

Molecular Epidemiology of SARS-CoV-2: The Dominant Role of Arginine in Mutations and Infectivity

 , , ,
, , ,  ,
,  ,
,  ,
,  and
and 
Abstract
1. Introduction
2. Materials and Methods
3. Results
3.1. Variant Distribution across EU/EEA Countries

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Country | Weeks of Data 1 | Data Source 2,3 | Number of Cases | Volume of Sequencing or Genotyping for Variant Detection | Total Known Variants Detected | BA.5 | BQ.1 | BA.2.75 | BA.4 | BA.2 | Other | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| n | % | Category | n | % | n | % | n | % | n | % | n | % | n | % | |||||
| Austria | 44–45 | GISAID | 54,210 | 4272 | 7.9 | L1a | 740 | 410 | 55.4 | 99 | 13.4 | 138 | 18.6 | 32 | 4.3 | 5 | 0.7 | 56 | 7.6 |
| Belgium | 44–45 | GISAID | 8953 | 235 | 2.6 | L2 | 235 | 122 | 51.9 | 91 | 38.7 | 7 | 3 | 2 | 0.9 | 5 | 2.1 | 8 | 3.4 |
| Bulgaria | 44 | TESSy | 2893 | 166 | 5.7 | L2 | 166 | 165 | 99.4 | 1 | 0.6 | ||||||||
| Croatia | GISAID | 0 | 0 | 0 | No data | 0 | |||||||||||||
| Cyprus | TESSy | 0 | 0 | 0 | No data | 0 | |||||||||||||
| Czechia | 44–45 | GISAID | 12,317 | 53 | 0.4 | L2 | 53 | 48 | 90.6 | 4 | 7.5 | 1 | 1.9 | ||||||
| Denmark | 44–45 | TESSy | 8450 | 5629 | 66.6 | L1a | 5629 | 3023 | 53.7 | 1852 | 32.9 | 560 | 9.9 | 57 | 1 | 137 | 2.4 | ||
| Estonia | 44–45 | TESSy | 1043 | 608 | 58.3 | L1b | 608 | 579 | 95.2 | 11 | 1.8 | 5 | 0.8 | 12 | 2 | 1 | 0.2 | ||
| Finland | GISAID | 0 | 0 | 0 | No data | 0 | |||||||||||||
| France | 44–45 | GISAID | 305,759 | 866 | 0.3 | L1c | 861 | 394 | 45.8 | 410 | 47.6 | 8 | 0.9 | 3 | 0.3 | 32 | 3.7 | 14 | 1.6 |
| Germany | 44–45 | TESSy | 443,282 | 4661 | 1.1 | L1a | 4661 | 4275 | 91.7 | 151 | 3.2 | 182 | 3.9 | 53 | 1.1 | ||||
| Greece | 44–45 | TESSy | 87,883 | 387 | 0.4 | L2 | 387 | 271 | 70 | 40 | 10.3 | 42 | 10.9 | 8 | 2.1 | 4 | 1 | 22 | 5.7 |
| Hungary | 45 | TESSy | 4431 | 168 | 3.8 | L2 | 168 | 165 | 98.2 | 1 | 0.6 | 2 | 1.2 | ||||||
| Iceland | 44–45 | GISAID | 615 | 145 | 23.6 | L2 | 145 | 51 | 35.2 | 62 | 42.8 | 19 | 13.1 | 7 | 4.8 | 6 | 4.1 | ||
| Ireland | 44–45 | TESSy | 3315 | 188 | 5.7 | L2 | 188 | 71 | 37.8 | 94 | 50 | 10 | 5.3 | 4 | 2.1 | 5 | 2.7 | 4 | 2.1 |
| Italy | 44–45 | GISAID | 298,878 | 1300 | 0.4 | L1b | 1290 | 835 | 64.7 | 373 | 28.9 | 27 | 2.1 | 12 | 0.9 | 22 | 1.7 | 21 | 1.6 |
| Latvia | 44–45 | TESSy | 5415 | 1086 | 20.1 | L1b | 1086 | 1072 | 98.7 | 5 | 0.5 | 5 | 0.5 | 3 | 0.3 | 1 | 0.1 | ||
| Liechtenstein | 44–45 | GISAID | 139 | 24 | 17.3 | L2 | 24 | 8 | 33.3 | 7 | 29.2 | 9 | 37.5 | ||||||
| Lithuania | GISAID | 0 | 0 | 0 | No data | 0 | |||||||||||||
| Luxembourg | 44–45 | TESSy | 2809 | 667 | 23.7 | L1c | 667 | 319 | 47.8 | 276 | 41.4 | 1 | 0.1 | 17 | 2.5 | 54 | 8.1 | ||
| Malta | TESSy | 0 | 0 | 0 | No data | 0 | |||||||||||||
| The Netherlands | 44 | TESSy | 8226 | 701 | 8.5 | L1b | 701 | 430 | 61.3 | 186 | 26.5 | 46 | 6.6 | 7 | 1 | 20 | 2.9 | 12 | 1.7 |
| Norway | 44–45 | TESSy | 1786 | 105 | 5.9 | L2 | 105 | 64 | 61 | 29 | 27.6 | 8 | 7.6 | 1 | 1 | 3 | 2.9 | ||
| Poland | 44–45 | GISAID | 5964 | 59 | 1 | L2 | 59 | 57 | 96.6 | 1 | 1.7 | 1 | 1.7 | ||||||
