1. Introduction
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is responsible for the global pandemic of COVID-19 disease and has spread around the world rapidly since its discovery in 2019. It was first reported in Wuhan, the capital city of Hubei province in China, and since then has led to over 6 million deaths globally (as of January 2023) (WHO Coronavirus (COVID-19 Dashboard) (WHO, 2020)). The virus affects the respiratory tract, and the most characteristic symptoms of COVID-19 are flu-like symptoms, such as fever, fatigue, dry cough, and loss of taste or smell. In severe life-threatening cases, patients with inflammation of the lungs can experience trouble breathing, persistent pressure in the chest, confusion, and require life support [
1]. The primary transmission mode is direct contact with the infected individual or by sneezing and coughing, leading to the transfer of nasal droplets [
2]. There have also been cases of asymptomatic COVID-19, which led to the silent transmission of the disease [
3].
SARS-CoV-2 is a positive sense single-stranded RNA virus belonging to the genus Betacoronaviridae. The genome of SARS-CoV-2 is ≈30 kb in length and encodes structural, non-structural, and accessory proteins [
4]. The genome has a 5′ cap and a 3′ poly-A sequence, and the 5′ and 3′ untranslated regions (UTR) of the genome are highly structured. Two-thirds of the genome encodes a single polypeptide called ORF1ab and consists of non-structural proteins. ORF1a encodes polypeptide 1a (pp1a); similarly, ORF1b encodes polypeptide 1b (pp1b). Polypeptide 1ab is a result of ribosomal frameshift during the translation of ORF1a. These polyproteins are processed by viral proteases and give rise to 16 non-structural proteins involved in virus replication and transcription [
5]. SARS-CoV-2 has four structural proteins—Spike (S), Membrane (M), Envelope (E), and Nucleocapsid (N). The function of the S protein is to bind to the host cell surface receptor, angiotensin-converting enzyme 2 (ACE2), and mediate virus entry. ACE2 is an integral membrane protein and serves as a receptor for SARS-CoV-2 by binding to the receptor binding domain of the S protein [
6]. The virus shares more than 80% nucleotide similarity with the SARS-CoV genome and more than 90% similarity in essential structural elements and proteins [
5].
The evolution of the SARS-CoV-2 genome has given rise to various variants, with several variants of concern (VOC) sequentially becoming dominant worldwide over the course of the pandemic (WHO Coronavirus (COVID-19) Dashboard, (WHO, 2020)). While rapid development and distribution of SARS-CoV-2 vaccines have reduced the risk of hospitalization, few treatments exist for those with severe disease. Monoclonal antibody therapies were effective in treating early SARS-CoV-2 variants, but most of these are no longer effective against Omicron due to the evolution of S mutations and immune escape.
Small molecule drug therapies are also currently limited to remdesivir (Veklury®), a combination of nirmatrelvir and ritonavir (brand name Pfizer-PaxlovidTM), and molnupiravir (LagevriTM) (Government of Canada, (Canada, 2022)). Cases of COVID-19 are still on the rise due to waning antibody response post-vaccination and immune escape of emerging variants, and more treatment options are needed. Effective antivirals for SARS-CoV-2 can be developed based on a better understanding of virus replication and will be aided by the development of novel virus and cell culture systems, including viruses with easy-to-monitor reporters. In this study, we have designed and constructed wild-type and modified SARS-CoV-2 genomes using reverse genetics and used them to generate wild-type and reporter viruses and a subgenomic replicon. We have also generated novel human cultured lung cells that are highly susceptible to SARS-CoV-2. These cell lines exhibited high cytopathic effects that are ideal for assays that use live–dead cell selection, such as CRISPR knockout screens, to identify host dependency factors. We also show the usefulness of the reporter virus and cells in antiviral drug screens. Together, these tools provide models for research to understand the mechanism of viral infection, discover new antiviral strategies, and screen effective antivirals.
2. Materials and Methods
Cell lines and maintenance: All the cells were maintained at 37 °C with 5% CO2. HEK293T cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM, with Sodium pyruvate) (HyClone, Ottawa, Canada, #SH30243.01) supplemented with 10% fetal bovine serum (FBS) (Gibco, Ottawa, Canada, #12483020) and 1x Penicillin-Streptomycin (PenStrep) (Gibco, Ottawa, Canada, #15140122). Vero76 cells were cultured in DMEM (without Sodium pyruvate) (Sigma, Markham, Canada, #D5796) supplemented with 10% FBS and PenStrep. A549 cells were a gift from Dr. Yan Zhou and were cultured in F-12K Medium (Kaighn’s Modification of Ham’s F-12 Medium) (ATCC #30-2004) supplemented with 10% FBS and 50 μg/mL Gentamycin Sulfate (BioBasic, Markham, Canada, #BS724). NCI-H23, NCI-H1703, HCC827, NCI-H226, and NCI-H520 were a gift from Dr. Deborah Anderson. The cells were cultured in Roswell Park Memorial Institute (RPMI) 1640 medium (Gibco, Ottawa, Canada, #11875093) supplemented with 10% FBS and 1x PenStrep. The cryomedia used for freezing the cells contained 45% complete media, 45% FBS, and 10% DMSO (MedChemExpress, Monmouth Junction, NJ, USA, #HY-N7060). To test for mycoplasma contamination in all the cell lines, a Mycoalert, Mycoplasma detection kit (Lonza, Kingston, Canada, #LT07-318) was used as per the manufacturer’s protocol.
