3.3. Introduced and Gained Genetic Diversity
The concatenated full genome ML tree (
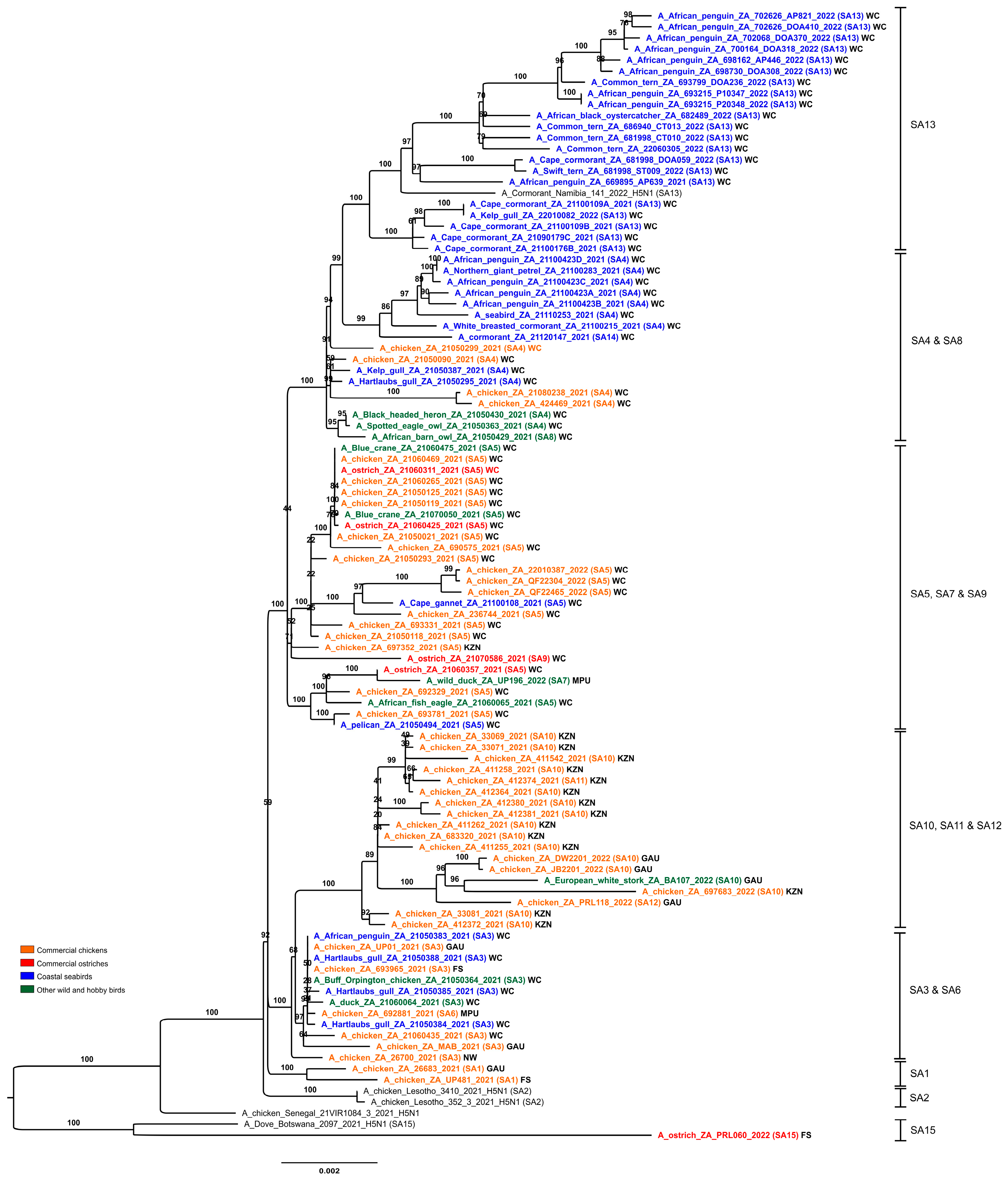
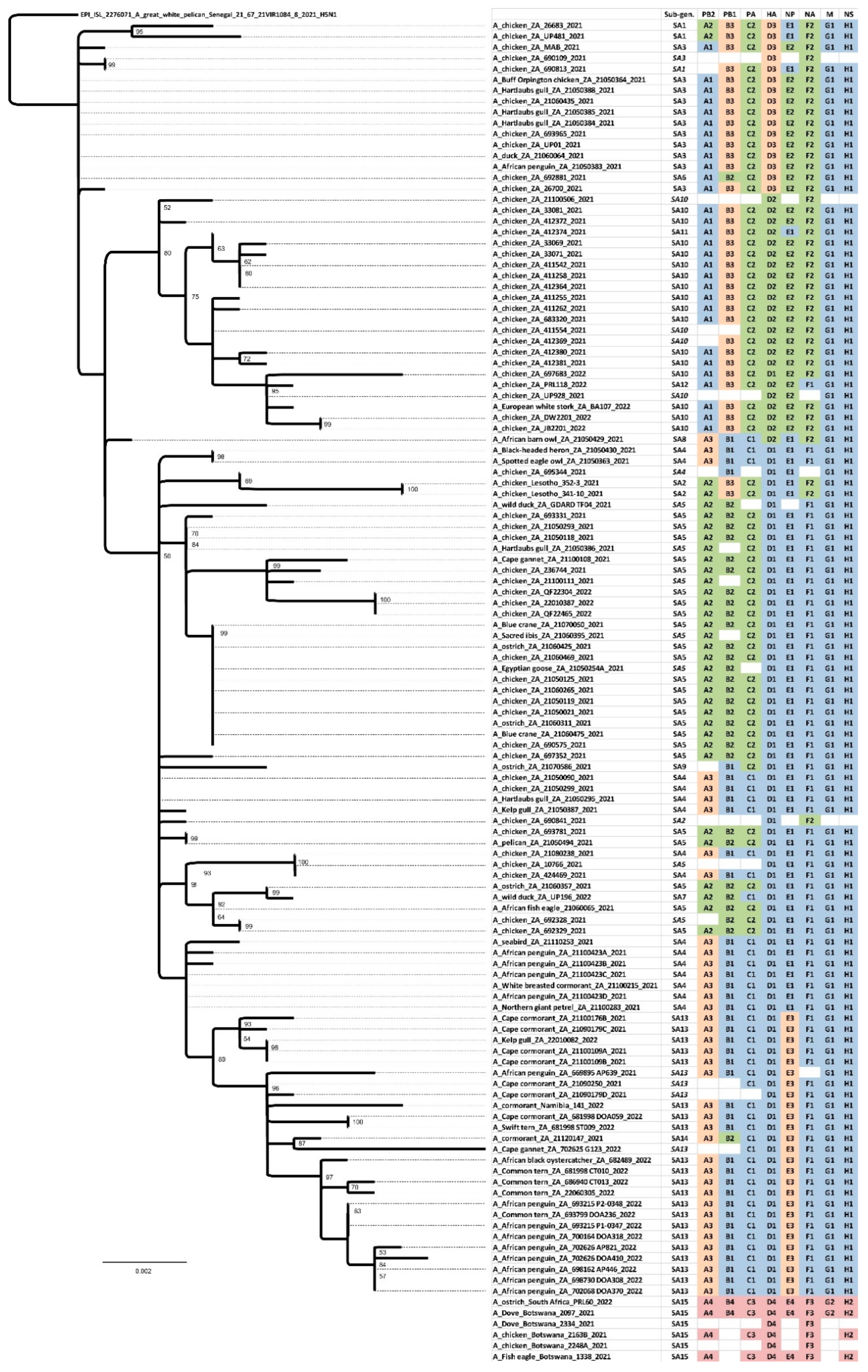
Figure 3) provides a useful overview of the total genetic distances and phylogenetic relationships between the southern African viruses, but in order to investigate the fine-scale epidemiological spread of H5N1 HPAI within the sub-region it was necessary to evaluate genomic reassortments between the viruses, including those for which only partial genomes were obtained. To achieve this, the radial views of the individual ML trees were examined and sub-clades were identified and assigned a unique letter and number (
Figure S3a–h). The sub-clades in the M and NS genes were less obvious and had low bootstrap values; therefore, only two sub-clades each were assigned here. Ultimately, sub-clades A1–4 (PB2), B1–4 (PB1), C1–3 (PA), D1–4 (HA), E1–4 (NA), G1 & 2 (M) and H1 & 2 (NS) were assigned, tabulated, and are presented for comparison in