| Portugal | 44–45 | TESSy | 11,310 | 242 | 2.1 | L2 | 242 | 225 | 93 | 14 | 5.8 | 3 | 1.2 | ||||||
| Romania | 44–45 | TESSy | 4995 | 189 | 3.8 | L2 | 189 | 154 | 81.5 | 1 | 0.5 | 3 | 1.6 | 2 | 1.1 | 1 | 0.5 | ||
| Slovakia | TESSy | 0 | 0 | 0 | No data | 0 | |||||||||||||
| Slovenia | 44 | GISAID | 4565 | 9 | 0.2 | L2 | 9 | 8 | 88.9 | 1 | 11.1 | ||||||||
| Spain | 44–45 | GISAID | 37,601 | 377 | 1 | L2 | 374 | 128 | 34.2 | 225 | 60.2 | 17 | 4.5 | 1 | 0.3 | 3 | 0.8 | ||
| Sweden | 44–45 | GISAID | 7151 | 756 | 10.6 | L1c | 756 | 461 | 61 | 233 | 30.8 | 19 | 2.5 | 4 | 0.5 | 14 | 1.9 | 25 | 3.3 |
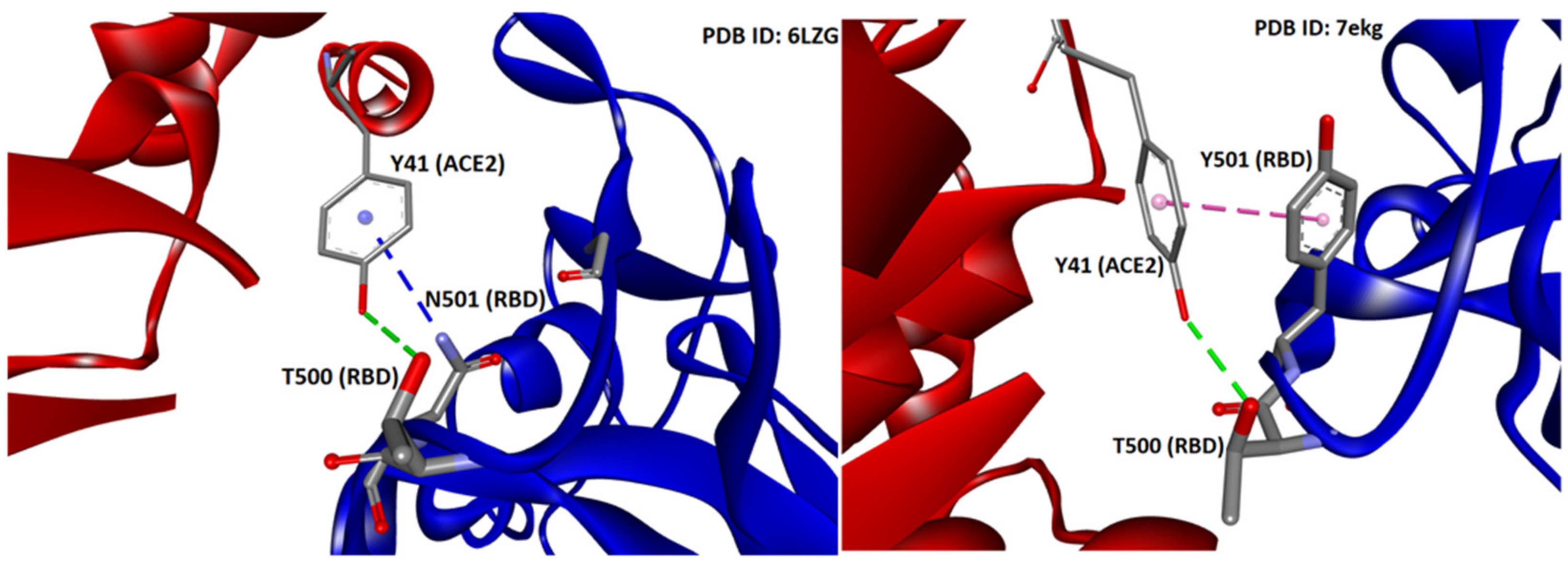
3.2. Arginine Mutations Stabilize Best the Conformation of S-Protein and the Complex with ACE2
4. Discussion
4.1. Variant Distribution across EU/EEA Countries
4.2. Polybasic Cleavage Sites of the SARS-CoV-2 S-Protein and 3CLpro Inhibition
4.3. Critical Mutations
4.3.1. The Triple Mutation E484K, K417N, N501Y (UK Variant)
4.3.2. The Triple Mutation L452R, E484Q, P681R (Indian Variant)
4.3.3. The E484K Mutation (UK, South Africa, and Brazil Variants)
4.3.4. Comparative Analysis of the E484Q and E484K Mutations
4.3.5. The K417N Mutation (South Africa Variant)
4.3.6. The P681H Mutation (Nigerian Variant) and the P681R (Indian Variant) in the Vicinity of the S1/S2 Spike Fusion Region
4.3.7. The D614G (India, South Africa, and Brazil Variants) and D614R Mutations
4.3.8. The R346T Mutation in Omicron Subvariants BQ.1 and BA.4.6
4.4. The Role of Tyrosine in Stabilizing Network Systems
4.4.1. The Example of AT1R/ARBs
4.4.2. The Role of Tyrosine in Triggering the Hypertensive Activity of Angiotensin II and Possibly the Proinflammatory Cytokine Storm in COVID-19
4.4.3. pi–pi Interactions Enhance Binding Affinity
4.4.4. RAS and ACE2 Are Targets for COVID-19 Antiviral Drugs
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Tian, D.; Nie, W.; Sun, Y.; Ye, Q. The Epidemiological Features of the SARS-CoV-2 Omicron Subvariant BA.5 and Its Evasion of the Neutralizing Activity of Vaccination and Prior Infection. Vaccines 2022, 10, 1699. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Yisimayi, A.; Jian, F.; Song, W.; Xiao, T.; Wang, L.; Du, S.; Wang, J.; Li, Q.; Chen, X.; et al. BA.2.12.1, BA.4 and BA.5 escape antibodies elicited by Omicron infection. Nature 2022, 608, 593–602. [Google Scholar] [CrossRef] [PubMed]
- Hachmann, N.P.; Miller, J.; Collier, A.-R.Y.; Barouch, D.H. Neutralization Escape by SARS-CoV-2 Omicron Subvariant BA.4.6. N. Engl. J. Med. 2022, 387, 1904–1906. [Google Scholar] [CrossRef] [PubMed]
- Fowler, P.W.; Moore, G.J. Calculation of the magnitude and orientation of electrostatic interactions between small aromatic rings in peptides and proteins: Implications for angiotensin II. Biochem. Biophys. Res. Commun. 1988, 153, 1296–1300. [Google Scholar] [CrossRef]
- Cherian, S.; Potdar, V.; Jadhav, S.; Yadav, P.; Gupta, N.; Das, M.; Rakshit, P.; Singh, S.; Abraham, P.; Panda, S.; et al. SARS-CoV-2 Spike Mutations, L452R, T478K, E484Q and P681R, in the Second Wave of COVID-19 in Maharashtra, India. Microorganisms 2021, 9, 1542. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, J.; Plante, K.S.; Plante, J.A.; Xie, X.; Zhang, X.; Ku, Z.; An, Z.; Scharton, D.; Schindewolf, C.; et al. The N501Y spike substitution enhances SARS-CoV-2 infection and transmission. Nature 2021, 602, 294–299. [Google Scholar] [CrossRef]