Virus stocks: Virus handling and related experiments were performed in the containment level 3 facility at the Vaccine and Infectious Disease Organization (VIDO). Most experiments with wild type virus were conducted using the P3 (passage #3) virus stock of SARS-CoV-2/Canada/ON/VIDO-01/2020 (hereafter referred to as SARS-CoV-2 VIDO-01). To prepare virus working stock, Vero76 cells were infected with the P2 virus at an MOI of ~0.01. Briefly, virus stock was diluted in 10 mL DMEM supplemented with 2% FBS and PenStrep and inoculated in a T175 flask of 80% confluent Vero76 cells seeded 24 h before infection. This was incubated at 37 °C for 1 h, gently rocking the flasks intermittently. After 1 h, 25 mL of complete media was added to the flask, followed by incubation at 37 °C for 3–4 days until cytopathic effect (CPE) was observed. Once there was sufficient virus-induced CPE (~70–80%), indicated by cell rounding and cell death, the supernatant was collected, centrifuged at 4000× g for 10 min, aliquoted in cryovials, and stored at −80 °C. The titer was determined by TCID50 (as described below).
Virus titration using TCID
50: Vero76 cells were seeded 24 h prior to infection in 96-well plates, such that they were 80% confluent the next day (1 × 10
4 cells/well). The virus to be titered was serially diluted 10-fold in a round bottom 96-well plate in DMEM supplemented with 2% FBS and PenStrep. Then, 50 µL of this was inoculated in the seeded Vero76 cells and incubated at 37 °C for 1 h. The virus inoculum was then removed and replaced with 100 µL DMEM supplemented with 2% FBS and 1x PenStrep. The plates were incubated at 37 °C, and CPE was noted by microscopy at 3–5 days post-infection. Titers were calculated using the Spearman–Karber algorithm [
7].
Virus titration using plaque assay: 24 h prior to infection, Vero76 cells were seeded in 12-well plates at 2.5 × 105 cells/well. A 400 µL volume of 10-fold serially diluted virus (in DMEM supplemented with 2% FBS and PenStrep) was inoculated per well and incubated at 37 °C for 1 h, with intermittent gentle rocking. After infection, the inoculum was replaced with 1.5 mL 0.75% carboxymethyl cellulose (CMC) (Sigma #21902) media and incubated at 37 °C. To prepare the plaquing media, autoclaved CMC was dissolved in plain DMEM (Sigma #D5796) by placing it on a magnetic stirrer overnight at 4 °C. Once the plaques of sufficient size developed (~72 h), the cells were fixed by adding 1.5 mL 10% buffered formalin (Sigma #HT501128-4L) over the plaquing media and incubating at room temperature for 30 min. The formalin-containing waste was discarded in Formalex® Green formalin neutralizer (Jones Scientific, Kitchener, Canada, #H-FORMG-CB). The plates were washed with water twice and then stained with 0.1% crystal violet (10% ethanol, 0.1% crystal violet, in water) for 30 min. The stain was washed with water and the visible plaques were counted to calculate the titer.
Design of SARS-CoV-2 molecular clones: Full-length SARS-CoV-2 Wuhan-1 (Wuhan seafood market pneumonia virus (2019-nCov) sequence NC_045512) cDNA genome cloned in a bacterial artificial chromosome (BAC) backbone was synthesized by CODEX DNA, Inc. The wild type full length (WTFL) (Codex DNA Inc, now known as Telesis Bio, SanDiego, CA, USA.. #SC2-FLSG-3333) construct is driven by the T7 promoter upstream of 5′ UTR, contains the D614G mutation, and was designed to have a unique SbfI enzyme restriction site downstream of 3′ UTR and poly-A. For making the reporter virus expressing nanoluciferase (NLuc) (NLucFL) (Codex DNA Inc.; custom order), the NLuc gene was inserted by replacing the complete ORF7a while keeping the respective TRS sequence intact [
8]. For making the SARS-CoV-2 sub-genomic replicon (SGR) (Codex DNA Inc.; custom order), all the genes from spike (S) through ORF8 were replaced with
Renilla luciferase (RLuc), Ubiquitin, and Neomycin resistance genes respectively, based on a SARS-CoV replicon [
9]. A plasmid encoding the Nucleocapsid gene (N gene) was a gift from Dr. Qiang Liu [
10]. Briefly, the codon-optimized N sequence is flanked by a T7 promoter on the 3′ end and a 3xFLAG tag on the 5′ end, directly followed by a unique XbaI restriction site.
