
Omicron Waves in Argentina: Dynamics of SARS-CoV-2 Lineages BA.1, BA.2 and the Emerging BA.2.12.1 and BA.4/BA.5
 , , ,
, , ,  ,
,  , , , , , ,
, , , , , ,  , , , add
Show full author list
, , , add
Show full author list
Abstract
1. Introduction
2. Materials and Methods
2.1. Samples
2.2. Sequencing
2.3. Statistical Analysis
2.4. Phylogenetic Analysis
2.5. Phylodynamic Analysis
2.6. Ethics Statement
3. Results
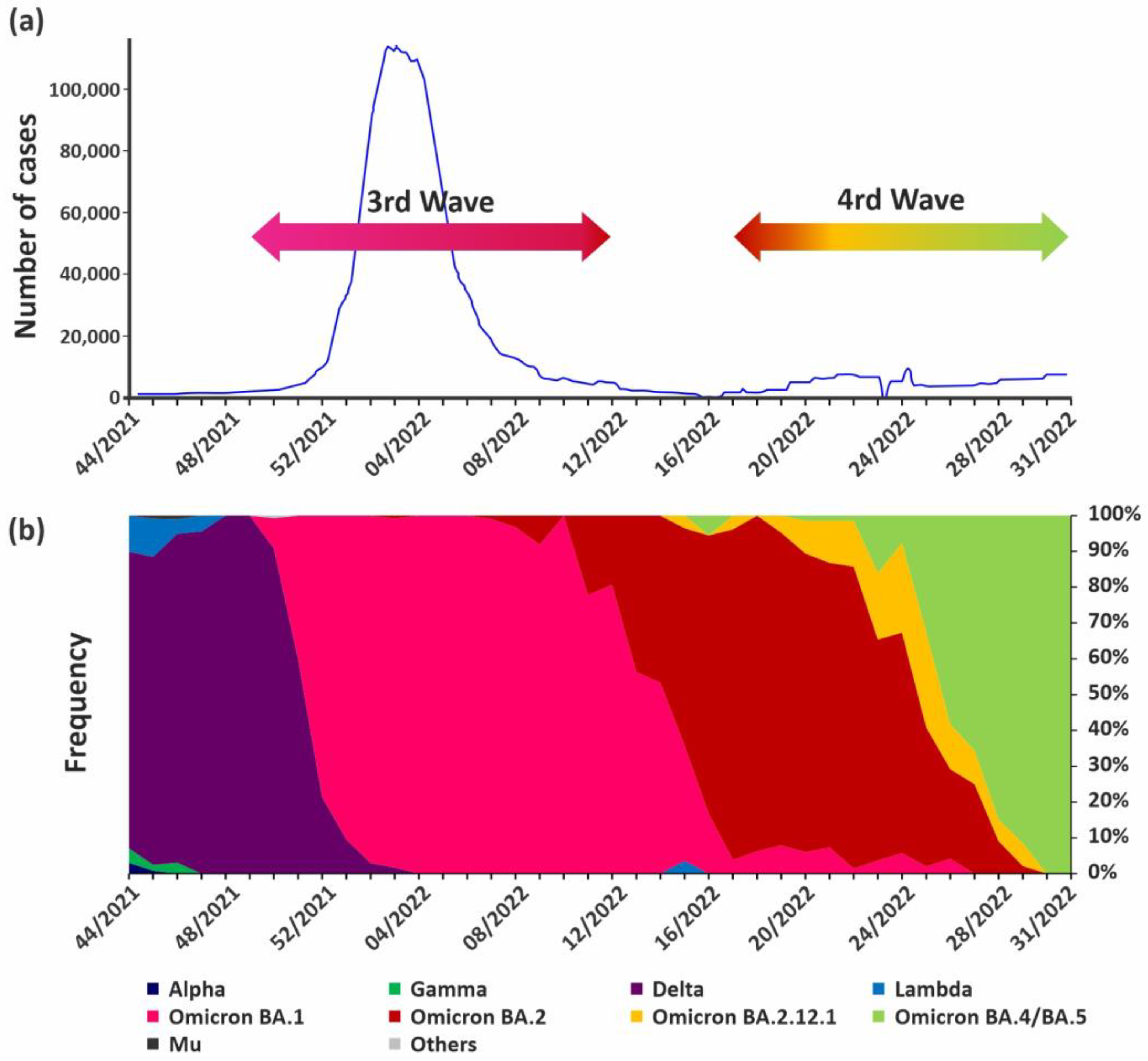
3.1. Molecular Surveillance of SARS-CoV-2 Variants in Argentina
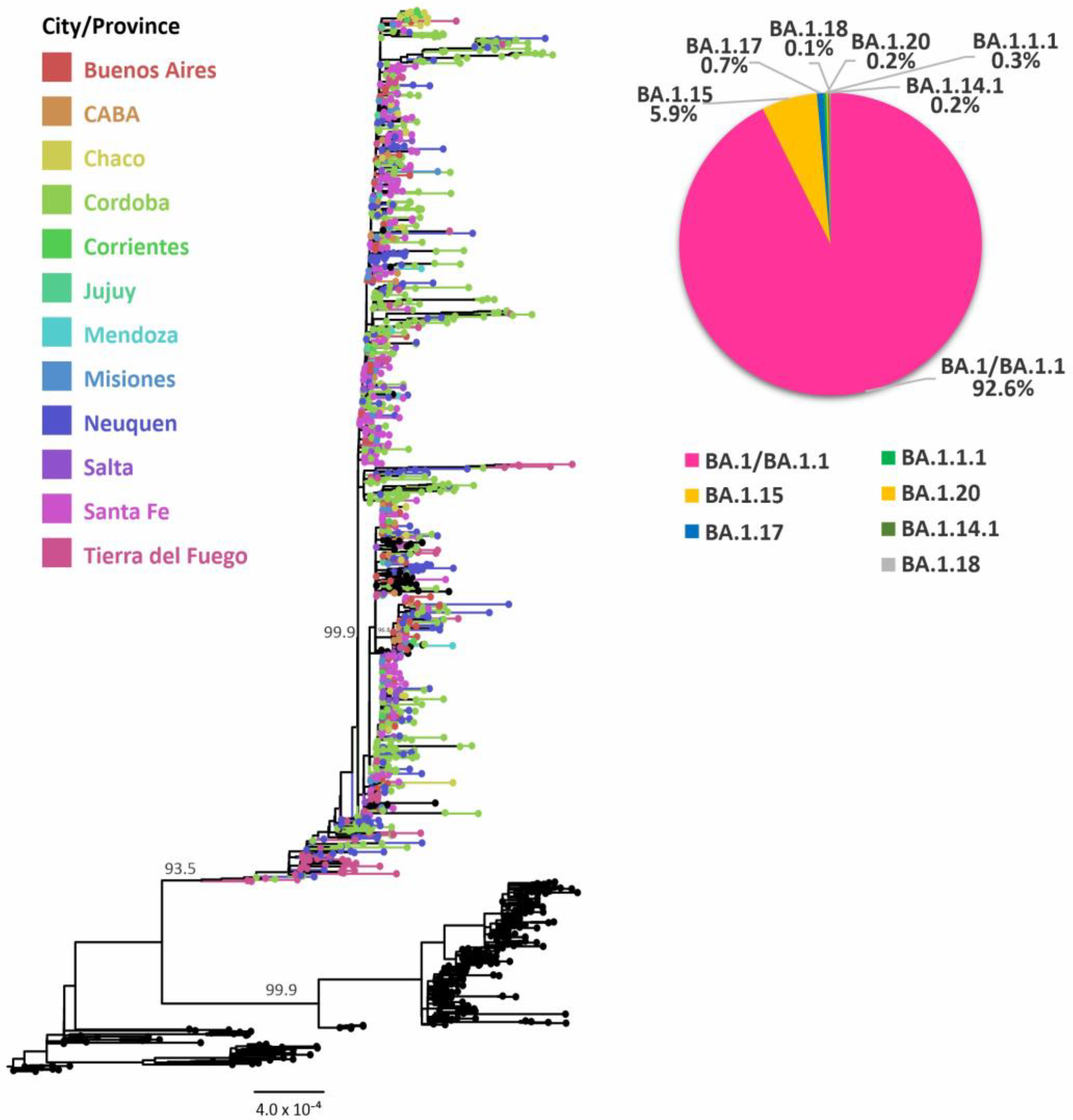
3.2. Evolutionary Analyses of Omicron BA.1 and its Sublineages
3.3. Evolutionary Analyses of Omicron BA.2 and its Sublineages