- Ali, F.; Kasry, A.; Amin, M. The new SARS-CoV-2 strain shows a stronger binding affinity to ACE2 due to N501Y mutant. Med. Drug Discov. 2021, 10, 100086. [Google Scholar] [CrossRef]
- Fratev, F. N501Y and K417N Mutations in the Spike Protein of SARS-CoV-2 Alter the Interactions with Both hACE2 and Human-Derived Antibody: A Free Energy of Perturbation Retrospective Study. J. Chem. Inf. Model. 2021, 61, 6079–6084. [Google Scholar] [CrossRef]
- Nelson, G.; Buzko, O.; Spilman, P.; Niazi, K.; Rabizadeh, S.; Soon-Shiong, P. Molecular dynamic simulation reveals E484K mutation enhances spike RBD-ACE2 affinity and the combination of E484K, K417N and N501Y mutations (501Y.V2 variant) induces conformational change greater than N501Y mutant alone, potentially resulting in an escape mutant. bioRxiv 2021. [Google Scholar] [CrossRef]
- Luan, B.; Wang, H.; Huynh, T. Enhanced binding of the N501Y-mutated SARS-CoV-2 spike protein to the human ACE2 receptor: Insights from molecular dynamics simulations. FEBS Lett. 2021, 595, 1454–1461. [Google Scholar] [CrossRef]
- Zhang, L.; Jackson, C.B.; Mou, H.; Ojha, A.; Peng, H.; Quinlan, B.D.; Rangarajan, E.S.; Pan, A.; Vanderheiden, A.; Suthar, M.S.; et al. SARS-CoV-2 spike-protein D614G mutation increases virion spike density and infectivity. Nat. Commun. 2020, 11, 6013. [Google Scholar] [CrossRef]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef]
- Ridgway, H.; Chasapis, C.T.; Kelaidonis, K.; Ligielli, I.; Moore, G.J.; Gadanec, L.K.; Zulli, A.; Apostolopoulos, V.; Mavromoustakos, T.; Matsoukas, J.M. Understanding the Driving Forces That Trigger Mutations in SARS-CoV-2: Mutational Energetics and the Role of Arginine Blockers in COVID-19 Therapy. Viruses 2022, 14, 1029. [Google Scholar] [CrossRef]
- Ridgway, H.; Moore, G.J.; Mavromoustakos, T.; Tsiodras, S.; Ligielli, I.; Kelaidonis, K.; Chasapis, C.T.; Gadanec, L.K.; Zulli, A.; Apostolopoulos, V.; et al. Discovery of a new generation of angiotensin receptor blocking drugs: Receptor mechanisms and in silico binding to enzymes relevant to SARS-CoV-2. Comput. Struct. Biotechnol. J. 2022, 20, 2091–2111. [Google Scholar] [CrossRef]
- Riley, K.E.; Hobza, P. On the Importance and Origin of Aromatic Interactions in Chemistry and Biodisciplines. Acc. Chem. Res. 2012, 46, 927–936. [Google Scholar] [CrossRef]
- Molčanov, K.; Kojić-Prodić, B. Towards understanding π-stacking interactions between non-aromatic rings. IUCrJ 2019, 6, 156–166. [Google Scholar] [CrossRef]
- Malenov, D.P.; Zarić, S.D. Stacking interactions of aromatic ligands in transition metal complexes. Coord. Chem. Rev. 2020, 419, 213338. [Google Scholar] [CrossRef]
- Chen, J.; Wang, R.; Gilby, N.B.; Wei, G.-W. Omicron Variant (B.1.1.529): Infectivity, Vaccine Breakthrough, and Antibody Resistance. J. Chem. Inf. Model. 2022, 62, 412–422. [Google Scholar] [CrossRef]
- Country Overview Report: 46th Week of 2022. Available online: https://www.ecdc.europa.eu/en/covid-19/country-overviews (accessed on 24 November 2022).