In vitro RNA transcription: For making in vitro transcribed RNA, 2.5 µg of WTFL and NLucFL or 1.5 µg of SGR purified genomic DNA was linearized with 1.5 µL of SbfI-HF
® (New England Biolabs, Whitby, Canada #R3642L) in 1x CutSmart buffer at 37 °C for 1 h, followed by addition of 1.5 µL of SbfI-HF
® again for 1 h. To remove the 3′ overhangs, the DNA was further treated with 1 µL of Mung Bean Nuclease (New England Biolabs #M0250L) for 1 h at 37 °C. The linearized DNA was extracted using phenol/chloroform and centrifugation in MaXtract High-density phase separation tubes (Qiagen, Toronto, Canada, #129046), followed by overnight precipitation with ammonium acetate and 100% ethanol at −20 °C. The next day the DNA was centrifuged at 14,000 rpm at 4 °C for 30 min, and the DNA pellet was dissolved in 6 µL of nuclease-free water. This linearized DNA was in vitro transcribed to RNA as per the mMessage mMachine
TM T7 kit protocol (Invitrogen, Burlington, Canada, #AM1344) with the following modifications. The total reaction volume was 50 µL made up by adding the linearized DNA template with 7.5 µL GTP (cap analog-to-GTP ratio of 1:1), 25 µL 2xNTP/CAP, 5 µL 10x reaction buffer, and 5 µL enzyme mix. The reaction was incubated at 32 °C for 5 h, followed by DNase treatment twice by adding 1 µL Turbo DNase (provided in the kit) and incubating for 20 min at 37 °C each time. The RNA was extracted using phenol/chloroform and centrifuged in phase separation tubes and precipitated using isopropanol [
8]. To prepare the N mRNA, 1 µg N gene plasmid DNA was linearized with XbaI enzyme for 1 h at 37 °C, followed by Mung Bean Nuclease treatment for 1 h at 37 °C. The purified and precipitated DNA was then used with mMessage mMachine
TM T7 Ultra kit (Invitrogen #AM1345) according to the manufacturer’s protocol; this gave us in vitro transcribed N gene mRNA with an added poly-A tail to mimic the SARS-CoV-2 subgenomic mRNA. To quantify the in vitro transcribed (IVT) RNA for SARS-CoV-2 SGR and N gene, Qubit™ RNA High Sensitivity (HS) kit (Invitrogen #Q32852) was used as per the manufacturer’s protocol. We recovered ~4–5 μg of SARS-CoV-2 SGR RNA from 1.5 μg of DNA template.
Electroporation and virus production: To make viruses from the in vitro transcribed RNA, SARS-CoV-2 RNA and N gene RNA were electroporated in Vero76 cells. N gene mRNA was added because it has been reported to enhance the infectivity of coronavirus transcripts and based on previously published methods to recover viruses [
8]. Briefly, cells were trypsinized and washed (with PBS) with centrifugation at 1000 rpm (RCF 201 g) for 5 min. The cells we resuspended in Ingenio
® Electroporation Solution (Mirus Bio, Madison, WI, USA. #MIR50114) such that the cell concentration was at 8 × 10
6 per electroporation in 800 μL final volume. Then, 1–2 preps of in vitro transcribed RNA and 5–10 µg N gene RNA were mixed with the prepared cell suspension in a 4 mm electroporation cuvette (VWR International, Aurora, CO, USA. #89047-210) and pulsed in a BioRad GenePusler using the exponential protocol with the following settings: Voltage—270 V, Capacitance—950 µF, Resistance—∞ (infinity). Electroporated cells were equilibrated at room temperature for 5 min and transferred to a T-75 flask with 12 mL DMEM supplemented with 10% FBS and 1X PenStrep. (Alternatively, 1 prep of in vitro transcribed RNA with ~5 µg N gene RNA can be electroporated in 4 × 10
6 cells in 400 μL final volume and plated in a T-25 flask with 5 mL DMEM supplemented with 10% FBS and PenStrep.) The electroporated cells were incubated at 37 °C and monitored daily until significant CPE was observed, 2–3 days post-electroporation. The virus stock was harvested as described above; this was considered passage 0 (P0) stock. To passage the virus, 1 mL of P0 virus stock was inoculated in a T-175 flask with 80% confluent Vero76 cells seeded 24 h before infection. Once sufficient CPE was observed, the supernatant was harvested and stored as P1 stock.
For transient replication assays with SARS-CoV-2 SGR RNA, 4 million Vero76 cells resuspended in 400 μL of Ingenio® Electroporation Solution were similarly electroporated with 1 prep of in vitro transcribed SGR RNA (prepared using 1.5 µg of SGR genomic DNA) and 5 µg of N gene RNA. After equilibrating at room temperature for 5 min, 100 μL of the electroporated cells were seeded in each well of a 6-well dish with 2 mL of DMEM supplemented with 10% FBS + Pen-Strep. The cells were incubated at 37 °C for 3 days, and 100 μL media was harvested daily to assess Renilla luciferase expression. For harvesting cells from 6-well dishes, the growth medium was removed, and cells were rinsed with PBS. Then, 200 μL/well of 1xRenilla lysis buffer was added to the well, and the cells were scraped off. The cell extracts were transferred to a microcentrifuge tube and stored at −80 °C until the luciferase assay was performed.
Reporter luciferase assays: The Nano-Glo® luciferase assay system (Promega, Madison, WI, USA, #N1120) was used to assess NLuc expression by SARS-CoV-2 NLucFL, as per the manufacturer’s protocol. Briefly, virus-infected 96-well plates were equilibrated at room temperature for ~5–10 min. The Nano-Glo® Luciferase Assay Reagent was prepared by combining one volume of Nano-Glo® Luciferase Assay Substrate with 50 volumes of Nano-Glo® Luciferase Assay Buffer and was also equilibrated to room temperature. Then, 100 µL of the reagent was added to each of the wells already containing 100 µL infected media. The components were mixed well, incubated at room temperature for 3 min, and then transferred to white cell culture plates (Corning, Tewksbury, MA, USA, #C3610). Luminescence was measured using the PromegaTM GloMax ® Explorer plate reader at 5 s of integration time. Luciferase assays to analyze SGR RNA replication were performed using the Renilla luciferase assay system (Promega #E2810) as per the manufacturer’s protocol with the following modifications. Before measuring the luminescence, cell extracts were centrifuged at 13,000 rpm for 10 min at 4 °C, and 10 μL of the solution was mixed with 30 μL of Luciferase Assay Reagent. Luminescence was measured in a PromegaTM GloMax® 20/20 Luminometer (Promega #E5311) with an integration time of 10 s.
