

An Early SARS-CoV-2 Omicron Outbreak in a Dormitory in Saint Petersburg, Russia
 , , , , , , , ,
, , , , , , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Methods
3. Results
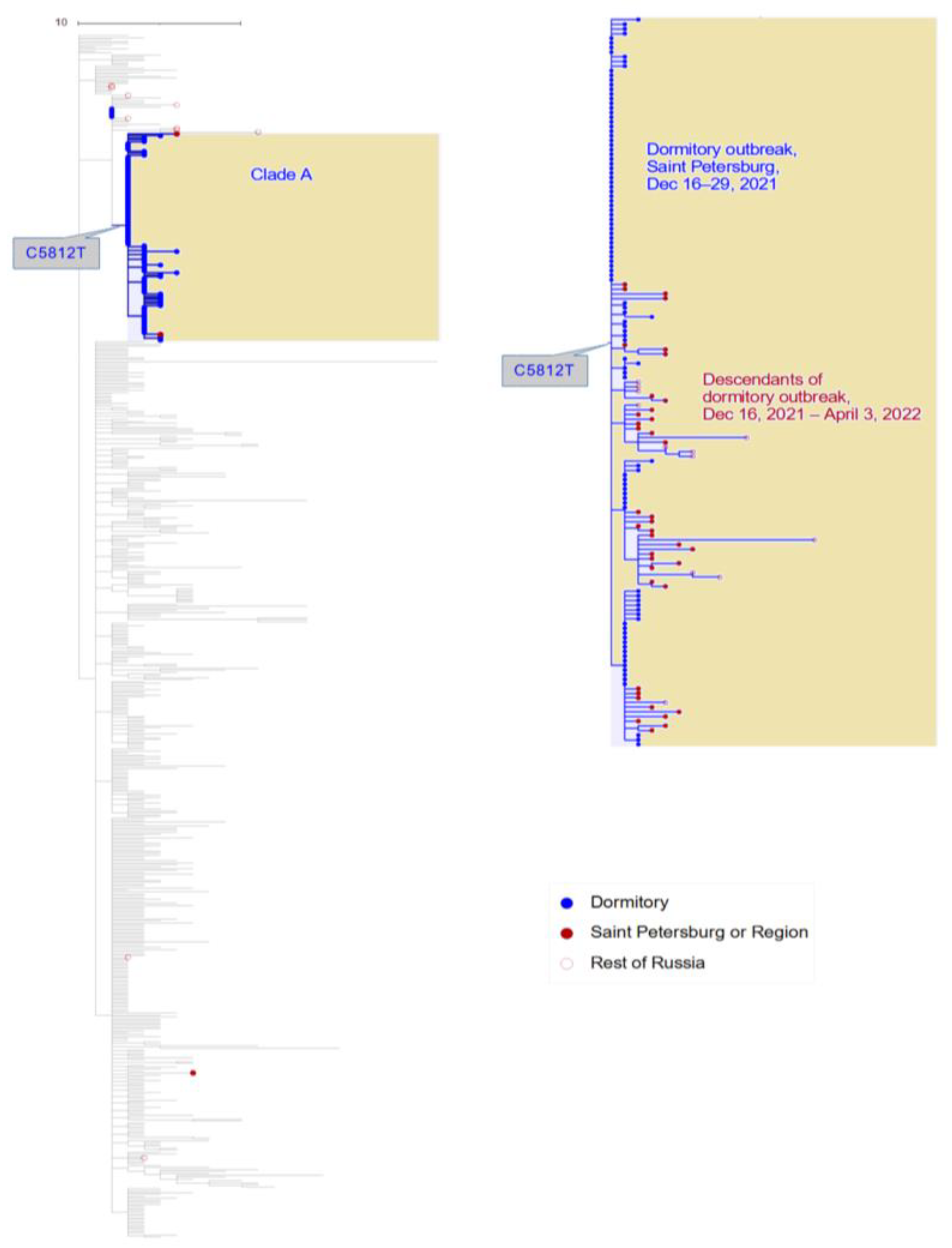
3.1. Phylogenetic Distribution of Samples Indicates a Single Introduction into the Dormitory
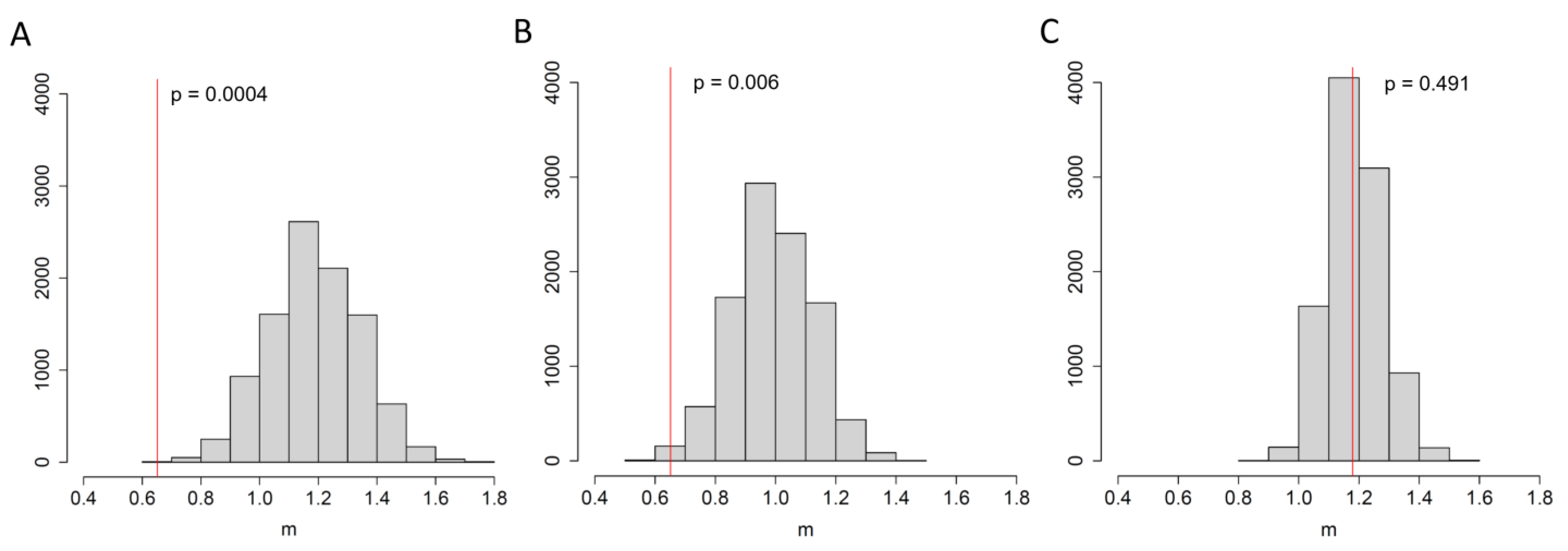
3.2. An Elevated Risk of Within-Room Transmission
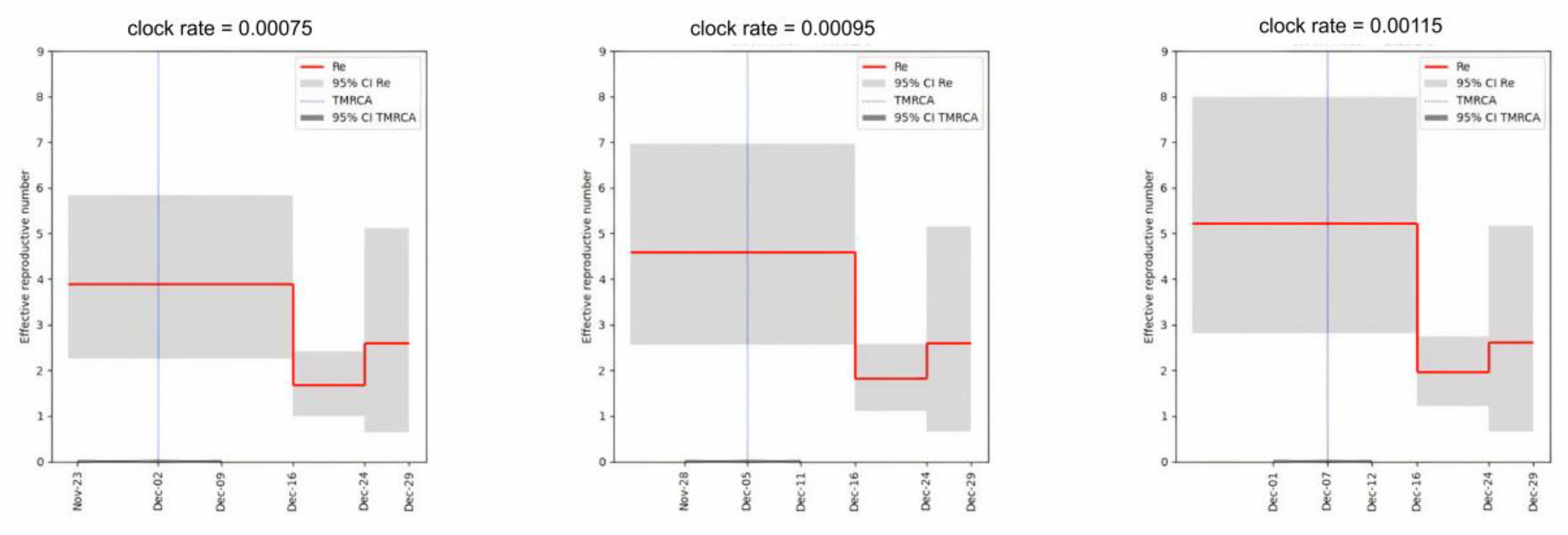
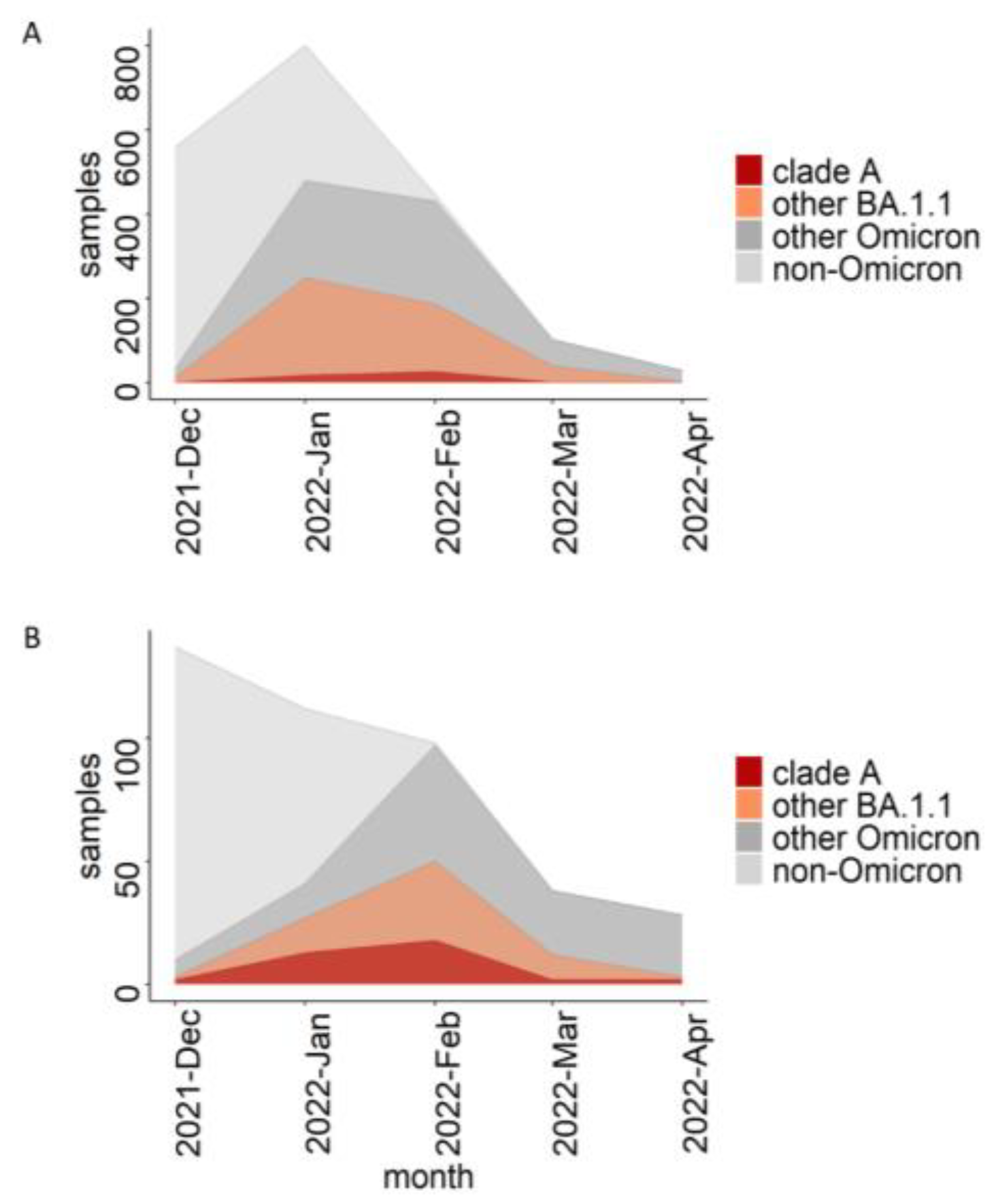
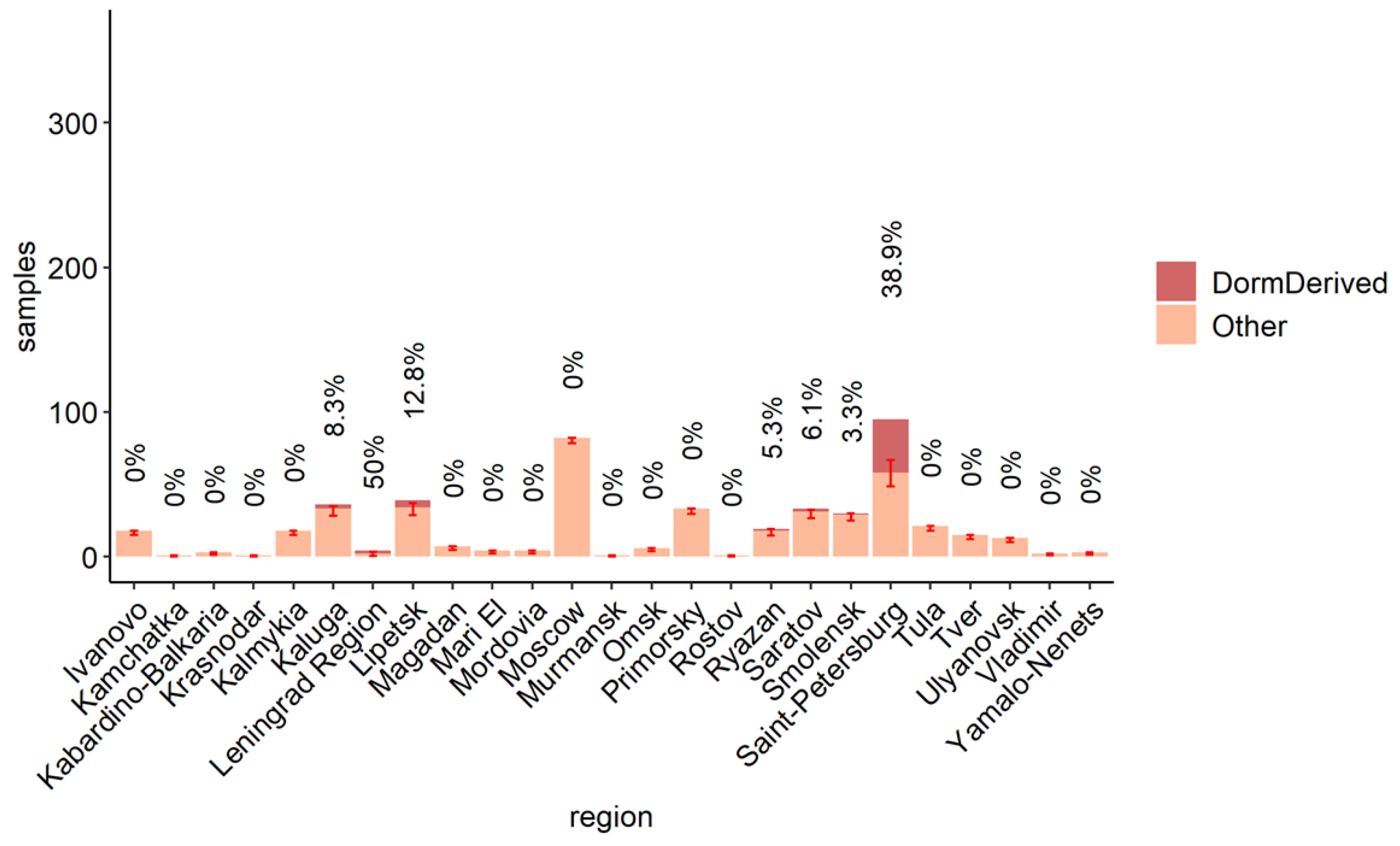
3.3. The Role of the Dormitory Outbreak in the Russian and Global Epidemic of Omicron