Figure 5, where the combination of sub-clades designates a sub-genotype. Thus, fifteen southern African (SA) sub-genotypes were identified that are also fully supported by the topology and bootstrap values > 90% in
Figure 3. The South Africa/Lesotho/Namibia sub-cluster is represented by sub-genotypes SA1 to SA14 and the unique SA/Botswana sub-cluster was designated as sub-genotype SA15. The insights gained from the genomic reassortment analysis read in conjunction with the temporal distributions (
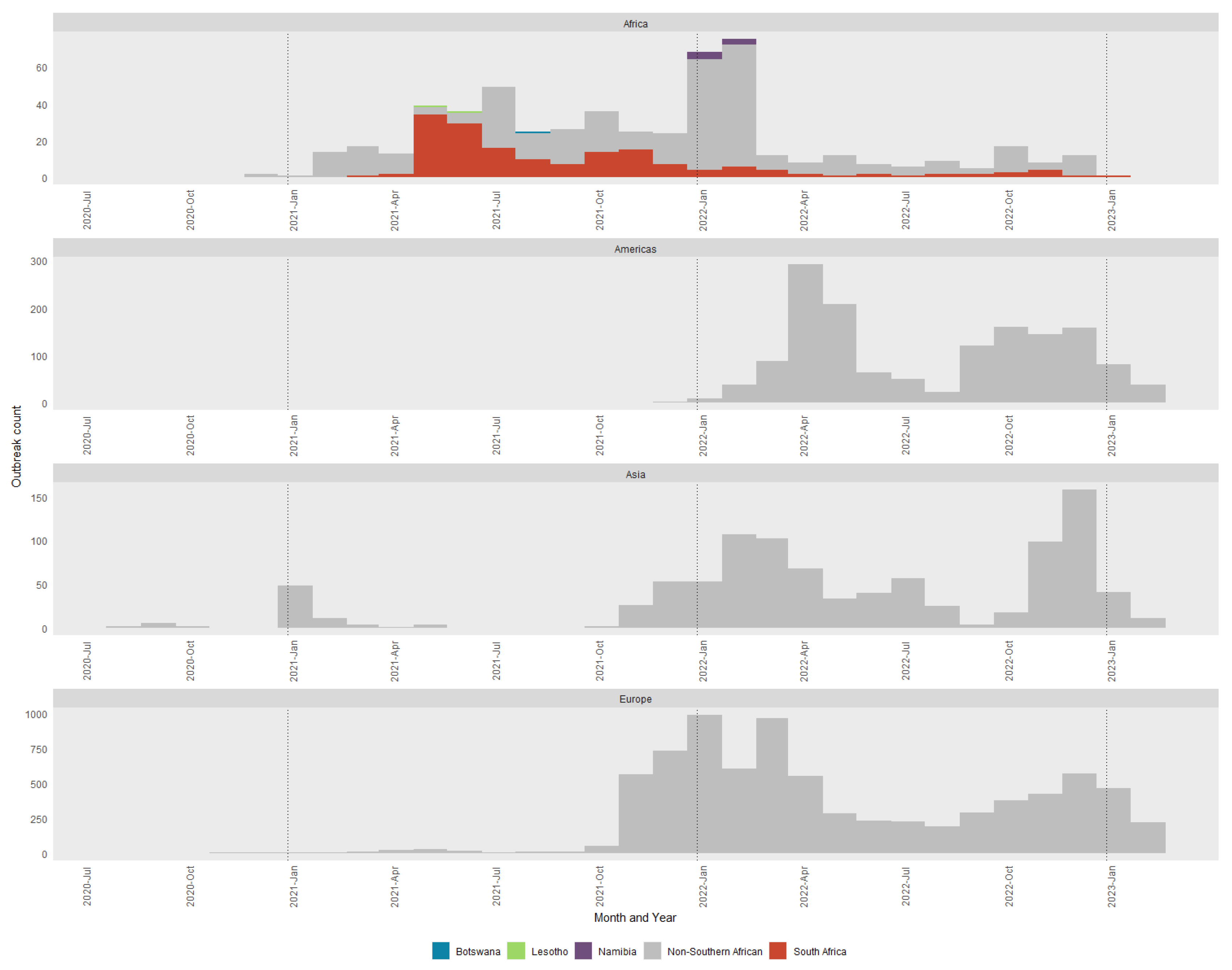
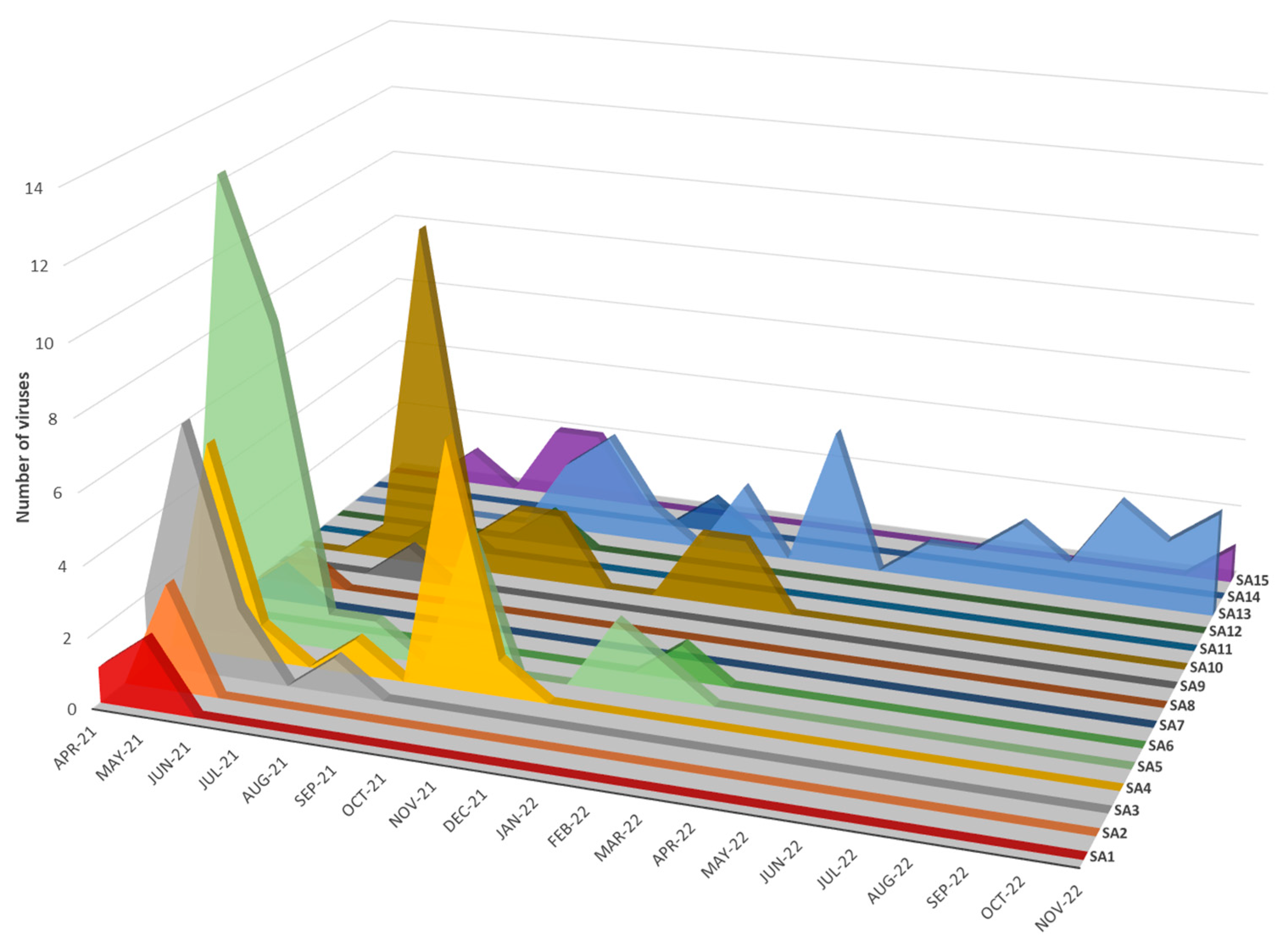
Figure 6) and mapped geographic locations (
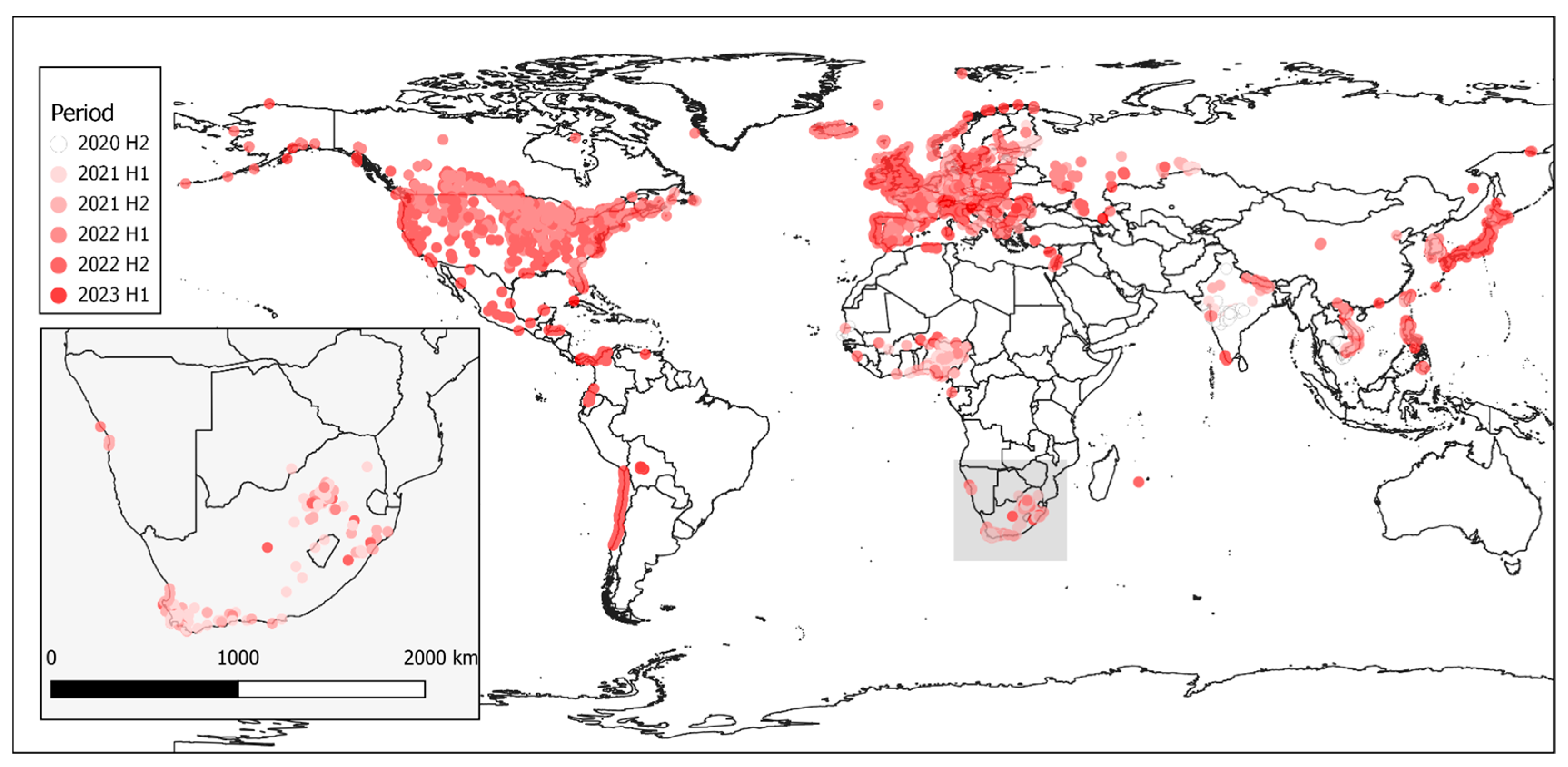
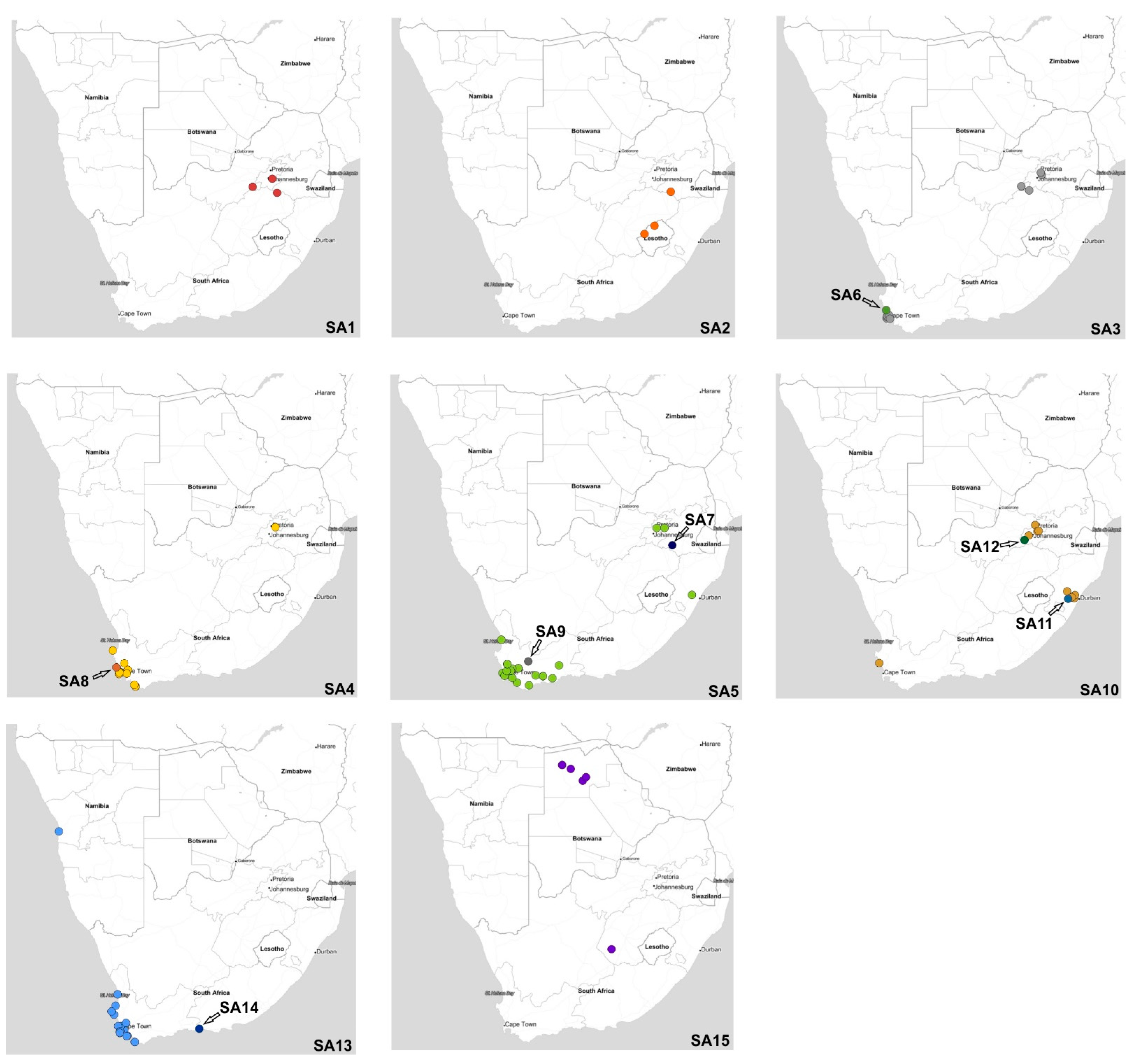
Figure 7) are discussed below.