Viral growth kinetics: To characterize the growth kinetics of patient and clone-derived SARS-CoV-2 stocks, supernatants from infected Vero76 cells were collected at different times post-infection and titered using TCID50. Briefly, cells were seeded 24 h prior to infection in 96-well plates at 1 × 104 cells/well; for infection, the virus was diluted in DMEM supplemented with 2% FBS and PenStrep at MOI 0.01 (50 µL infection volume/well). The cells were infected for 1 h at 37 °C, after which the 50 µL virus inoculum was replaced with 100 µL fresh media (DMEM supplemented with 2% FBS and 1x PenStrep). This marks the 0-h post-infection timepoint. The supernatant was collected at 0, 2, 4, 6, 8, 24, 48, and 72 h post-infection. For harvesting at different time points, 100 µL supernatant from respective wells was collected in a round bottom 96-well plate, sealed with a sealing tape, and stored at −80 °C in Ziploc bags until they were to be used for TCID50. The TCID50 was performed in Vero76 cells in 3–4 replicates. For assessing the luciferase kinetics, an identical 96-well plate was set up for infection in white cell culture plates (Corning #C3610). At the required time intervals, the Nano-Glo® luciferase assay was performed as described above.
Antiviral assay: To evaluate the antiviral effects of remdesivir on the wild type and reporter virus stocks, Vero76 cells were seeded 24 h before infection in 96-well cell culture plates at 1 × 104 cells/well; for infection, the virus was diluted to a concentration of 1 × 102 TCID50 per 50 µL in DMEM supplemented with 2% FBS and PenStrep. Remdesivir (MedChemExpress #HY-104077) was reconstituted in DMSO to make the main stock. To generate a drug dose–response curve, remdesivir was 3-fold serially diluted in DMEM supplemented with 2% FBS, PenStrep, and 0.1% DMSO. The cells were infected for 1 h at 37 °C with 50 µL of the diluted virus at an MOI 0.01, after which the virus inoculum was replaced with 100 µL media containing the serially diluted remdesivir. The cells were incubated at 37 °C for 48 h, and the viral supernatants were harvested and titrated using TCID50. The drug treatment was performed in triplicates, followed by an equal number of TCID50 for each of the replicates in Vero76 cells. In parallel, uninfected plates were treated with remdesivir to assess the cell viability under treatment. After 48 h of drug treatment, the cell viability was assessed using the CellTiter 96® AQueous One Solution Proliferation assay (Promega #G3580). Briefly, 20 µL reagent was added to each well, incubated for 2 h at 37 °C, and absorbance was read at 490 nm as an endpoint assay on the Bio-Rad xMarkTM Microplate Absorbance Spectrophotometer. For assessing the antiviral potency of compounds using the NLuc reporter system, an identical antiviral assay was set up in parallel in white cell culture plates (Corning #C3610). After 48 h of incubation, the Nano-Glo® Luciferase assay was performed on the compound-treated infected wells, as described above. The EC50 of the compound and 50% cytotoxicity concentration (CC50) were determined in GraphPad Prism9 using the non-linear regression analysis. The cell viability was normalized to untreated cells depicting 100% viability, whereas the virus inhibition was normalized to untreated infected cells, depicting 100% virus luciferase expression.
Generation of ACE-2 lentivirus particles: The lentivirus vectors containing the ACE2 gene were generated by co-transfecting psPAX2, pMD2.G, and EX-U1285-Lv197 (GeneCopoeia, Rockville, MD, USA), an ACE2 gene containing lentivirus expression vector that also contained a Blasticidin selection gene. The plasmids were transfected into HEK293T cells using X-tremeGENE 9 (Roche, Mississauga, Canada, #XTG9-RO) as per the manufacturer’s instructions. At 18 h post-transfection, the media was replaced with DMEM containing 2% (w/v) bovine serum albumin (BSA), and then lentiviruses were collected after 24 and 48 h [
11].
ACE2 transduction of cell lines and monoclonal cell selection: To make ACE2-expressing cell lines, ACE2 lentiviruses were filtered through a 0.45-micron filter and used to transduce respective cell lines using the reverse transduction method (Addgene, 2019). Briefly, 1–1.5 mL of filtered virus particles were added to the cell suspension of 50,000 cells in the appropriate media supplemented with 10% FBS and 8–10 μg/mL polybrene (Sigma #TR-1003-G) (without any antibiotics) and plated in 6-well cell culture plates with a final volume of 2.5–3 mL. At 72 h post-transduction, fresh media supplemented with 10% FBS and Blasticidin S HCl (Gibco #R21001) was added to cells. Cells were expanded in increasingly larger cell culture plates, and ACE2 expression was confirmed with flow cytometry and based on susceptibility to infection with the SARS-CoV-2 VIDO-01 virus. The selection concentration of Blasticidin S HCl for HEK293T and A549 cells was 5 μg/mL, for NCI-H23, HCC827, and NCI-H1703, it was 4 μg/mL, and for NCI-H520 and NCI-H226, it was 2 μg/mL. After selection and 3–4 passages, the cells were maintained at half the selection concentration of Blasticidin S HCl. Single clone isolation from the HEK293T
ACE2, A549
ACE2, and NCI-H23
ACE2 transduced cell pools was carried out using the array dilution method in 96-well plates [
12]. Cell colonies from single clones were collected 2–3 weeks after seeding and were expanded in increasingly larger cell culture plates.