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Viana, R.; Moyo, S.; Amoako, D.G.; Tegally, H.; Scheepers, C.; Althaus, C.L.; Anyaneji, U.J.; Bester, P.A.; Boni, M.F.; Chand, M.; et al. Rapid Epidemic Expansion of the SARS-CoV-2 Omicron Variant in Southern Africa. Nature 2022, 603, 679–686. [Google Scholar] [CrossRef]
- World Health Organization. Classification of Omicron (B.1.1.529): SARS-CoV-2 Variant of Concern 2021. Available online: https://www.who.int/news/item/26-11-2021-classification-of-omicron-(b.1.1.529)-sars-cov-2-variant-of-concern (accessed on 26 November 2021).
- Figgins, M.D.; Bedford, T. SARS-CoV-2 Variant Dynamics across US States Show Consistent Differences in Effective Reproduction Numbers. medRxiv 2022. [Google Scholar] [CrossRef]
- Klink, G.V.; Safina, K.R.; Nabieva, E.; Shvyrev, N.; Garushyants, S.; Alekseeva, E.; Komissarov, A.B.; Danilenko, D.M.; Pochtovyi, A.A.; Divisenko, E.V.; et al. The Rise and Spread of the SARS-CoV-2 AY.122 Lineage in Russia. Virus Evol. 2022, 8, veac017. [Google Scholar] [CrossRef] [PubMed]
- Turakhia, Y.; Thornlow, B.; Hinrichs, A.S.; De Maio, N.; Gozashti, L.; Lanfear, R.; Haussler, D.; Corbett-Detig, R. Ultrafast Sample Placement on Existing TRees (UShER) Enables Real-Time Phylogenetics for the SARS-CoV-2 Pandemic. Nat. Genet. 