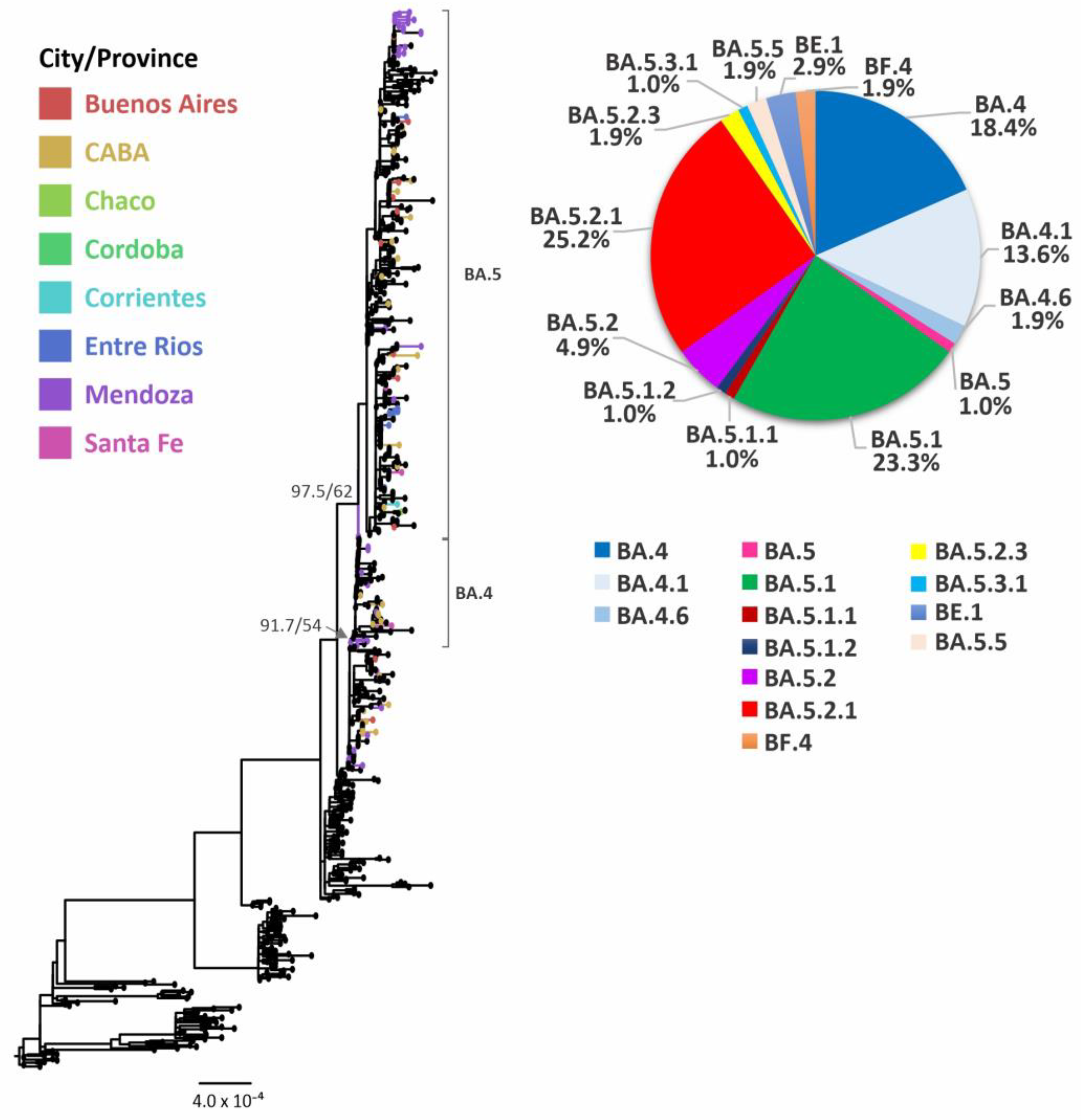
3.4. Evolutionary Analyses of Omicron BA.4/BA.5 and its Sublineages
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- World Health Organization Tracking SARS-CoV-2 Variants. Available online: https://www.who.int/en/activities/tracking-SARS-CoV-2-variants/ (accessed on 1 December 2022).