Analysis of cell surface ACE2 by flow cytometry: The cells to be analyzed were seeded in a 96-well plate at 1 × 105 cells per well. At 70–80% confluency, healthy cells were detached from the monolayer using 0.5 mM EDTA (Gibco #15575020) in PBS and centrifuged at 1500 rpm for 3 min. The cell pellet was stained for 1 h at 4 °C with primary ACE2 antibody (R&D systems #AF933, used at a concentration of 0.25 µg/1 million cells). The cells were washed thrice with 100 µL flow wash buffer (2% FBS in PBS), followed by staining with secondary Goat IgG APC conjugated antibody (R&D systems #F0108, at the recommended volume of 10 µL/1 million cells) and 1000x live–dead viability stain (Invitrogen #L34958, used as 1x final concentration) at 4 °C for 1 h in the dark. The cells were washed with flow wash buffer before fixing with 2% paraformaldehyde (PFA) (diluted in flow wash buffer) for at least 20 min or overnight. The cells were read on a Beckman CytoFLEX Flow Cytometer.
Infectivity assay to assess cell susceptibility: To assess whether the untransduced and ACE2 transduced cell lines were susceptible to SARS-CoV-2 infection leading to cytopathic effect, the cell lines were infected with SARS-CoV-2 VIDO and SARS-CoV-2-NLucFL. At 24 h before infection, 1–1.6 × 104 cells were seeded in 96-well plates such that they were 80% confluent the next day. The viruses were diluted to an MOI 0.5 in corresponding media supplemented with 2% FBS and 1x PenStrep, and the cells were infected for 1 h at 37 °C with a volume of 50 µL virus inoculum. After infection, the virus inoculum was replaced with 100 µL fresh media supplemented with 2% FBS and PenStrep. The CPE from the SARS-CoV-2 VIDO-01 virus and NLuc expression from SARS-CoV-2 NLucFL was evaluated at 6, 24, 48, and 72 h post-infection. The NLuc expression was assessed using the Nano-Glo® luciferase assay, as described above. The virus-induced CPE was assessed using the Viral ToxGloTM assay (Promega #G8943) according to the manufacturer’s protocol. Briefly, infected cells were equilibrated to room temperature for 5–10 min. One hundred microlitres of ATP detection reagent (prepared by adding ATP detection buffer to the ATP detection substrate) was added to the infected wells, incubated at room temperature for 10 min, and the luminescence was read at 5 s integration time on a PromegaTM GloMax ® Explorer plate reader. The luciferase values were normalized to blank readings (ATP detection reagent added to plain media).
Data analysis and graphs: All data were analyzed and plotted in GraphPad Prism 9 software, and the graphs are represented as mean and errir bars showing standard deviation.
4. Discussion
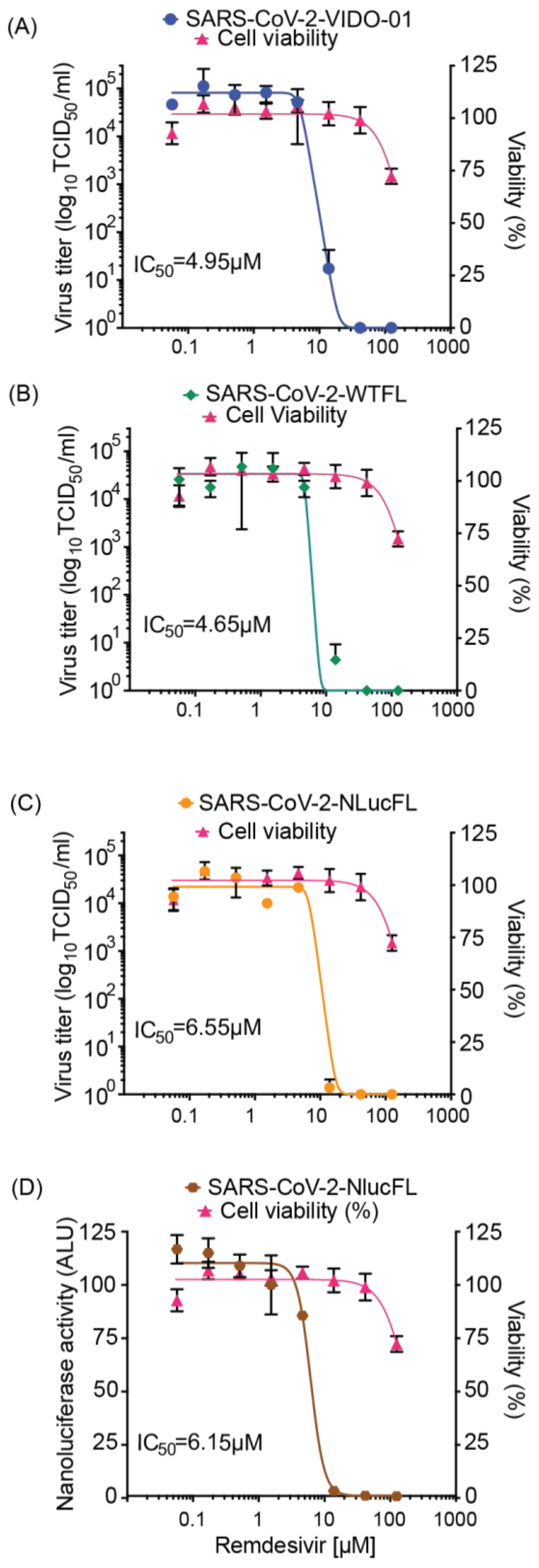
In this study, we report the generation of virus clones and cell lines as replication tools to study SARS-CoV-2 virus–host interactions and rapidly screen for antiviral agents. We have successfully used reverse genetics to generate wild type and reporter SARS-CoV-2 viruses. Using synthetic cDNA clones of SARS-CoV-2 sequences followed by in vitro RNA transcription, we recovered wild type and recombinant viruses in Vero76 cells and successfully characterized them. We compared the wild type full-length clone (WTFL) and the Nluc expressing reporter clone (NlucFL) with the SARS-CoV-2 VIDO-01 clinical isolate, and the resulting CPE, plaque morphology, and replication kinetics between the three viruses were found to be comparable (
Figure 1B,D). This highlights that the virus obtained from molecular clones recapitulates the replication properties of the wild type clinical isolate and can be used as equivalent to clinical isolates for in vitro studies on SARS-CoV-2. Our system differs from others in that it requires us to simply order fully assembled SARS-CoV-2 genomes which were used to generate viruses, without having to amplify the DNA in bacteria or assemble multiple fragments as is required for other SARS-CoV-2 reverse genetic systems [