2021, 53, 809–816. [Google Scholar] [CrossRef]
- Komissarov, A.B.; Safina, K.R.; Garushyants, S.K.; Fadeev, A.V.; Sergeeva, M.V.; Ivanova, A.A.; Danilenko, D.M.; Lioznov, D.; Shneider, O.V.; Shvyrev, N.; et al. Genomic Epidemiology of the Early Stages of the SARS-CoV-2 Outbreak in Russia. Nat. Commun. 2021, 12, 649. [Google Scholar] [CrossRef]
- Yolshin, N.; Varchenko, K.; Komissarova, K.; Danilenko, D.; Komissarov, A.; Lioznov, D. One-Step RT-PCR Ins214EPE Assay for Omicron (B.1.1.529) Variant Detection. Protocols. Io. 2021. [Google Scholar] [CrossRef]
- Yolshin, N.D.; Komissarov, A.B.; Varchenko, K.V.; Musaeva, T.D.; Fadeev, A.V.; Lioznov, D.A. Detection of the Omicron SARS-CoV-2 Lineage and Its BA.1 Variant with Multiplex RT-QPCR. Int. J. Mol. Sci. 2022, 23, 16153. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree Of Life (ITOL) v5: An Online Tool for Phylogenetic Tree Display and Annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2021. [Google Scholar]
- Harrell, F.E., Jr.; Dupont, C. Hmisc: Harrell Miscellaneous; R Package Version. 2008, Volume 3, p. 437. Available online: https://cran.r-project.org/web/packages/Hmisc/index.html (accessed on 1 June 2023).