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2009, 2, 455–461. [Google Scholar] [CrossRef]
- Duan, Y.; Wu, C.; Chowdhury, S.; Lee, M.C.; Xiong, G.; Zhang, W.; Yang, R.; Cieplak, P.; Luo, R.; Lee, T.; et al. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J. Comput. Chem. 2003, 24, 1999–2012. [Google Scholar] [CrossRef]
- Arnautova, Y.A.; Jagielska, A.; Scheraga, H.A. A New Force Field (ECEPP-05) for Peptides, Proteins, and Organic Molecules. J. Phys. Chem. B 2006, 110, 5025–5044. [Google Scholar] [CrossRef] [PubMed]
- Arnautova, Y.A.; Vorobjev, Y.N.; Vila, J.A.; Scheraga, H.A. Identifying native-like protein structures with scoring functions based on all-atom ECEPP force fields, implicit solvent models and structure relaxation. Proteins Struct. Funct. Bioinform. 2009, 77, 38–51. [Google Scholar] [CrossRef] [PubMed]
- Krieger, E.; Koraimann, G.; Vriend, G. Increasing the precision of comparative models with YASARA NOVA-a self-parameterizing force field. Proteins Struct. Funct. Bioinform. 2002, 47, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Krieger, E.; Vriend, G. YASARA View—Molecular graphics for all devices—From smartphones to workstations. Bioinformatics 2014, 30, 2981–2982. [Google Scholar] [CrossRef] [PubMed]
- Krieger, E.; Nielsen, J.E.; Spronk, C.A.E.M.; Vriend, G. Fast empirical pKa prediction by Ewald summation. J. Mol. Graph. Model. 2006, 25, 481–486. [Google Scholar] [CrossRef]
- Krieger, E.; Dunbrack, R.L.; Hooft, R.W.W.; Krieger, B. Assignment of Protonation States in Proteins and Ligands: Combining pKa Prediction with Hydrogen Bonding Network Optimization; Springer: New York, NY, USA, 2012; Volume 819, pp. 405–421. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Jakalian, A.; Jack, D.B.; Bayly, C.I. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. J. Comput. Chem. 2002, 23, 1623–1641. [Google Scholar] [CrossRef]
- Hornak, V.; Abel, R.; Okur, A.; Strockbine, B.; Roitberg, A.; Simmerling, C. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins Struct. Funct. Bioinform. 2006, 65, 712–725. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Krieger, E.; Vriend, G. New ways to boost molecular dynamics simulations. J. Comput. Chem. 2015, 36, 996–1007. [Google Scholar] [CrossRef]
- Aleem, A.; Akbar Samad, A.B.; Slenker, A.K. Emerging Variants of SARS-CoV-2 And Novel Therapeutics Against Coronavirus (COVID-19). In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Tracking SARS-CoV-2 Variants. Available online: https://www.who.int/activities/tracking-SARS-CoV-2-variants (accessed on 24 November 2022).
- Chen, J.; Wang, R.; Hozumi, Y.; Liu, G.; Qiu, Y.; Wei, X.; Wei, G.W. Emerging dominant SARS-CoV-2 variants. arXiv 2022. [Google Scholar] [CrossRef] [PubMed]
- Spread of the SARS-CoV-2 Omicron Variant Sub-Lineage BQ.1 in the EU/EEA. Available online: https://www.ecdc.europa.eu/en/publications-data/spread-sars-cov-2-omicron-variant-sub-lineage-bq1-eueea (accessed on 24 November 2022).
- GISAID Database. Available online: https://gisaid.org/ (accessed on 24 November 2022).
- The European Surveillance System (TESSy). Available online: https://www.ecdc.europa.eu/en/publications-data/european-surveillance-system-tessy (accessed on 24 November 2022).