4,
8,
15,
16]. This strategy is beneficial during pandemics because it allows virology labs to generate wild type, mutant, or reporter viruses quickly using only sequence information.
We have also confirmed the potential to use the reporter virus to study virus replication kinetics and drug screens. NLuc expression during replication of the reporter virus SARS-CoV-2 NLucFL corresponded to its replication kinetics, indicating that the luciferase assay can be used as a proxy system to detect virus replication (
Figure 1E). Furthermore, the use of a reporter virus reduces the turnaround time of virus detection by simply assessing luciferase values from five days to 24–48 h. The NLuc signals, even at low MOIs, were robust and stable; however, this also led the signal to bleed in neighboring wells. This limitation was overcome by including appropriate mock-infected controls.
In addition, we have confirmed the potential to use the reporter virus in high throughput drug screens through proof-of-principle analysis of inhibition by remdesivir. The design of our reporter construct was based on similar NLuc SARS-CoV-2 viruses used for rapid screening of antiviral drugs effective against COVID-19 [
8]. We have assessed and optimized the use of our molecular clones for use in high throughput drug screens and confirmed results similar to ones conducted in parallel using a clinical virus isolate (VIDO-01) when treated with remdesivir in a dose-dependent manner. The resulting EC
50 of remdesivir for VIDO-01 and WTFL were comparable at 4.95 and 4.65 µM, respectively. Similarly, the EC
50 for NLucFL using titrations was calculated as 6.55 µM, whereas using the NLuc assay, it was 6.15 µM. In conclusion, we successfully optimized the antiviral reporter assay using the NLucFL virus. Moreover, this further emphasized that molecular clones can be effectively used as efficient counterparts to clinical isolates for testing drugs.
Since working in biosafety level (BSL) 3 facilities to study SARS-CoV-2 is expensive, cumbersome, and sometimes not available, we aimed to design a replication assay system that can be used in conventional BSL2 facilities. We designed the SARS-CoV-2 sub-genomic replicon (SGR) based on previous SGR constructs used to study and discover inhibitors for HCV and SARS-CoV [
9,
17]. While the SGR showed evidence of replication based on an increase in luciferase expression over time which peaked at 24 h, the replication assays were hindered by replicon-induced CPE, leading to a rapid decline in luciferase counts. Similar luciferase expression patterns were observed in both Vero76 and Huh7.5 cells, but in general, the results using the replicon system were inconsistent. The inconsistent luciferase numbers could be a result of varying viable RNA yields and quality obtained from in vitro transcription. Thus, although accessible as a BSL2 system, the transient assays were found to be inefficient and difficult to reproduce. Furthermore, we were unable to make stably expressing cell lines in Vero76 and Huh7.5 cells through selection with G418. In another report, the successful selection of a BHK21 cell line stably harboring autonomously replicating SARS-CoV-2 replicon RNA replicon cells required two attenuating mutations in NSP1 to reduce virus-induced cellular toxicity [
18]. Other strategies to improve the ease of use of replicon systems include the use of a CMV promoter to remove the need for the in vitro transcription step and the generation of virus particles through trans-complementation that can be used for a single round of replication assays [
19].