Seven distinct H5N1 HPAI sub-genotypes (SA1 to SA5, SA7 and SA8) were detected within the first six weeks of the outbreaks in South Africa in April and May 2021 (
Figure 6 and
Figure 7), and this high genetic diversity suggests that multiple relatively heterogenous viruses were introduced almost simultaneously. The index case in commercial layer hens near Brakpan, Gauteng province on 9 April 2021 (26683/21) is an SA1 strain, whereas the second outbreak on 19 April in Potchefstroom, North West province (26700/21) is an SA3 strain. Sub-genotypes SA1 and SA2 were only found in the north-central regions of South Africa and Lesotho (
Figure 7) and were not detected again after early June 2021, whereas SA3 (
n = 12), SA4 (
n = 16) and SA5 (
n = 31) viruses were found in both the north-central regions and much further south in the Western Cape province in the first 6 weeks of the epizootic. SA3 circulated in both the northern and southern regions until August 2021, and an SA3 virus was likely the progenitor of SA6, which differs by a PB1 reassortment and is represented by a single virus (692881/21) that caused an outbreak in commercial chickens near Wellington in the Western Cape in mid-May 2021.
SA4 was found almost exclusively in the Western Cape province where it caused outbreaks in commercial chickens, coastal seabirds and other wild birds from May to November 2021. The only exception was 695344/2021 from an outbreak in commercial chickens in the Pretoria region, Gauteng province on 8 June. However, since only a partial genome could be sequenced the designation of 695344/2021 as SA4 is tentative. SA8, with a sole representative in African barn owl/21050429/21, was the result of reassortment between an SA4 virus (from which it differs by a double HA and NA reassortment) and SA10, with which it shares the most closely related HA and NA genes.