- Courvoisier, E.; Williams, P.A.; Lim, G.K.; Hughes, C.E.; Harris, K.D.M. The crystal structure of l-arginine. Chem. Commun. 2012, 48, 2761. [Google Scholar] [CrossRef]
- Marvin Sketch Software. Available online: https://chemaxon.com/marvin (accessed on 24 November 2022).
- Zhang, Y.; Zhang, L.; Wu, J.; Yu, Y.; Liu, S.; Li, T.; Li, Q.; Ding, R.; Wang, H.; Nie, J.; et al. A second functional furin site in the SARS-CoV-2 spike protein. Emerg. Microbes Infect. 2022, 11, 182–194. [Google Scholar] [CrossRef]
- Yu, S.; Zheng, X.; Zhou, B.; Li, J.; Chen, M.; Deng, R.; Wong, G.; Lavillette, D.; Meng, G. SARS-CoV-2 spike engagement of ACE2 primes S2′ site cleavage and fusion initiation. Proc. Natl. Acad. Sci. USA 2021, 119, e2111199119. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e278. [Google Scholar] [CrossRef]
- Kim, Y.; Jang, G.; Lee, D.; Kim, N.; Seon, J.W.; Kim, Y.-h.; Lee, C. Trypsin enhances SARS-CoV-2 infection by facilitating viral entry. Arch. Virol. 2022, 167, 441–458. [Google Scholar] [CrossRef]
- Essalmani, R.; Jain, J.; Susan-Resiga, D.; Andréo, U.; Evagelidis, A.; Derbali, R.M.; Huynh, D.N.; Dallaire, F.; Laporte, M.; Delpal, A.; et al. Distinctive Roles of Furin and TMPRSS2 in SARS-CoV-2 Infectivity. J. Virol. 2022, 96, e00128-22. [Google Scholar] [CrossRef]
- Xia, S.; Liu, M.; Wang, C.; Xu, W.; Lan, Q.; Feng, S.; Qi, F.; Bao, L.; Du, L.; Liu, S.; et al. Inhibition of SARS-CoV-2 (previously 2019-nCoV) infection by a highly potent pan-coronavirus fusion inhibitor targeting its spike protein that harbors a high capacity to mediate membrane fusion. Cell Res. 2020, 30, 343–355. [Google Scholar] [CrossRef]
- Jaimes, J.A.; André, N.M.; Chappie, J.S.; Millet, J.K.; Whittaker, G.R. Phylogenetic Analysis and Structural Modeling of SARS-CoV-2 Spike Protein Reveals an Evolutionary Distinct and Proteolytically Sensitive Activation Loop. J. Mol. Biol. 2020, 432, 3309–3325. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Pöhlmann, S. A Multibasic Cleavage Site in the Spike Protein of SARS-CoV-2 Is Essential for Infection of Human Lung Cells. Mol. Cell 2020, 78, 779–784.e775. [Google Scholar] [CrossRef] [PubMed]
- Mody, V.; Ho, J.; Wills, S.; Mawri, A.; Lawson, L.; Ebert, M.C.C.J.C.; Fortin, G.M.; Rayalam, S.; Taval, S. Identification of 3-chymotrypsin like protease (3CLPro) inhibitors as potential anti-SARS-CoV-2 agents. Commun. Biol. 2021, 4, 93. [Google Scholar] [CrossRef] [PubMed]
- García-Iriepa, C.; Hognon, C.; Francés-Monerris, A.; Iriepa, I.; Miclot, T.; Barone, G.; Monari, A.; Marazzi, M. Thermodynamics of the Interaction between the Spike Protein of Severe Acute Respiratory Syndrome Coronavirus-2 and the Receptor of Human Angiotensin-Converting Enzyme 2. Effects of Possible Ligands. J. Phys. Chem. Lett. 2020, 11, 9272–9281. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Wang, R.; Wang, M.; Wei, G.W. Mutations Strengthened SARS-CoV-2 Infectivity. J. Mol. Biol. 2020, 432, 5212–5226. [Google Scholar] [CrossRef]
- Muchtaridi, M.; Fauzi, M.; Khairul Ikram, N.K.; Mohd Gazzali, A.; Wahab, H.A. Natural Flavonoids as Potential Angiotensin-Converting Enzyme 2 Inhibitors for Anti-SARS-CoV-2. Molecules 2020, 25, 3980. [Google Scholar] [CrossRef]
- Peter, E.K.; Schug, A. The inhibitory effect of a coronavirus spike protein fragment with ACE2. Biophys. J. 2021, 120, 1001–1010. [Google Scholar] [CrossRef]
- Unni, S.; Aouti, S.; Thiyagarajan, S.; Padmanabhan, B. Identification of a repurposed drug as an inhibitor of Spike protein of human coronavirus SARS-CoV-2 by computational methods. J. Biosci. 2020, 45, 130. [Google Scholar] [CrossRef]
- BIOVIA, Dassault Systèmes, Discovery Studio Visualizer, v21.1.0.20298; Dassault Systèmes: San Diego, CA, USA, 2021.
- SARS-CoV-2 Variants of Concern. Available online: https://www.ecdc.europa.eu/en/covid-19/variants-concern (accessed on 24 November 2022).