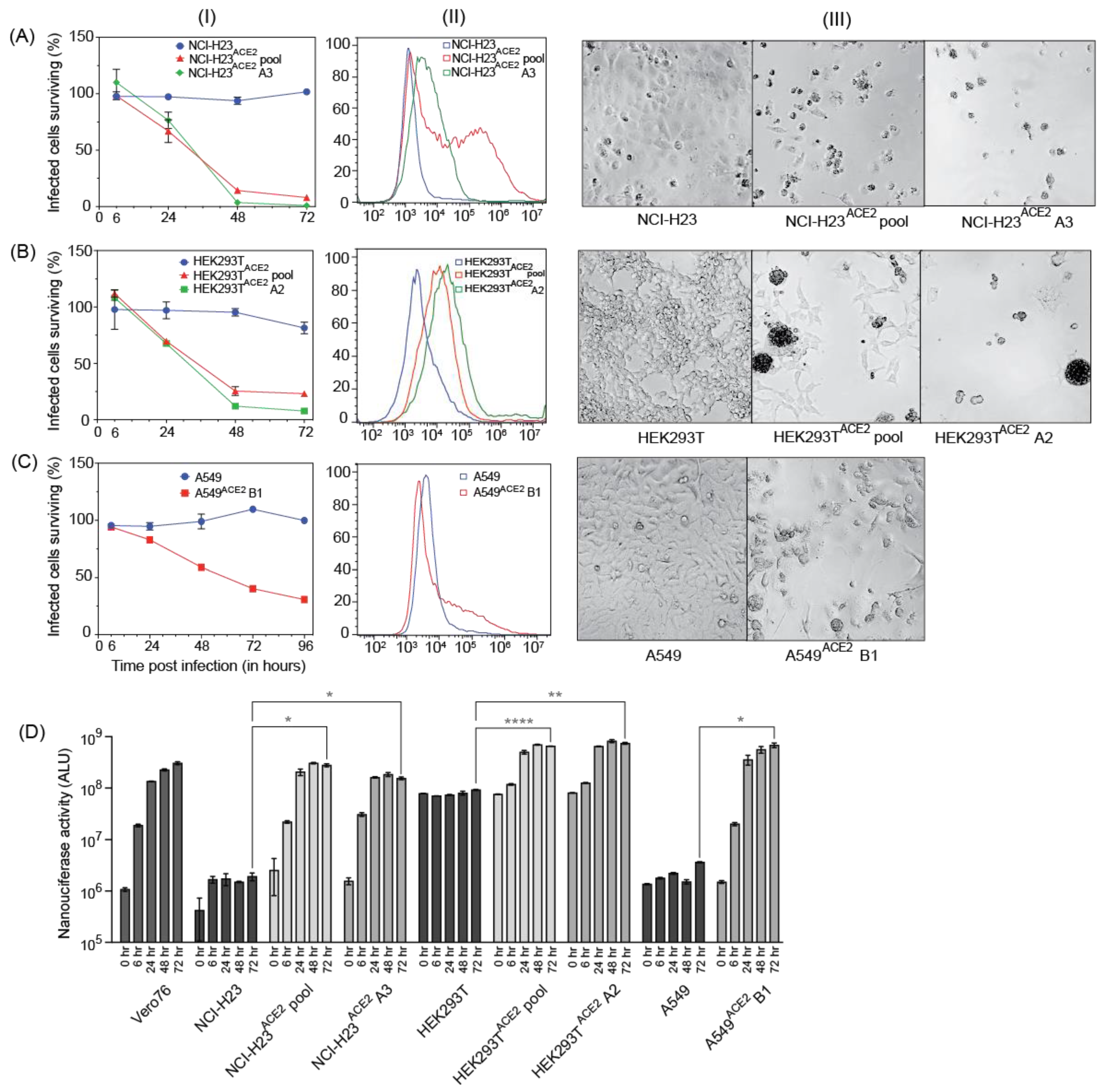
In addition, we have generated novel human lung cell lines that can be used to evaluate SARS-CoV-2 replication, virus–host interactions, and screen drugs. Our goal was to generate cell lines that support SARS-CoV-2 and that are also readily killed by the infection so that they can be used in genetic screens that rely on live/dead selection. We also wished to identify other cell lines supporting SARS-CoV-2 replication with less CPE such that they might be amenable to the development of stable replicon cells. We tested several human lung cell lines since they are most likely to mimic virus–host interactions in the lung and possibly provide insight into mechanisms of lung pathogenesis. We also used HEK293T, a human kidney cell line that was permissive for SARS-CoV-2 replication as a positive control [
20], and A549, a commonly used lung cell line. All the cell lines tested required transduction with ACE2 to become permissive to SARS-CoV-2. Three cell lines, NCI-H23
ACE2, A549
ACE2, and HEK293T
ACE2, were notably successful in supporting virus infection while exhibiting more than >70% cell death post-infection (
Figure 3). Clones from these cell lines were also generated, and NCI-H23
ACE2 clone A3 exhibited nearly 100% CPE following infection, making it ideal for genetic screens such as CRISPR knockout and activation screens (
Figure 3(AI)). The HEK293T
ACE2 monoclonal cell line clone A2 (
Figure 3(BI)) displayed CPE of approximately 90% following infection with SARS-CoV-2. These cell lines also showed robust NLuc expression after infection with SARS-CoV-2 NLucFL (
Figure 3D). The monoclonal cell line A549
ACE2 clone B1 showed about 70% CPE on day 4 post-infection (
Figure 3(CI)) with SARS-CoV-2 VIDO-01 isolate as well as increasing NLuc expression with SARS-CoV-2 NLucFL infection (
Figure 3D). However, flow cytometry data suggest that these cells expressed less ACE2 (
Figure 3(CII)), and this may explain the delayed and less dramatic virus-induced cell death, but these cells could further be used for drug screening.