SA5 was predominant in the Western Cape province from early May 2021 until February 2022 where it caused multiple outbreaks in commercial birds, especially layer hens, but it was also detected in commercial ostriches as well as a wide variety of wild birds including Egyptian geese, Hartlaub’s gulls, Sacred ibis, an African fish eagle, blue cranes and Cape gannets. In late June 2021, SA5 appeared in Ashburton, KZN province where it caused an outbreak in commercial chickens (697352/2021), and in August 2021 in an outbreak in 68-week-old commercial layers in Pretoria, Gauteng province (10766/21). The virus may have persisted in the north-central region until at least October 2021 when it was detected during active surveillance of wild ducks in the Bronkhorstspruit region (GDARD TF04/2021; [
17]), but since only partial genomes were available for the latter two cases their classification as SA5 is tentative. Sub-genotypes SA7 and SA9 are represented by single viruses that are related to SA5; SA7 differs from SA5 in a PA reassortment, and the sole representative, UP196/21, was collected during active surveillance of wild ducks near Standerton, Mpumalanga province in mid-February 2022. SA9 (2107586/2021) was detected in ostriches near Touwsrivier, Western Cape province differing from SA5 in the PA gene, but other unique features of this virus are discussed later on.
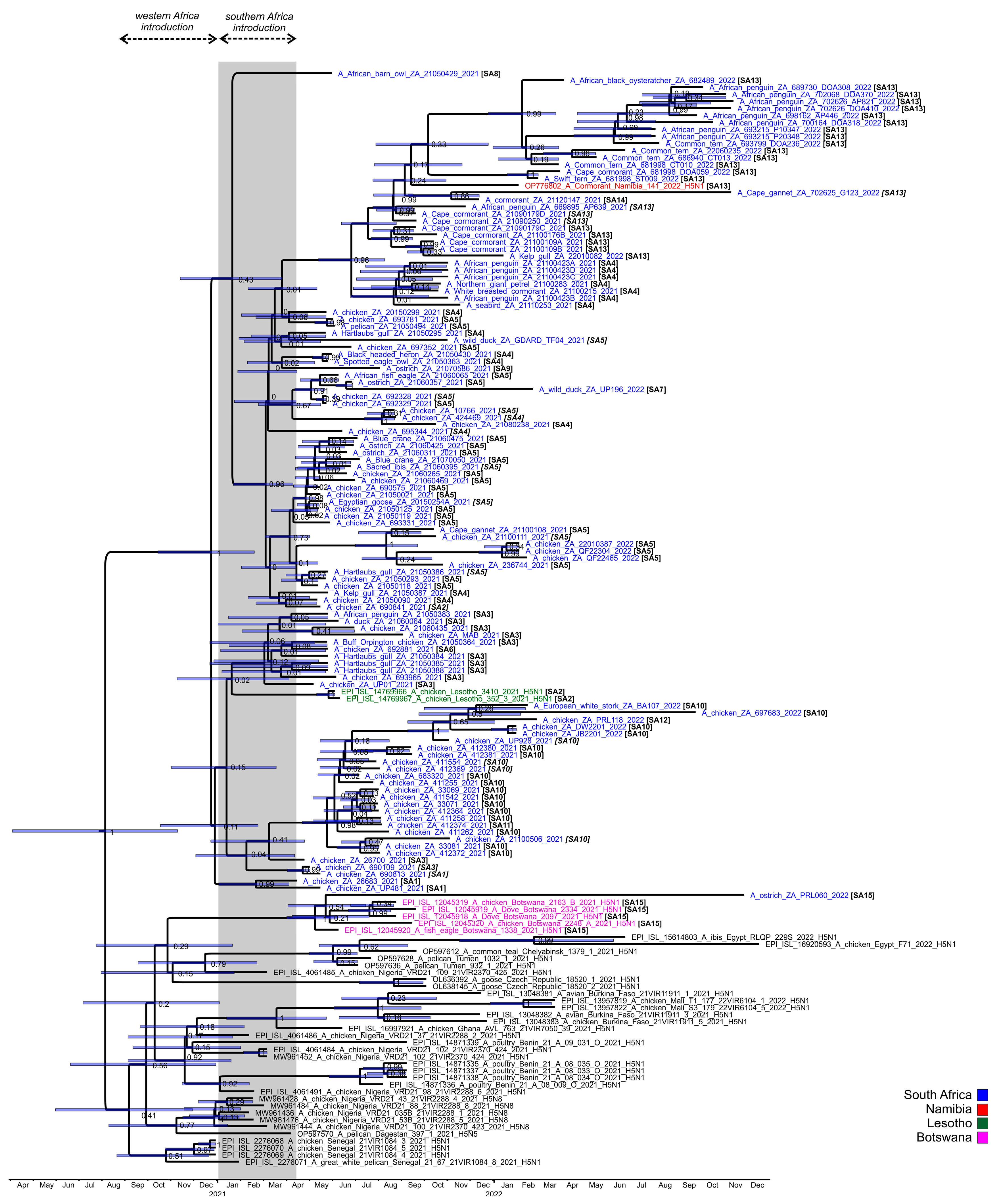
The ancestral nodes of these early sub-genotypes in South Africa (SA1, SA2, SA3/SA6, SA4/SA8 and SA5/SA7/SA9) pre-date the 9 April index case, but the posterior probabilities in the HA gene MCC tree are generally low (
Figure 4). Therefore, precise dating of when they arose was not possible, but their progenitor emerged sometime between late December 2020 and March 2021. The sub-genotypes detected after May 2021, viz. SA10 to SA14, likely emerged within South Africa and are discussed along with SA15 in further detail below.
3.5. Outbreaks of H5N1 HPAI in Commercial Ostriches Were Point Introductions
Commercial ostriches are regularly screened to detect exposure to avian influenza using serological tests, and the detection of any influenza A antibodies triggers swab collection for virus detection. Under experimental conditions, ostriches infected with clade 2.3.4.4B H5 HPAI may appear clinically healthy whilst shedding high amounts of virus, but if they ingest an excessive viral load from the environment, or if they are stressed and immune-compromised, they can develop typical HPAI-related neurological and respiratory signs and die within a few days [
25]. Overall, there were fewer reported cases of H5N1 HPAI in commercial ostriches in 2021/2022 compared to the 2017/2018 H5N8 outbreaks, with only five ostrich-origin viral genomes available for analysis this time.