- Rambaut, A.; Holmes, E.C.; O’Toole, Á.; Hill, V.; McCrone, J.T.; Ruis, C.; du Plessis, L.; Pybus, O.G. A Dynamic Nomenclature Proposal for SARS-CoV-2 Lineages to Assist Genomic Epidemiology. Nat. Microbiol. 2020, 5, 1403–1407. [Google Scholar] [CrossRef] [PubMed]
- Webster, H.H.; Nyberg, T.; Sinnathamby, M.A.; Aziz, N.A.; Ferguson, N.; Seghezzo, G.; Blomquist, P.B.; Bridgen, J.; Chand, M.; Groves, N.; et al. Hospitalisation and Mortality Risk of SARS-COV-2 Variant Omicron Sub-Lineage BA.2 Compared to BA.1 in England. Nat. Commun. 2022, 13, 6053. [Google Scholar] [CrossRef] [PubMed]
- Strasser, Z.H.; Greifer, N.; Hadavand, A.; Murphy, S.N.; Estiri, H. Estimates of SARS-CoV-2 Omicron BA.2 Subvariant Severity in New England. JAMA Netw. Open 2022, 5, e2238354. [Google Scholar] [CrossRef] [PubMed]
- Kirsebom, F.C.M.; Andrews, N.; Stowe, J.; Toffa, S.; Sachdeva, R.; Gallagher, E.; Groves, N.; O’Connell, A.M.; Chand, M.; Ramsay, M.; et al. COVID-19 Vaccine Effectiveness against the Omicron (BA.2) Variant in England. Lancet Infect. Dis. 2022, 22, 931–933. [Google Scholar] [CrossRef] [PubMed]
- Khan, K.; Karim, F.; Ganga, Y.; Bernstein, M.; Jule, Z.; Reedoy, K.; Cele, S.; Lustig, G.; Amoako, D.; Wolter, N.; et al. Omicron BA.4/BA.5 Escape Neutralizing Immunity Elicited by BA.1 Infection. Nat. Commun. 2022, 13, 4686. [Google Scholar] [CrossRef]
- Cao, Y.; Yisimayi, A.; Jian, F.; Song, W.; Xiao, T.; Wang, L.; Du, S.; Wang, J.; Li, Q.; Chen, X.; et al. BA.2.12.1, BA.4 and BA.5 Escape Antibodies Elicited by Omicron Infection. Nature 2022, 608, 593–602. [Google Scholar] [CrossRef]
- World Health Organization. COVID-19 Weekly Epidemiological Update. Edition 125 Published 11 January 2023. Available online: https://www.who.int/publications/m/item/weekly-epidemiological-update-on-covid-19---11-january-2023 (accessed on 1 December 2022).
- Khare, S.; Gurry, C.; Freitas, L.; Schultz, M.B.; Bach, G.; Diallo, A.; Akite, N.; Ho, J.; Lee, R.T.C.; Yeo, W.; et al. GISAID’s Role in Pandemic Response. China CDC Wkly. 2021, 3, 1049–1051. [Google Scholar] [CrossRef]
- Proyecto PAIS. Proyecto Argentino Interinstitucional de Genómica de SARS-CoV-2. 2022. Available online: http://pais.qb.fcen.uba.ar/about.php (accessed on 1 December 2022).