- Pommié, C.; Levadoux, S.; Sabatier, R.; Lefranc, G.; Lefranc, M.-P. IMGT standardized criteria for statistical analysis of immunoglobulin V-REGION amino acid properties. J. Mol. Recognit. 2004, 17, 17–32. [Google Scholar] [CrossRef]
- Bozdaganyan, M.E.; Shaitan, K.V.; Kirpichnikov, M.P.; Sokolova, O.S.; Orekhov, P.S. Computational Analysis of Mutations in the Receptor-Binding Domain of SARS-CoV-2 Spike and Their Effects on Antibody Binding. Viruses 2022, 14, 295. [Google Scholar] [CrossRef]
- Happi, A.N.; Ugwu, C.A.; Happi, C.T. Tracking the emergence of new SARS-CoV-2 variants in South Africa. Nat. Med. 2021, 27, 372–373. [Google Scholar] [CrossRef]
- Othman, H.; Bouslama, Z.; Brandenburg, J.-T.; da Rocha, J.; Hamdi, Y.; Ghedira, K.; Srairi-Abid, N.; Hazelhurst, S. Interaction of the spike protein RBD from SARS-CoV-2 with ACE2: Similarity with SARS-CoV, hot-spot analysis and effect of the receptor polymorphism. Biochem. Biophys. Res. Commun. 2020, 527, 702–708. [Google Scholar] [CrossRef]
- Piplani, S.; Singh, P.K.; Winkler, D.A.; Petrovsky, N. In silico comparison of SARS-CoV-2 spike protein-ACE2 binding affinities across species and implications for virus origin. Sci. Rep. 2021, 11, 13063. [Google Scholar] [CrossRef]
- Moore, G.J.; Matsoukas, J.M. Angiotensin as a model for hormone—Receptor interactions. Biosci. Rep. 1985, 5, 407–416. [Google Scholar] [CrossRef]
- Moore, G.J.; Scanlon, M.N. Methods for analyzing and interpreting cooperativity in dose-response curves—I. Antagonist effects on angiotensin receptors in smooth muscle. Gen. Pharmacol. Vasc. Syst. 1989, 20, 193–198. [Google Scholar] [CrossRef]
- Blow, D.M.; Birktoft, J.J.; Hartley, B.S. Role of a Buried Acid Group in the Mechanism of Action of Chymotrypsin. Nature 1969, 221, 337–340. [Google Scholar] [CrossRef]
- Zhang, S.-J.; Wang, G.-D.; Ma, P.; Zhang, L.-L.; Yin, T.-T.; Liu, Y.-H.; Otecko, N.O.; Wang, M.; Ma, Y.-P.; Wang, L.; et al. Genomic regions under selection in the feralization of the dingoes. Nat. Commun. 2020, 11, 671. [Google Scholar] [CrossRef]
- TAG-VE Statement on Omicron Sublineages BQ.1 and XBB. Available online: https://www.who.int/news/item/27-10-2022-tag-ve-statement-on-omicron-sublineages-bq.1-and-xbb (accessed on 24 November 2022).
- Hachmann, N.P.; Miller, J.; Collier, A.-R.Y.; Ventura, J.D.; Yu, J.; Rowe, M.; Bondzie, E.A.; Powers, O.; Surve, N.; Hall, K.; et al. Neutralization Escape by SARS-CoV-2 Omicron Subvariants BA.2.12.1, BA.4, and BA.5. N. Engl. J. Med. 2022, 387, 86–88. [Google Scholar] [CrossRef]
- Zhang, H.; Unal, H.; Desnoyer, R.; Han, G.W.; Patel, N.; Katritch, V.; Karnik, S.S.; Cherezov, V.; Stevens, R.C. Structural Basis for Ligand Recognition and Functional Selectivity at Angiotensin Receptor. J. Biol. Chem. 2015, 290, 29127–29139. [Google Scholar] [CrossRef]
- Zhang, H.; Unal, H.; Gati, C.; Han, G.W.; Liu, W.; Zatsepin, N.A.; James, D.; Wang, D.; Nelson, G.; Weierstall, U.; et al. Structure of the Angiotensin Receptor Revealed by Serial Femtosecond Crystallography. Cell 2015, 161, 833–844. [Google Scholar] [CrossRef]
- Meng, J.; Xiao, G.; Zhang, J.; He, X.; Ou, M.; Bi, J.; Yang, R.; Di, W.; Wang, Z.; Li, Z.; et al. Renin-angiotensin system inhibitors improve the clinical outcomes of COVID-19 patients with hypertension. Emerg. Microbes Infect. 2020, 9, 757–760. [Google Scholar] [CrossRef] [PubMed]
- Qaradakhi, T.; Gadanec, L.; Matsoukas, J.; Apostolopoulos, V.; Zulli, A. Could DIZE be the answer to COVID-19? Maturitas 2020, 140, 83–84. [Google Scholar] [CrossRef] [PubMed]
- Gadanec, L.K.; McSweeney, K.R.; Qaradakhi, T.; Ali, B.; Zulli, A.; Apostolopoulos, V. Can SARS-CoV-2 Virus Use Multiple Receptors to Enter Host Cells? Int. J. Mol. Sci. 2021, 22, 992. [Google Scholar] [CrossRef] [PubMed]