A sub-genotype SA5 strain, 21060357/2021, was recovered on 17 June 2021 from an outbreak in 8-month-old commercial ostriches near Swellendam, Western Cape. At least a third of the flock died in this outbreak, with congested organs seen in one bird post-mortem. This virus was genetically most closely related to UP197/21 (SA7), detected seven months later and 1440 km away in environmental wild duck fecal swabs collected at a dam near Standerton, Mpumalanga province (
Figure 3). Then, 21060425/2021, detected on the same ostrich farm six days later, and 21060311/2021, from an outbreak near Albertinia, were similarly classified as the SA5 sub-genotype that predominated in the Western Cape at the time. Although these two ostrich viruses are genetically similar across the full genome (0.0001 nucleotide substitutions per site;
Table S1), the farms were approximately 75 km apart with no known epidemiological links. Notably, 21060311 was identical in the consensus genome sequence to those of five other H5N1 viruses from the Western Cape in May and June in commercial and backyard chickens and wild blue cranes that succumbed to the disease (blue crane/21060475/2021, chicken/21050119/2021, chicken/21050125/2021, chicken/21060265/2021 and chicken/21060469/2021; see
Figure 3;
Supplementary Table S1), pointing to the wide dissemination of this particular strain in the Western Cape’s wild birds and poultry sector preceding and coinciding with these ostrich outbreaks.
Next, 21070586/2021, a unique sub-genotype SA9 strain, was detected in 5–8-month-old ostriches (grower phase) on 28 July 2021 on a farm near Touwsrivier, Western Cape. A mortality rate of approximately 15% was recorded in June and July and samples taken post-mortem on 12 July had tested HP H5 rRT-PCR-positive. Affected birds appeared to be in good body condition but showed clinical signs including depression, recumbency, anorexia and sudden death within 24 h. Other clinical signs included neon-green feces, apparent sinusitis with purulent ocular discharge and severe ulcerative stomatitis. Post-mortem signs included petechial hemorrhage on the serosal surfaces of the small intestines, in the trachea and on the epicardium. The farm housed multi-age ostriches (breeders, chicks and slaughter birds) and Touwsrivier, located in the driest part of the Klein Karoo semi-desert, is a veritable oasis attracting wild birds to its irrigated lucerne pastures and dams. Blood tests from the grower bird epidemiological group (7 weeks to slaughter age) sampled on 9 June 2021 were AI-antibody negative, but by 19 July, serological testing indicated that approximately 54.4% of the grower flock had seroconverted, most with clade 2.3.4.4 H5-specific antibody hemagglutination inhibition titers reaching 1:256 (A. Olivier, pers. comm.). The primary virus introduction could have occurred any time from mid-May 2021, and, notably, this virus contained an E627K mutation in the PB2 protein, which is associated with mammalian adaptation [
26], but is also a known marker of ostrich adaptation that emerges after a period of transmission within a flock [
27].
Interestingly, PRL060/2022, the SA15 virus from the outbreak near Fauresmith, Free State province in November 2022 that shared an RCA with the Botswanan strains, also contained the ostrich-specific E627K mutation in the PB2-protein (but E627K was absent in three PB2 sequences available from Botswana). The virus was similarly detected in 6–8-week-old ostrich chicks, but these birds displayed classical neurological signs including incoordination, “star gazing” and sudden collapse when excited. The birds preferred to be recumbent, and when disturbed seemed to be blind, disorientated and uncoordinated. Good rains in the higher-lying Orania/Hopetown region transformed what is normally a dry catchment area at the bottom of a valley where Fauresmith is situated into a wetland for large parts of the spring season. Large numbers of waders and other smaller wetland birds as well as larger species such as Sacred ibises were observed there, and passerine bridging hosts are suspected to have introduced the virus as fomites into the chick camps (A. Olivier, pers. comm.). The HA tMRCA of the SA/Botswana sub-clade was dated to early May 2021 (95% HPD Feb–June 2021) (
Figure 4) but no SA15 or similar viruses were detected in South Africa throughout 2021 or most of 2022, and therefore this sub-genotype was likely a new introduction into the region just weeks before its detection in the ostriches, and the timing is consistent with a southward spring migration of the unknown wild hosts.
3.6. Limited Farm-to-Farm Spread in South African Commercial Chicken Outbreaks
Despite their intensified efforts to maintain strict biosecurity, poultry producers were heavily affected by H5N1 HPAI outbreaks in 2021, with sporadic cases in commercial, small-scale and backyard chickens continuing into the first month of 2022. IAV whole genome data was essential to determine whether outbreaks were due to point introductions (a localized biosecurity break where the external environment was heavily contaminated by wild bird-origin H5N1 viruses) or were caused by farm-to-farm spread by contaminated vehicles, people, feed and equipment. In many cases, the genetic data was unequivocal that outbreaks in commercial chickens were caused by point introductions. For example, in the Western Cape province between the 6 and 10 May 2021, almost simultaneous outbreaks occurred in a single large producer’s operations near Malmesbury (21050090/2021) and Worcester (21050118/2021, 21050119/2021, 21050125/2021 and 21050021/2021). Thus, 21050090/2021 (Malmesbury; 6 May) was phylogenetically most closely related to coastal seabird viruses kelp gull/21050387/2021 and Hartlaub’s gull/21050295/2021 (all sub-genotype SA4) (
Figure 3), whereas all the cases near Worcester in that same week were caused by SA5 strains. Among the latter, 21050118/2021 was phylogenetically distinct from the other three outbreaks in this producer and shared an RCA with 693331/2021 (an outbreak in layers near Wellington on 23 May, from a different producer), 236744/2021 (the layers of yet another producer near Paarl on 19 October), and a Cape gannet virus (21100108/2021) sampled at Lambert’s Bay in early October. The affected farm is 10 km south of the other three outbreaks, which were on different sites of the same farm.
Other sub-genotype SA5 viruses in the Western Cape that caused chicken outbreaks grouped most closely with wild bird viruses. Indeed, 693781/2021 caused an outbreak in 14-week-old layers near Malmesbury on 27 May, and its closest relative was detected in a dead pelican (21050494/2021) found two days later in the same area, and 692329/2021, from an outbreak near Grabouw in mid-May, shared a most recent common ancestor with an African fish eagle virus (21060065/2021) diagnosed in a weak bird found near Bredasdorp at the end of June. Later on, 21060435/2021 was associated with an outbreak causing 100% mortality in backyard chickens in Phillipi, Cape Town on 24 June, but this is an SA3 strain. Next, 21050299/2021, the cause of the 18 May outbreak near Piketberg, an SA4-type virus, was genetically distinct from all other chicken outbreaks in the Western Cape at the time and shared ancestry with what would eventually emerge as the SA13 coastal seabird-specific lineage that is discussed in the next section. Viruses from outbreaks in other provinces were also genetically distinct, and therefore point introductions into those operations. Examples include UP481/2021 (SA1, broiler outbreak in Villiers, Free State province on 10 May) and MAB/2021 (SA3, Rietvlei near Pretoria, Gauteng province on 27 August).
