CIERRE DE FRONTERAS. Decisión Administrativa 268/2021. Available online: https://www.argentina.gob.ar/normativa/nacional/decisi%c3%b3n_administrativa-268-2021-348256 (accessed on 15 February 2023).
- Torjesen, I. Covid-19: Delta Variant Is Now UK’s Most Dominant Strain and Spreading through Schools. BMJ 2021, n1445. [Google Scholar] [CrossRef] [PubMed]
- Bolze, A.; Luo, S.; White, S.; Cirulli, E.T.; Wyman, D.; Dei Rossi, A.; Machado, H.; Cassens, T.; Jacobs, S.; Schiabor Barrett, K.M.; et al. SARS-CoV-2 Variant Delta Rapidly Displaced Variant Alpha in the United States and Led to Higher Viral Loads. Cell Rep. Med. 2022, 3, 100564. [Google Scholar] [CrossRef] [PubMed]
- Tegally, H.; Wilkinson, E.; Althaus, C.L.; Giovanetti, M.; San, J.E.; Giandhari, J.; Pillay, S.; Naidoo, Y.; Ramphal, U.; Msomi, N.; et al. Rapid Replacement of the Beta Variant by the Delta Variant in South. Africa. Epidemiology. 2021. Available online: https://www.medrxiv.org/content/10.1101/2021.09.23.21264018v1 (accessed on 15 February 2023).
- Giovanetti, M.; Fonseca, V.; Wilkinson, E.; Tegally, H.; San, E.J.; Althaus, C.L.; Xavier, J.; Nanev Slavov, S.; Viala, V.L.; Ranieri Jerônimo Lima, A.; et al. Replacement of the Gamma by the Delta Variant in Brazil: Impact of Lineage Displacement on the Ongoing Pandemic. Virus Evol. 2022, 8, veac024. [Google Scholar] [CrossRef]
- Kannan, S.R.; Spratt, A.N.; Cohen, A.R.; Naqvi, S.H.; Chand, H.S.; Quinn, T.P.; Lorson, C.L.; Byrareddy, S.N.; Singh, K. Evolutionary Analysis of the Delta and Delta Plus Variants of the SARS-CoV-2 Viruses. J. Autoimmun. 2021, 124, 102715. [Google Scholar] [CrossRef]
- Focosi, D.; Quiroga, R.; McConnell, S.; Johnson, M.C.; Casadevall, A. Convergent Evolution in SARS-CoV-2 Spike Creates a Variant Soup from Which New COVID-19 Waves Emerge. IJMS 2023, 24, 2264. [Google Scholar] [CrossRef]
- Kimura, I.; Kosugi, Y.; Wu, J.; Zahradnik, J.; Yamasoba, D.; Butlertanaka, E.P.; Tanaka, Y.L.; Uriu, K.; Liu, Y.; Morizako, N.; et al. The SARS-CoV-2 Lambda Variant Exhibits Enhanced Infectivity and Immune Resistance. Cell Rep. 2022, 38, 110218. [Google Scholar] [CrossRef]
- Chen, J.; Wang, R.; Wang, M.; Wei, G.-W. Mutations Strengthened SARS-CoV-2 Infectivity. J. Mol. Biol. 2020, 432, 5212–5226. [Google Scholar] [CrossRef]
- Liu, Z.; VanBlargan, L.A.; Bloyet, L.-M.; Rothlauf, P.W.; Chen, R.E.; Stumpf, S.; Zhao, H.; Errico, J.M.; Theel, E.S.; Liebeskind, M.J.; et al. Identification of SARS-CoV-2 Spike Mutations That Attenuate Monoclonal and Serum Antibody Neutralization. Cell Host Microbe 2021, 29, 477–488.e4. [Google Scholar] [CrossRef]
- Venkatakrishnan, A.J.; Anand, P.; Lenehan, P.; Ghosh, P.; Suratekar, R.; Siroha, A.; Chowdhury, D.R.; O’Horo, J.C.; Yao, J.D.; Pritt, B.S.; et al. Antigenic Minimalism of SARS-CoV-2 Is Linked to Surges in COVID-19 Community Transmission and Vaccine Breakthrough Infections. Infectious Diseases (except HIV/AIDS). 2021. Available online: https://www.medrxiv.org/content/10.1101/2021.05.23.21257668v3.full (accessed on 15 February 2023).