- Wickham, H.; Averick, M.; Bryan, J.; Chang, W.; McGowan, L.; François, R.; Grolemund, G.; Hayes, A.; Henry, L.; Hester, J.; et al. Welcome to the Tidyverse. JOSS 2019, 4, 1686. [Google Scholar] [CrossRef]
- Ahlmann-Eltze, C.; Patil, I. Ggsignif: R Package for Displaying Significance Brackets for “Ggplot2”. PsyArXiv 2021. [Google Scholar] [CrossRef]
- Badr, H.S.; Zaitchik, B.F.; Kerr, G.H.; Nguyen, N.-L.H.; Chen, Y.-T.; Hinson, P.; Colston, J.M.; Kosek, M.N.; Dong, E.; Du, H.; et al. Unified Real-Time Environmental-Epidemiological Data for Multiscale Modeling of the COVID-19 Pandemic. Sci. Data 2023, 10, 367. [Google Scholar] [CrossRef] [PubMed]
- Jian, F.; Yu, Y.; Song, W.; Yisimayi, A.; Yu, L.; Gao, Y.; Zhang, N.; Wang, Y.; Shao, F.; Hao, X.; et al. Further Humoral Immunity Evasion of Emerging SARS-CoV-2 BA.4 and BA.5 Subvariants. Lancet Infect. Dis. 2022, 22, 1535–1537. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.P.; Lytras, S.; Lucaci, A.G.; Maier, W.; Grüning, B.; Shank, S.D.; Weaver, S.; MacLean, O.A.; Orton, R.J.; Lemey, P.; et al. Selection Analysis Identifies Clusters of Unusual Mutational Changes in Omicron Lineage BA.1 That Likely Impact Spike Function. Mol. Biol. Evol. 2022, 39, msac061. [Google Scholar] [CrossRef]
- Obermeyer, F.; Jankowiak, M.; Barkas, N.; Schaffner, S.F.; Pyle, J.D.; Yurkovetskiy, L.; Bosso, M.; Park, D.J.; Babadi, M.; MacInnis, B.L.; et al. Analysis of 6.4 Million SARS-CoV-2 Genomes Identifies Mutations Associated with Fitness. Science 2022, 376, 1327–1332. [Google Scholar] [CrossRef]
- Stadler, T.; Kühnert, D.; Bonhoeffer, S.; Drummond, A.J. Birth-Death Skyline Plot Reveals Temporal Changes of Epidemic Spread in HIV and Hepatitis C Virus (HCV). Proc. Natl. Acad. Sci. USA 2013, 110, 228–233. [Google Scholar] [CrossRef]
- Bouckaert, R.; Vaughan, T.G.; Barido-Sottani, J.; Duchêne, S.; Fourment, M.; Gavryushkina, A.; Heled, J.; Jones, G.; Kühnert, D.; De Maio, N.; et al. BEAST 2.5: An Advanced Software Platform for Bayesian Evolutionary Analysis. PLoS Comput. Biol. 2019, 15, e1006650. [Google Scholar] [CrossRef]
- du Plessis, L.; McCrone, J.T.; Zarebski, A.E.; Hill, V.; Ruis, C.; Gutierrez, B.; Raghwani, J.; Ashworth, J.; Colquhoun, R.; Connor, T.R.; et al. Establishment and Lineage Dynamics of the SARS-CoV-2 Epidemic in the UK. Science 2021, 371, 708–712. [Google Scholar] [CrossRef]
- Chaguza, C.; Coppi, A.; Earnest, R.; Ferguson, D.; Kerantzas, N.; Warner, F.; Young, H.P.; Breban, M.I.; Billig, K.; Koch, R.T.; et al. Rapid Emergence of SARS-CoV-2 Omicron Variant Is Associated with an Infection Advantage over Delta in Vaccinated Persons. Med 2022, 3, 325–334.e4. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Peng, P.; Cao, X.; Wu, K.; Chen, J.; Wang, K.; Tang, N.; Huang, A.-L. Increased Immune Escape of the New SARS-CoV-2 Variant of Concern Omicron. Cell. Mol. Immunol. 2022, 19, 293–295. [Google Scholar] [CrossRef]