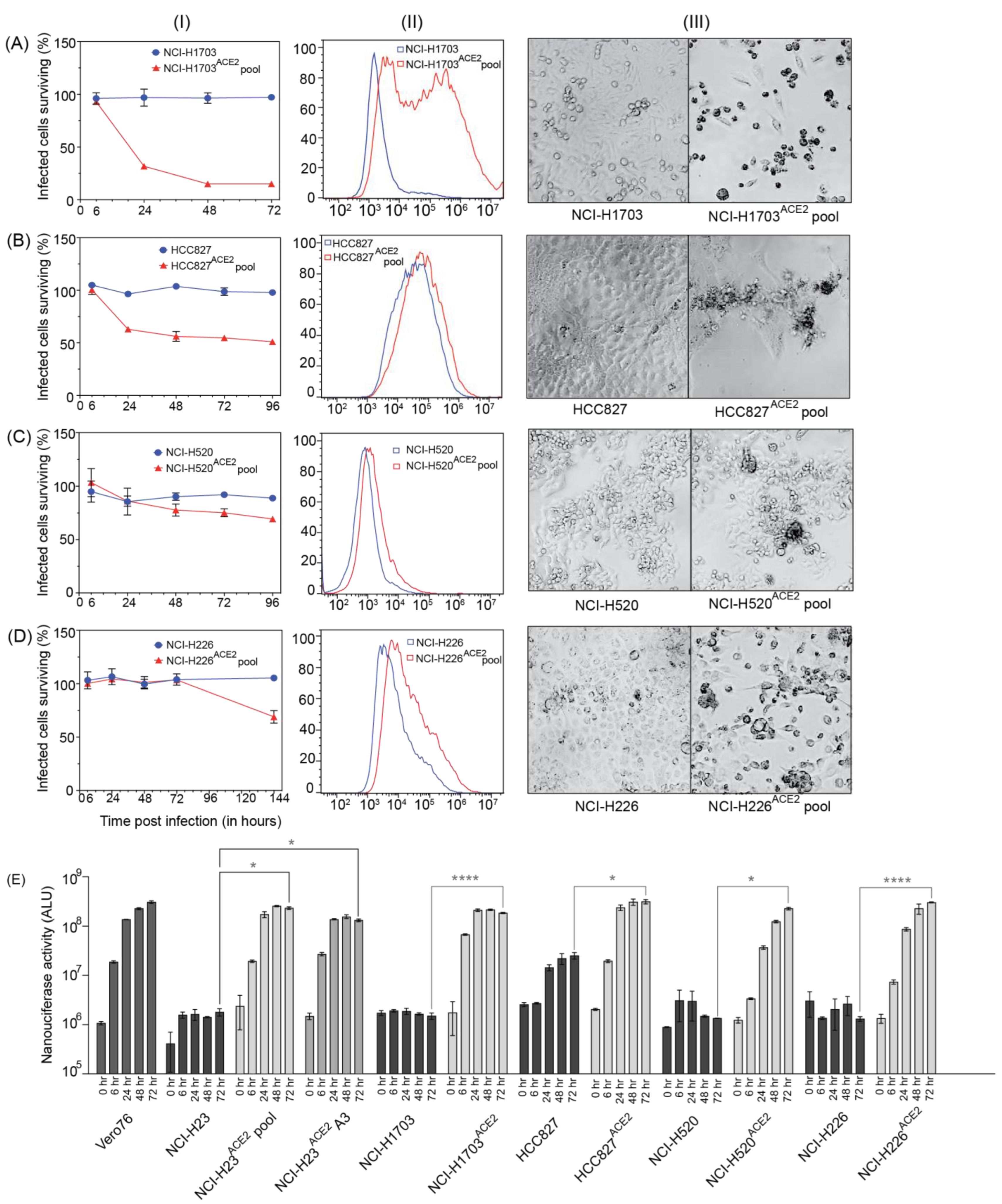
Transduction by ACE2 also allowed SARS-CoV-2 to infect NCI-H1703, NCI-H226, NCI-H520, and HCC827. Of these, NCI-H1703
ACE2 exhibited ~80% CPE. This cell line can be further looked at if another novel cell line is required to study SARS-CoV-2. Other cell lines transduced with ACE2 did not exhibit high CPE or overexpression of ACE2. Interestingly, these cell lines supported the growth of SARS-CoV-2 NLucFL assessed by NLuc expression compared to their untransduced counterparts (
Figure 4). These cell lines could be useful in studying infections leading to less CPE.
Thus, we have generated viruses and tools that are valuable for the study of SARS-CoV-2 virus–host interactions, and NCI-H23
ACE2 clone A3 has already been used to assess the effects of an inhibitor on virus replication [
14]. These molecular clones can be further extended to study the impact of specific mutations on the phenotypes of SARS-CoV-2 variants of concern.




 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