- Kate Gadanec, L.; Qaradakhi, T.; Renee McSweeney, K.; Ashiana Ali, B.; Zulli, A.; Apostolopoulos, V. Dual targeting of Toll-like receptor 4 and angiotensin-converting enzyme 2: A proposed approach to SARS-CoV-2 treatment. Future Microbiol. 2021, 16, 205–209. [Google Scholar] [CrossRef]
- Verdecchia, P.; Cavallini, C.; Spanevello, A.; Angeli, F. The pivotal link between ACE2 deficiency and SARS-CoV-2 infection. Eur. J. Intern. Med. 2020, 76, 14–20. [Google Scholar] [CrossRef]
- Duarte, M.; Pelorosso, F.; Nicolosi, L.N.; Victoria Salgado, M.; Vetulli, H.; Aquieri, A.; Azzato, F.; Castro, M.; Coyle, J.; Davolos, I.; et al. Telmisartan for treatment of COVID-19 patients: An open multicenter randomized clinical trial. EClinicalMedicine 2021, 37, 100962. [Google Scholar] [CrossRef]
- Zhang, P.; Zhu, L.; Cai, J.; Lei, F.; Qin, J.-J.; Xie, J.; Liu, Y.-M.; Zhao, Y.-C.; Huang, X.; Lin, L.; et al. Association of Inpatient Use of Angiotensin-Converting Enzyme Inhibitors and Angiotensin II Receptor Blockers With Mortality Among Patients With Hypertension Hospitalized With COVID-19. Circ. Res. 2020, 126, 1671–1681. [Google Scholar] [CrossRef]
- Rysz, S.; Al-Saadi, J.; Sjöström, A.; Farm, M.; Campoccia Jalde, F.; Plattén, M.; Eriksson, H.; Klein, M.; Vargas-Paris, R.; Nyrén, S.; et al. COVID-19 pathophysiology may be driven by an imbalance in the renin-angiotensin-aldosterone system. Nat. Commun. 2021, 12, 2417. [Google Scholar] [CrossRef] [PubMed]
- Almutlaq, M.; Mansour, F.A.; Alghamdi, J.; Alhendi, Y.; Alamro, A.A.; Alghamdi, A.A.; Alamri, H.S.; Alroqi, F.; Barhoumi, T. Angiotensin II Exaggerates SARS-CoV-2 Specific T-Cell Response in Convalescent Individuals following COVID-19. Int. J. Mol. Sci. 2022, 23, 8669. [Google Scholar] [CrossRef]
- Platten, M.; Youssef, S.; Hur, E.M.; Ho, P.P.; Han, M.H.; Lanz, T.V.; Phillips, L.K.; Goldstein, M.J.; Bhat, R.; Raine, C.S.; et al. Blocking angiotensin-converting enzyme induces potent regulatory T cells and modulates TH1- and TH17-mediated autoimmunity. Proc. Natl. Acad. Sci. USA 2009, 106, 14948–14953. [Google Scholar] [CrossRef]
- Dargahi, N.; Matsoukas, J.; Apostolopoulos, V. Streptococcus thermophilus ST285 Alters Pro-Inflammatory to Anti-Inflammatory Cytokine Secretion against Multiple Sclerosis Peptide in Mice. Brain Sci. 2020, 10, 126. [Google Scholar] [CrossRef]
- Stegbauer, J.; Lee, D.-H.; Seubert, S.; Ellrichmann, G.; Manzel, A.; Kvakan, H.; Muller, D.N.; Gaupp, S.; Rump, L.C.; Gold, R.; et al. Role of the renin-angiotensin system in autoimmune inflammation of the central nervous system. Proc. Natl. Acad. Sci. USA 2009, 106, 14942–14947. [Google Scholar] [CrossRef] [PubMed]
- Matsoukas, J.; Cordopatis, P.; Belte, U.; Goghari, M.H.; Ganter, R.C.; Franklin, K.J.; Moore, G.J. Importance of the N-terminal domain of the type II angiotensin antagonist sarmesin for receptor blockade. J. Med. Chem. 1988, 31, 1418–1421. [Google Scholar] [CrossRef] [PubMed]
- Matsoukas, J.M.; Agelis, G.; Hondrelis, J.; Yamdagni, R.; Wu, Q.; Ganter, R.; Moore, D.; Moore, G.J.; Smith, J.R. Synthesis and biological activities of angiotensin II, sarilesin, and sarmesin analogs containing Aze or Pip at position 7. J. Med. Chem. 1993, 36, 904–911. [Google Scholar] [CrossRef] [PubMed]
- Matsoukas, J.M.; Hondrelis, J.; Keramida, M.; Mavromoustakos, T.; Makriyannis, A.; Yamdagni, R.; Wu, Q.; Moore, G.J. Role of the NH2-terminal domain of angiotensin II (ANG II) and [Sar1]angiotensin II on conformation and activity. NMR evidence for aromatic ring clustering and peptide backbone folding compared with [des-1,2,3]angiotensin II. J. Biol. Chem. 1994, 269, 5303–5312. [Google Scholar] [CrossRef] [PubMed]