- Chen, P.Z.; Koopmans, M.; Fisman, D.N.; Gu, F.X. Understanding Why Superspreading Drives the COVID-19 Pandemic but Not the H1N1 Pandemic. Lancet Infect. Dis. 2021, 21, 1203–1204. [Google Scholar] [CrossRef]
- Lewis, D. Superspreading Drives the COVID Pandemic—And Could Help to Tame It. Nature 2021, 590, 544–546. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Zhao, S.; Lee, S.S.; Mok, C.K.P.; Wong, N.S.; Wang, J.; Jia, K.M.; Wang, M.H.; Yam, C.H.K.; Chow, T.Y.; et al. Superspreading Potential of COVID-19 Outbreak Seeded by Omicron Variants of SARS-CoV-2 in Hong Kong. J. Travel Med. 2022, 29, taac049. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.-C.; Au, A.K.-W.; Chen, H.; Yuen, L.L.-H.; Li, X.; Lung, D.C.; Chu, A.W.-H.; Ip, J.D.; Chan, W.-M.; Tsoi, H.-W.; et al. Transmission of Omicron (B.1.1.529)—SARS-CoV-2 Variant of Concern in a Designated Quarantine Hotel for Travelers: A Challenge of Elimination Strategy of COVID-19. Lancet Reg. Health—West Pac. 2022, 18, 100360. [Google Scholar] [CrossRef]
- Cegolon, L.; Ronchese, F.; Ricci, F.; Negro, C.; Larese-Filon, F. SARS-CoV-2 Infection in Health Care Workers of Trieste (North-Eastern Italy), 1 October 2020–7 February 2022: Occupational Risk and the Impact of the Omicron Variant. Viruses 2022, 14, 1663. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Klink, G.V.; Danilenko, D.; Komissarov, A.B.; Yolshin, N.; Shneider, O.; Shcherbak, S.; Nabieva, E.; Shvyrev, N.; Konovalova, N.; Zheltukhina, A.; et al. An Early SARS-CoV-2 Omicron Outbreak in a Dormitory in Saint Petersburg, Russia. Viruses 2023, 15, 1415. https://doi.org/10.3390/v15071415
Klink GV, Danilenko D, Komissarov AB, Yolshin N, Shneider O, Shcherbak S, Nabieva E, Shvyrev N, Konovalova N, Zheltukhina A, et al. An Early SARS-CoV-2 Omicron Outbreak in a Dormitory in Saint Petersburg, Russia. Viruses. 2023; 15(7):1415. https://doi.org/10.3390/v15071415
Chicago/Turabian StyleKlink, Galya V., Daria Danilenko, Andrey B. Komissarov, Nikita Yolshin, Olga Shneider, Sergey Shcherbak, Elena Nabieva, Nikita Shvyrev, Nadezhda Konovalova, Alyona Zheltukhina, and et al. 2023. "An Early SARS-CoV-2 Omicron Outbreak in a Dormitory in Saint Petersburg, Russia" Viruses 15, no. 7: 1415. https://doi.org/10.3390/v15071415
APA StyleKlink, G. V., Danilenko, D., Komissarov, A. B., Yolshin, N., Shneider, O., Shcherbak, S., Nabieva, E., Shvyrev, N., Konovalova, N., Zheltukhina, A., Fadeev, A., Komissarova, K., Ksenafontov, A., Musaeva, T., Eder, V., Pisareva, M., Nekrasov, P., Shchur, V., Bazykin, G. A., & Lioznov, D. (2023). An Early SARS-CoV-2 Omicron Outbreak in a Dormitory in Saint Petersburg, Russia. Viruses, 15(7), 1415. https://doi.org/10.3390/v15071415