- Paden, C.; Tao, Y.; Queen, K.; Zhang, J.; Li, Y.; Uehara, A.; Tong, S. Rapid, Sensitive, Full-Genome Sequencing of Severe Acute Respiratory Syndrome Coronavirus 2. Emerg. Infect. Dis. J. 2020, 26, 2401. [Google Scholar] [CrossRef]
- Torres, C.; Mojsiejczuk, L.; Acuña, D.; Alexay, S.; Amadio, A.; Aulicino, P.; Debat, H.; Fay, F.; Fernández, F.; Giri, A.A.; et al. Cost-Effective Method to Perform SARS-CoV-2 Variant Surveillance: Detection of Alpha, Gamma, Lambda, Delta, Epsilon, and Zeta in Argentina. Front. Med. 2021, 8, 755463. [Google Scholar] [CrossRef]
- Quick, J. NCoV-2019 Sequencing Protocol v3 (LoCost) V.3. Available online: https://www.protocols.io/view/ncov-2019-sequencing-protocol-v3-locost-bh42j8ye (accessed on 1 December 2022).
- Freed, N.; Silander, O. SARS-CoV2 Genome Sequencing Protocol (1200 bp Amplicon “Midnight” Primer Set, Using Nanopore Rapid Kit) V.6. Available online: https://www.protocols.io/view/sars-cov2-genome-sequencing-protocol-1200bp-amplic-rm7vz8q64vx1/v6 (accessed on 1 December 2022).
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [PubMed]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; Von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast Model Selection for Accurate Phylogenetic Estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing the Performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef]
- Drummond, A.J.; Suchard, M.A.; Xie, D.; Rambaut, A. Bayesian Phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 2012, 29, 1969–1973. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A.; Lam, T.T.; Carvalho, L.M.; Pybus, O.G. Exploring the Temporal Structure of Heterochronous Sequences Using TempEst (Formerly Path-O-Gen). Virus Evol. 2016, 2, vew007. [Google Scholar] [CrossRef] [PubMed]
- Viana, R.; Moyo, S.; Amoako, D.G.; Tegally, H.; Scheepers, C.; Althaus, C.L.; Anyaneji, U.J.; Bester, P.A.; Boni, M.F.; Chand, M.; et al. Rapid Epidemic Expansion of the SARS-CoV-2 Omicron Variant in Southern Africa. Nature 2022, 603, 679–686. [Google Scholar] [CrossRef] [PubMed]
- Tsui, J.L.-H.; Lambert, B.; Bajaj, S.; McCrone, J.T.; Inward, R.P.D.; Bosetti, P.; Hill, V.; Pena, R.E.; Zarebski, A.E.; Peacock, T.P.; et al. Genomic Assessment of Invasion Dynamics of SARS-CoV-2 Omicron BA.1. medRxiv 2023. [Google Scholar] [CrossRef]
- Zambrana Montaño, R.; Culasso, A.C.A.; Fernández, F.; Marquez, N.; Debat, H.; Salmerón, M.; Zamora, A.M.; Ruíz de Huidobro, G.; Costas, D.; Alabarse, G.; et al. Evolution of SARS-CoV-2 during the First Year of the COVID-19 Pandemic in Northwestern Argentina. Virus Res. 2023, 323, 198936. [Google Scholar] [CrossRef] [PubMed]
- Grabowski, F.; Kochańczyk, M.; Lipniacki, T. The Spread of SARS-CoV-2 Variant Omicron with a Doubling Time of 2.0–3.3 Days Can Be Explained by Immune Evasion. Viruses 2022, 14, 294. [Google Scholar] [CrossRef]
- Rearte, A.; Castelli, J.M.; Rearte, R.; Fuentes, N.; Pennini, V.; Pesce, M.; Barbeira, P.B.; Iummato, L.E.; Laurora, M.; Bartolomeu, M.L.; et al. Effectiveness of RAd26-RAd5, ChAdOx1 NCoV-19, and BBIBP-CorV Vaccines for Risk of Infection with SARS-CoV-2 and Death Due to COVID-19 in People Older than 60 Years in Argentina: A Test-Negative, Case-Control, and Retrospective Longitudinal Study. Lancet 2022, 399, 1254–1264. [Google Scholar] [CrossRef]
- Our World in Data. Vaccination in Argentina. Available online: https://ourworldindata.org/covid-vaccinations (accessed on 1 December 2022).
- UK Health Security Agency. SARS-CoV-2 Variants of Concern and Variants under Investigation in England: Technical Briefing 42; UK Health Security Agency: London, UK, 2022.