- Hondrelis, J.; Lonergan, G.; Voliotis, S.; Matsoukas, J. One pot synthesis and conformation of N-t-butyloxycarbonyl, O-Phenacyl derivatives of proline and other secondary amino acids. Tetrahedron 1990, 46, 565–576. [Google Scholar] [CrossRef]
- Polevaya, L.; Mavromoustakos, T.; Zoumboulakis, P.; Grdadolnik, S.G.; Roumelioti, P.; Giatas, N.; Mutule, I.; Keivish, T.; Vlahakos, D.V.; Iliodromitis, E.K.; et al. Synthesis and study of a cyclic angiotensin II antagonist analogue reveals the role of π*–π* interactions in the C-terminal aromatic residue for agonist activity and its structure resemblance with AT1 non-peptide antagonists. Bioorganic Med. Chem. 2001, 9, 1639–1647. [Google Scholar] [CrossRef]
- Osman, E.E.A.; Rehemtulla, A.; Neamati, N. Why All the Fury over Furin? J. Med. Chem. 2021, 65, 2747–2784. [Google Scholar] [CrossRef]
- Wu, C.; Zheng, M.; Yang, Y.; Gu, X.; Yang, K.; Li, M.; Liu, Y.; Zhang, Q.; Zhang, P.; Wang, Y.; et al. Furin: A Potential Therapeutic Target for COVID-19. iScience 2020, 23, 101642. [Google Scholar] [CrossRef]
- Cheng, Y.-W.; Chao, T.-L.; Li, C.-L.; Chiu, M.-F.; Kao, H.-C.; Wang, S.-H.; Pang, Y.-H.; Lin, C.-H.; Tsai, Y.-M.; Lee, W.-H.; et al. Furin Inhibitors Block SARS-CoV-2 Spike Protein Cleavage to Suppress Virus Production and Cytopathic Effects. Cell Rep. 2020, 33, 108254. [Google Scholar] [CrossRef]
- Brevini, T.; Maes, M.; Webb, G.J.; John, B.V.; Fuchs, C.D.; Buescher, G.; Wang, L.; Griffiths, C.; Brown, M.L.; Scott, W.E.; et al. FXR inhibition may protect from SARS-CoV-2 infection by reducing ACE2. Nature 2022, in press. [Google Scholar] [CrossRef]
- Zhang, F.; Jenkins, J.; de Carvalho, R.V.H.; Nakandakari-Higa, S.; Chen, T.; Abernathy, M.E.; Nyakatura, E.; Andrew, D.; Lebedeva, I.; Lorenz, I.C.; et al. Human anti-ACE2 monoclonal antibodies as pan-sarbecovirus prophylactic agents. bioRxiv 2022. [Google Scholar] [CrossRef]










| Volume Classes | “Hydropathy “ Classes | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Hydrophobic | Neutral | Hydrophilic | ||||||||
| very large | F | W | Y | |||||||
| large | I | L | M | K | R | |||||
| medium | H | |||||||||
| small | V | C | P | T | E | Q | ||||
| very small | A | G | S | D | N | |||||
| aliphatic | sulfur | hydroxyl | basic | acidic | amide | |||||
| uncharged | charged | uncharged | ||||||||
| Non-polar | Polar | |||||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ridgway, H.; Ntallis, C.; Chasapis, C.T.; Kelaidonis, K.; Matsoukas, M.-T.; Plotas, P.; Apostolopoulos, V.; Moore, G.; Tsiodras, S.; Paraskevis, D.; et al. Molecular Epidemiology of SARS-CoV-2: The Dominant Role of Arginine in Mutations and Infectivity. Viruses 2023, 15, 309. https://doi.org/10.3390/v15020309
Ridgway H, Ntallis C, Chasapis CT, Kelaidonis K, Matsoukas M-T, Plotas P, Apostolopoulos V, Moore G, Tsiodras S, Paraskevis D, et al. Molecular Epidemiology of SARS-CoV-2: The Dominant Role of Arginine in Mutations and Infectivity. Viruses. 2023; 15(2):309. https://doi.org/10.3390/v15020309
Chicago/Turabian StyleRidgway, Harry, Charalampos Ntallis, Christos T. Chasapis, Konstantinos Kelaidonis, Minos-Timotheos Matsoukas, Panagiotis Plotas, Vasso Apostolopoulos, Graham Moore, Sotirios Tsiodras, Dimitrios Paraskevis, and et al. 2023. "Molecular Epidemiology of SARS-CoV-2: The Dominant Role of Arginine in Mutations and Infectivity" Viruses 15, no. 2: 309. https://doi.org/10.3390/v15020309
APA StyleRidgway, H., Ntallis, C., Chasapis, C. T., Kelaidonis, K., Matsoukas, M.-T., Plotas, P., Apostolopoulos, V., Moore, G., Tsiodras, S., Paraskevis, D., Mavromoustakos, T., & Matsoukas, J. M. (2023). Molecular Epidemiology of SARS-CoV-2: The Dominant Role of Arginine in Mutations and Infectivity. Viruses, 15(2), 309. https://doi.org/10.3390/v15020309