- McCallum, M.; De Marco, A.; Lempp, F.A.; Tortorici, M.A.; Pinto, D.; Walls, A.C.; Beltramello, M.; Chen, A.; Liu, Z.; Zatta, F.; et al. N-Terminal Domain Antigenic Mapping Reveals a Site of Vulnerability for SARS-CoV-2. Cell 2021, 184, 2332–2347.e16. [Google Scholar] [CrossRef] [PubMed]
- Cerutti, G.; Guo, Y.; Zhou, T.; Gorman, J.; Lee, M.; Rapp, M.; Reddem, E.R.; Yu, J.; Bahna, F.; Bimela, J.; et al. Potent SARS-CoV-2 Neutralizing Antibodies Directed against Spike N-Terminal Domain Target a Single Supersite. Cell Host Microbe 2021, 29, 819–833.e7. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, K.R.; Rennick, L.J.; Nambulli, S.; Robinson-McCarthy, L.R.; Bain, W.G.; Haidar, G.; Duprex, W.P. Recurrent Deletions in the SARS-CoV-2 Spike Glycoprotein Drive Antibody Escape. Science 2021, 371, 1139–1142. [Google Scholar] [CrossRef] [PubMed]
- Vogels, C.B.F.; Breban, M.I.; Ott, I.M.; Alpert, T.; Petrone, M.E.; Watkins, A.E.; Kalinich, C.C.; Earnest, R.; Rothman, J.E.; Goes De Jesus, J.; et al. Multiplex QPCR Discriminates Variants of Concern to Enhance Global Surveillance of SARS-CoV-2. PLoS Biol. 2021, 19, e3001236. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Chan, J.F.-W.; Liu, H.; Liu, Y.; Chai, Y.; Shi, J.; Shuai, H.; Hou, Y.; Huang, X.; Yuen, T.T.-T.; et al. Spike Mutations Contributing to the Altered Entry Preference of SARS-CoV-2 Omicron BA.1 and BA.2. Emerg. Microbes Infect. 2022, 11, 2275–2287. [Google Scholar] [CrossRef] [PubMed]
- Meng, B.; Kemp, S.A.; Papa, G.; Datir, R.; Ferreira, I.A.T.M.; Marelli, S.; Harvey, W.T.; Lytras, S.; Mohamed, A.; Gallo, G.; et al. Recurrent Emergence of SARS-CoV-2 Spike Deletion H69/V70 and Its Role in the Alpha Variant, B.1.1.7. Cell Rep. 2021, 35, 109292. [Google Scholar] [CrossRef]
- Shapira, G.; Patalon, T.; Gazit, S.; Shomron, N. Immunosuppression as a Hub for SARS-CoV-2 Mutational Drift. Viruses 2023, 15, 855. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nabaes Jodar, M.S.; Torres, C.; Mojsiejczuk, L.; Acuña, D.; Valinotto, L.E.; Goya, S.; Natale, M.; Lusso, S.; Alexay, S.; Amadio, A.; et al. The Lambda Variant in Argentina: Analyzing the Evolution and Spread of SARS-CoV-2 Lineage C.37. Viruses 2023, 15, 1382. https://doi.org/10.3390/v15061382
Nabaes Jodar MS, Torres C, Mojsiejczuk L, Acuña D, Valinotto LE, Goya S, Natale M, Lusso S, Alexay S, Amadio A, et al. The Lambda Variant in Argentina: Analyzing the Evolution and Spread of SARS-CoV-2 Lineage C.37. Viruses. 2023; 15(6):1382. https://doi.org/10.3390/v15061382
Chicago/Turabian StyleNabaes Jodar, Mercedes Soledad, Carolina Torres, Laura Mojsiejczuk, Dolores Acuña, Laura Elena Valinotto, Stephanie Goya, Monica Natale, Silvina Lusso, Sofia Alexay, Ariel Amadio, and et al. 2023. "The Lambda Variant in Argentina: Analyzing the Evolution and Spread of SARS-CoV-2 Lineage C.37" Viruses 15, no. 6: 1382. https://doi.org/10.3390/v15061382
APA StyleNabaes Jodar, M. S., Torres, C., Mojsiejczuk, L., Acuña, D., Valinotto, L. E., Goya, S., Natale, M., Lusso, S., Alexay, S., Amadio, A., Irazoqui, M., Fernandez, F., Acevedo, M. E., Alvarez Lopez, C., Angelletti, A., Aulicino, P., Bolatti, E., Brusés, B., Cacciahue, M., ... Viegas, M. (2023). The Lambda Variant in Argentina: Analyzing the Evolution and Spread of SARS-CoV-2 Lineage C.37. Viruses, 15(6), 1382. https://doi.org/10.3390/v15061382