- Akerman, A.; Milogiannakis, V.; Jean, T.; Esneu, C.; Silva, M.R.; Ison, T.; Fitcher, C.; Lopez, J.A.; Chandra, D.; Naing, Z.; et al. Emergence and Antibody Evasion of BQ and BA.2.75 SARS-CoV-2 Sublineages in the Face of Maturing Antibody Breadth at the Population Level. medRxiv 2022. [Google Scholar] [CrossRef]
- Uraki, R.; Ito, M.; Furusawa, Y.; Yamayoshi, S.; Iwatsuki-Horimoto, K.; Adachi, E.; Saito, M.; Koga, M.; Tsutsumi, T.; Yamamoto, S.; et al. Humoral Immune Evasion of the Omicron Subvariants BQ.1.1 and XBB. Lancet. Infect. Dis. 2022, 23, 2022–2024. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lineage | Rate of Evolution [s/s/y] (HPD95%) | Ancestral Date (HPD95%) | Doubling Time [days] (HPD95%) | Exponential Growth Rate [days−1] (HPD95%) |
|---|---|---|---|---|
| BA.1 | 9.3 × 10−4 * (6.9 × 10−4–1.2 × 10−3) | 12 November 2021 (25 October–25 November) | 3.3 (1.6–5.4) | 0.213 (0.127–0.441) |
| BA.2 | 1.0 × 10−3 * (8.7 × 10−4–1.1 × 10−3) | 3 February 2022 (11 January–22 February) | 9.9 (7.3–13.2) | 0.070 (0.053–0.096) |
| 5.5 × 10−4 (3.1 × 10−4–7.7 × 10−4) | 24 November 2021 (23 August –26 January) | 16.5 (9.5–26.4) | 0.042 (0.026–0.073) | |
| BA.4/BA.5 | 8.5 × 10−4 * (6.4 × 10−4–1.0 × 10−3) | 25 March 2022 (15 February–24 April ) | 10.8 (6.1–17.3) | 0.064 (0.040–0.114) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Torres, C.; Nabaes Jodar, M.; Acuña, D.; Montaño, R.M.Z.; Culasso, A.C.A.; Amadio, A.F.; Aulicino, P.; Ceballos, S.; Cacciabue, M.; Debat, H.; et al. Omicron Waves in Argentina: Dynamics of SARS-CoV-2 Lineages BA.1, BA.2 and the Emerging BA.2.12.1 and BA.4/BA.5. Viruses 2023, 15, 312. https://doi.org/10.3390/v15020312
Torres C, Nabaes Jodar M, Acuña D, Montaño RMZ, Culasso ACA, Amadio AF, Aulicino P, Ceballos S, Cacciabue M, Debat H, et al. Omicron Waves in Argentina: Dynamics of SARS-CoV-2 Lineages BA.1, BA.2 and the Emerging BA.2.12.1 and BA.4/BA.5. Viruses. 2023; 15(2):312. https://doi.org/10.3390/v15020312
Chicago/Turabian StyleTorres, Carolina, Mercedes Nabaes Jodar, Dolores Acuña, Romina Micaela Zambrana Montaño, Andrés Carlos Alberto Culasso, Ariel Fernando Amadio, Paula Aulicino, Santiago Ceballos, Marco Cacciabue, Humberto Debat, and et al. 2023. "Omicron Waves in Argentina: Dynamics of SARS-CoV-2 Lineages BA.1, BA.2 and the Emerging BA.2.12.1 and BA.4/BA.5" Viruses 15, no. 2: 312. https://doi.org/10.3390/v15020312
APA StyleTorres, C., Nabaes Jodar, M., Acuña, D., Montaño, R. M. Z., Culasso, A. C. A., Amadio, A. F., Aulicino, P., Ceballos, S., Cacciabue, M., Debat, H., Dus Santos, M. J., Eberhardt, M. F., Espul, C., Fay, F., Fernández, M. A., Fernández, F., Muñoz, J. M. F., Ferrini, F., Gallego, F., ... Viegas, M. (2023). Omicron Waves in Argentina: Dynamics of SARS-CoV-2 Lineages BA.1, BA.2 and the Emerging BA.2.12.1 and BA.4/BA.5. Viruses, 15(2), 312. https://doi.org/10.3390/v15020312