To evaluate possible secondary spread in cases within the same sub-genotype where the genetic distances were smaller, the phylogenetic grouping in the concatenated genome tree (
Figure 3) was used in conjunction with the distance matrix (
Table S1). In the distance matrix, we considered a cutoff value of ≤0.001 nucleotide substitutions per site between two sequences to be a possible secondary spread, unless closely related or identical wild bird viruses were located in the same sub-clade as the chicken virus (a point introduction cannot be ruled out). Outbreaks in poultry in geographically separated regions with no known epidemiological links were also disregarded.
Of the 44 chicken-origin complete genomes available for analysis, in three cases the sequenced viruses were sampled from the same farm on the same day. In addition, 21080238/2021 and 424469/2021 were sampled at the same time from an outbreak in a single free-range layer producer’s layer hen operation near Stellenbosch but tested at different laboratories. An interesting case was an outbreak in broiler breeders in Worcester, Western Cape province in early May 2021: 21050021/2021 and 690575/2021 were sampled from different houses on the same farm, on the same day, but 690575/2021 was located on a relatively long branch in the phylogenetic tree (
Figure 3), and the distance between these two viruses was 0.0011 nucleotide substitutions per site. Such genetic distance indicates the diversity of viruses in the external natural environment at the time the biosecurity breach occurred. In contrast, 22010387/2022 and QF22304/2022, also sampled from a single outbreak one day apart (layer hens, Wellington, late January 2022), were highly similar across the genome (0.0002 nucleotide substitutions per site). QF22465/2022 was sampled from a different house on the same site one week later, with 0.0008 nucleotide substitutions per site (
Supplemental Table S1) and, therefore, the second outbreak was likely a secondary spread within the producer’s operations.
Sub-genotypes SA10, SA11 and SA12 form a distinct and strongly supported sub-clade in
Figure 3, comprising viruses from a cluster of outbreaks in the Kwa-Zulu-Natal province between the end of June to early September 2021, and that later on appeared in Gauteng. The MCC HA gene analysis (
Figure 4) dated the RCA of sub-clades SA10/SA11/SA12 to May 2021 (95% HPD April to June), i.e., after the index cases, signifying that the ancestral SA10/11/12 virus probably emerged within South Africa’s borders. The only SA10 virus from the Western Cape province, 21100506/2021, from an outbreak in layer hens near Yzerfontein at the end of October, is a tentative assignment to SA10 because only the HA and NA genes were available for analysis (it is equally likely to be a unique sub-genotype). The partial genome availability was due to an H9N2 co-infecting virus. The closest relatives (>97% nucleotide sequence identity) to the partial H9 and N2-specific genes as determined by BLAST analysis were the Eurasian-type H9N2 strains A/Mallard(
Anas platyrhynchos)/South Korea/KNU2021-41/2021(H9N2) (accession number ON505892) and A/Bean Goose(
Anser fabalis)/South Korea/KNU 2019-16/2019(H9N2) (accession number MW380632), respectively, providing supporting evidence that the Yzerfontein outbreak was caused by a point introduction from wild birds. Nonetheless, the homology in the H5N1 HA and NA genes suggests an epidemiological link between the Western Cape and KwaZulu-Natal viruses, and that wild birds may have spread the virus north-eastwards, potentially driven from the southern Cape region by an extreme cold front in June 2021.
The Camperdown region of the KZN province has a high poultry density, dominated by a single large producer that operates multiple broiler breeder and layer hen sites there. The outbreaks in the Camperdown region started in late June 2021 and lasted until early September 2021. Since the large producer was most affected and outbreaks occurred within a relatively short space of time, the farm-to-farm spread was assumed, but the molecular evidence shows otherwise. Firstly, SA11 (412374/2021), which is an NP reassortant, can unequivocally be identified as a point introduction. A sub-cluster comprising 33069/2021, 33071/2021, 411542/2021, 411258/2021, 412374/2021 and 412364/2021, all sampled between 20 to 30 July 2021, cannot be ruled out as farm-to-farm spread because of their close phylogenetic relationships (
Figure 3). However, as a cluster supported by a 99% bootstrap value, they collectively represent a second point introduction. Then, 412380/2021 and 412381/2021 were isolated from the same site on consecutive days during December 2021; the viruses are not identical (0.0006 nucleotide substitutions per site;
Supplemental Table S1) and their location on a separate branch with 100% support, plus 0.0011–0.0032 nucleotide substitution differences with other viruses in the general cluster, indicates a third primary introduction to the region. To be sure, 411255/2021 contains sufficient changes across the genome (0.0015 nucleotide substitutions per site) to designate it as a fourth-point introduction; 411262/2021 and 683320/2021 are phylogenetically basal to the others and cannot be separated or designated as a unique introduction but 683320/2021 caused an outbreak in the hens of a different producer; 33081/2021 and 412372/2021 (sub-clade supported by a high bootstrap value) had a distance within the cutoff of 0.0009 nucleotide substitutions per site. These two SA10 viruses were collected from simultaneous outbreaks in different producers, located 50 km apart in Howick and Camperdown, respectively, with no known epidemiological link, and were therefore not considered a secondary spread event and represent the fifth-point introduction to KZN. The ecological factors causing the high levels of H5N1 environmental contamination at the time in the KwaZulu-Natal province remain unknown, and no large die-offs in wild birds were reported here.
The SA10/SA11 sub-cluster from KZN and the SA10/SA12 sub-cluster detected later in Gauteng shared an RCA that was dated June 2021 (95% HPD May–Aug 2021) (
Figure 3). The first SA10-associated outbreaks in poultry in the Gauteng province are represented by DW2201/2022 (Kempton Park) and JB2201/2022 (Elandsfontein), in simultaneous outbreaks in late January 2022 in the layers of different producers located 32 km apart. Although the genetic distance between these viruses was only 0.0002 nucleotide substitutions per site, epidemiological tracing could not establish a link between these farms. Subsequently, PRL118/2022 was the cause of an outbreak in “speculator” chickens near Fochville on 20 February 2022, but the significant genetic distance (
Figure 3) as well as the NA reassortment that designates this virus as a unique SA12 sub-genotype rules out the Kempton Park and Elandsfontein outbreaks as the source. The most recent SA10 virus sequenced, 697683/2022, was recovered from an outbreak in 31-week-old commercial layers in Cato Ridge, KwaZulu-Natal province in mid-September 2022. Its closest relative, however, was BA107/2022, sampled from a sick European white stork caught during active wild bird surveillance at Bon Accord dam near Pretoria at the beginning of February 2022 [
4]. The phylogenetic data suggest SA10/SA12-like viruses persisted in the Gauteng province wild bird reservoir until late 2022, which then spread the virus back to KwaZulu-Natal because the Cato Ridge strain was evidently unrelated to the 2021 outbreaks in KwaZulu-Natal.
3.8. Emergence of a Coastal Seabird-Specific H5N1 Sub-Lineage in South Africa That Spread to Namibia
H5N1 HPAI appeared relatively early in Western Cape coastal seabirds compared with the H5N8 HPAI epizootic, and at least two distinct sub-genotypes were co-circulating in the gull populations early on. Hartlaub’s gulls were among the first species affected in and around Cape Town, with the first case found on 14 May 2021. SA3 virus strains were identified in three Hartlaub’s gulls (21050388/2021, 21050385/2021 and 21050384/2021), a kelp gull (21050387/2021) and an African penguin (21050383/2021). These are genetically similar to the other SA3/SA6 strains that were being diagnosed in poultry and terrestrial wild birds in outbreaks in the Western Cape and more northern provinces in a similar period (
Figure 3,
Figure 5 and
Figure 7). Hartlaub’s gull/21050295/2021 and kelp gull/21050299/2021, also from around Cape Town, were, however, identified as SA4 strains. SA5 viruses also spilled into coastal seabirds at some point. One of 40 dead pelicans (21050494/2021) was diagnosed from a farm dam near Malmesbury at the end of May; the only mass mortality recorded in pelicans and SA5 also caused mortalities in Cape cormorants (21100108/2021), and presumably in Cape gannets, in Lambert’s Bay in October.
A distinct cluster of SA4 viruses in coastal seabirds in
Figure 3, supported by a high bootstrap value, would ultimately give rise to the seabird-restricted sub-lineage of viruses, SA13. Basal to this cluster of ancestral SA4 viruses is cormorant/21120147/2021 (Nature’s Valley Beach, 6 December), the sole representative of SA14, which differs from SA13 by a PB1 reassortment (
Figure 5). The cluster of progenitor viruses, sampled from mid-October to November 2021, comprise a white-breasted cormorant (21100215/2021), a bank cormorant (21110253/2021), a northern giant petrel (21100283/2021) and viruses in African penguins (21100423A/2021, 21100423B, 21100423C and 21100423D) on Dyer Island. The latter viruses were associated with mass mortality in both African penguins (at least 200 deaths) and, presumably, Cape cormorants (at least 15,000 deaths) at the same time, during the peak Cape cormorant breeding season. The viruses were all sampled from the Dyer Island colony on 26 October but, interestingly, they are not identical. Most notably, 21100423D/2021 was homologous with the northern giant petrel virus sampled 10 days earlier at St Helena Bay, 320 km away. The genetic distance between this cluster of African penguin H5N1 viruses ranged from 0.002 to 0.011 nucleotide substitutions per site, with the high genetic diversity indicating the likelihood of multiple-point introductions to the island.
Chronologically the next cluster of related viruses, this time detected predominantly in Cape cormorants, represents the emergence of the seabird-restricted sub-genotype SA13. SA4 and SA13 differ by an NP gene reassortment, and the specific NP gene of SA14 was unique to the coastal seabird lineage. The variation in the NP gene was limited to the nucleotide sequence level, which is expected since the nucleocapsid protein is not associated with host specificity or adaptation. None of the unique protein markers that distinguished the South African/Namibian coastal seabird H5N8 HPAI sub-lineage in 2017/2018, viz., N11K and T29S in HA, R95K in M1 and P559A in PA [
5], were present in the SA4/SA13/SA14 viruses.
The first example of the newly emerged SA13 viruses was first detected in Cape cormorants in mid-September 2021 at Glencairn, Cape Town (21090179C/2021), then they appeared three weeks later in Cape Cormorants in Lambert’s Bay (21100109A and 21100109B/2021), in Betty’s Bay in mid-October (21100176B/2021) (900 dead chicks were found at this site in mid-November) and, finally, caused a kelp gull mortality in Muizenberg in early January 2022 (22010082/2022). Lambert’s Bay and Muizenberg were epidemiologically linked by highly similar viruses 21100109A/2021 and 22010082/2022. The tMRCA of the SA4/SA13 sub-clade progenitor was dated June 2021 (95% HPD May–August 2021) and the SA13 progenitor July 2021 (95% HPD June–August 2021), the latter indicating that the SA13 virus was probably already circulating in coastal seabirds for several weeks before the increased mortalities in Cape cormorants started.
Our phylogenetic evidence shows that the Cape cormorant virus detected at Bird Island, Walvis Bay in Namibia in December 2021, shared an RCA with the viruses detected in South Africa’s Cape Cormorants from September 2021. Indeed, the RCA to the Namibian virus in
Figure 4 was dated September 2021 (95% HPD Jul–Nov 2021, albeit with a low posterior probability). This evidence points towards a spread of H5N1 up the southwestern African coast to Namibia between September and December in an unknown seabird host, but no deaths in other species were reported along the Namibian coastline or in Walvis Bay at the time of the outbreak [
7].
H5N1 HPAI outbreaks in coastal seabirds in South Africa continued in 2022. The molecular data established an epidemiological link between swift tern/681998 ST009/2022, sampled near Stellenbosch on 22 March 2022, and Cape cormorant/681998 DOA059/2022 from Fish Hoek on 23 March 2022, and these viruses, in turn, shared an RCA with an earlier African penguin case (a chick with no clinical signs) at Stony Point on 18 November 2021. Sporadic detections in common terns and an African black oystercatcher occurred throughout March and into June 2022, with the viruses (22060305/2022, 681998 CT010/2022, 686940 CT013/2022 and 682489/2022) exhibiting continuing genetic drift in the SA13 sub-genotype.
The most recent cluster of seabird outbreaks affected African penguins, starting with at least twenty cases on Dyer Island at the end of July 2022 (including 693215-P10347 and P10348/2022) before spreading in September to the Simon’s Town colony, with a related common tern virus sampled in the interim at Macassar Beach (693799 DOA236/2022). Viruses detected among at least 60 penguin deaths in Simon’s Town in September and October (698730 DOA308/2022, 698162 AP446/2022, 700164 DOA318/2022 and 702068 DOA370/2022) were phylogenetically related to those causing the death of a penguin in November at Stony Point (702626 DOA410/2022) and one at Strandfontein (702626 AP821/2022), both within swimming distance of Simon’s Town.


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






