
Mitochondrial Oxidative Phosphorylation in Viral Infections


Abstract
:1. Introduction
2. Physiology of OxPhos
3. Complex I of the ETC (NADH Dehydrogenase)
4. Complex II (Succinate Dehydrogenase, SDH)
5. Complex III (Cytochrome bc1 Complex, CIII)
6. Complex IV (Cytochrome c Oxidase, COX)
7. Complex V (ATP Synthase, CV)
8. Supercomplexes
9. Viruses and Oxidative Phosphorylation
10. Viruses and Mitochondrial Reactive Oxygen Species
11. Summary
Author Contributions
Funding
Conflicts of Interest
References
- Siekevitz, P. Powerhouse of the Cell. Sci. Am. 1957, 197, 131–144. [Google Scholar] [CrossRef]
- Yoo, H.C.; Yu, Y.C.; Sung, Y.; Han, J.M. Glutamine reliance in cell metabolism. Exp. Mol. Med. 2020, 52, 1496–1516. [Google Scholar] [CrossRef] [PubMed]
- Thaker, S.K.; Ch’Ng, J.; Christofk, H.R. Viral hijacking of cellular metabolism. BMC Biol. 2019, 17, 59. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; King, M.S.; Yu, M.; Klipcan, L.; Leslie, A.G.; Hirst, J. Structure of subcomplex Ibeta of mammalian respiratory complex I leads to new supernumerary subunit assignments. Proc. Natl. Acad. Sci. USA 2015, 112, 12087–12092. [Google Scholar] [CrossRef] [PubMed]
- Ernster, L.; Dallner, G. Biochemical, physiological and medical aspects of ubiquinone function. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 1995, 1271, 195–204. [Google Scholar] [CrossRef]
- Ndi, M.; Marin-Buera, L.; Salvatori, R.; Singh, A.P.; Ott, M. Biogenesis of the bc1 Complex of the Mitochondrial Respiratory Chain. J. Mol. Biol. 2018, 430, 3892–3905. [Google Scholar] [CrossRef]
- Turrens, J.F.; Alexandre, A.; Lehninger, A.L. Ubisemiquinone is the electron donor for superoxide formation by complex III of heart mitochondria. Arch. Biochem. Biophys. 1985, 237, 408–414. [Google Scholar] [CrossRef]
- Muller, F.L.; Liu, Y.; Van Remmen, H. Complex III Releases Superoxide to Both Sides of the Inner Mitochondrial Membrane. J. Biol. Chem. 2004, 279, 49064–49073. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, R.; Purhonen, J.; Kallijärvi, J. The mitochondrial coenzyme Q junction and complex III: Biochemistry and pathophysiology. FEBS J. 2021, 289, 6936–6958. [Google Scholar] [CrossRef] [PubMed]
- Arnold, S. The power of life—Cytochrome c oxidase takes center stage in metabolic control, cell signalling and survival. Mitochondrion 2012, 12, 46–56. [Google Scholar] [CrossRef]
- Napiwotzki, J.; Kadenbach, B. Extramitochondrial ATP/ADP-Ratios Regulate Cytochrome c Oxidase Activity via Binding to the Cytosolic Domain of Subunit IV. Biol. Chem. 1998, 379, 335–340. [Google Scholar] [CrossRef]
- Hüttemann, M.; Kadenbach, B.; Grossman, L.I. Mammalian subunit IV isoforms of cytochrome c oxidase. Gene 2001, 267, 111–123. [Google Scholar] [CrossRef] [PubMed]
- Steenaart, N.A.; Shore, G.C. Mitochondrial cytochrome c oxidase subunit IV is phosphorylated by an endogenous kinase. FEBS Lett. 1997, 415, 294–298. [Google Scholar] [CrossRef]
- Clayton, S.A.; Daley, K.K.; MacDonald, L.; Fernandez-Vizarra, E.; Bottegoni, G.; O’Neil, J.D.; Major, T.; Griffin, D.; Zhuang, Q.; Adewoye, A.B.; et al. Inflammation causes remodeling of mitochondrial cytochrome c oxidase mediated by the bifunctional gene C15orf48. Sci. Adv. 2021, 7, eabl5182. [Google Scholar] [CrossRef] [PubMed]
- Angireddy, R.; Kazmi, H.R.; Srinivasan, S.; Sun, L.; Iqbal, J.; Fuchs, S.Y.; Guha, M.; Kijima, T.; Yuen, T.; Zaidi, M.; et al. Cytochrome c oxidase dysfunction enhances phagocytic function and osteoclast formation in macrophages. FASEB J. 2019, 33, 9167–9181. [Google Scholar] [CrossRef] [PubMed]
- Mookerjee, S.A.; Gerencser, A.A.; Nicholls, D.G.; Brand, M.D. Quantifying intracellular rates of glycolytic and oxidative ATP production and consumption using extracellular flux measurements. J. Biol. Chem. 2017, 292, 7189–7207. [Google Scholar] [CrossRef]
- Caruana, N.J.; Stroud, D.A. The road to the structure of the mitochondrial respiratory chain supercomplex. Biochem. Soc. Trans. 2020, 48, 621–629. [Google Scholar] [CrossRef] [PubMed]
- Milenkovic, D.; Blaza, J.N.; Larsson, N.-G.; Hirst, J. The Enigma of the Respiratory Chain Supercomplex. Cell Metab. 2017, 25, 765–776. [Google Scholar] [CrossRef] [PubMed]
- Lapuente-Brun, E.; Moreno-Loshuertos, R.; Acín-Pérez, R.; Latorre-Pellicer, A.; Colás, C.; Balsa, E.; Perales-Clemente, E.; Quirós, P.M.; Calvo, E.; Rodríguez-Hernández, M.A.; et al. Supercomplex Assembly Determines Electron Flux in the Mitochondrial Electron Transport Chain. Science 2013, 340, 1567–1570. [Google Scholar] [CrossRef] [PubMed]
- Milenkovic, D.; Misic, J.; Hevler, J.F.; Molinié, T.; Chung, I.; Atanassov, I.; Li, X.; Filograna, R.; Mesaros, A.; Mourier, A.; et al. Preserved respiratory chain capacity and physiology in mice with profoundly reduced levels of mitochondrial respirasomes. Cell Metab. 2023, 35, 1799–1813.e7. [Google Scholar] [CrossRef]
- Letts, J.A.; Sazanov, L.A. Clarifying the supercomplex: The higher-order organization of the mitochondrial electron transport chain. Nat. Struct. Mol. Biol. 2017, 24, 800–808. [Google Scholar] [CrossRef] [PubMed]
- Schagger, H.; Pfeiffer, K. The Ratio of Oxidative Phosphorylation Complexes I–V in Bovine Heart Mitochondria and the Composition of Respiratory Chain Supercomplexes. J. Biol. Chem. 2001, 276, 37861–37867. [Google Scholar] [CrossRef] [PubMed]
- Letts, J.A.; Fiedorczuk, K.; Sazanov, L.A. The architecture of respiratory supercomplexes. Nature 2016, 537, 644–648. [Google Scholar] [CrossRef] [PubMed]
- Gray, M.W. Mitochondrial evolution. Cold Spring Harb. Perspect. Biol. 2012, 4, a011403. [Google Scholar] [CrossRef] [PubMed]
- Rathore, S.; Berndtsson, J.; Marin-Buera, L.; Conrad, J.; Carroni, M.; Brzezinski, P.; Ott, M. Cryo-EM structure of the yeast respiratory supercomplex. Nat. Struct. Mol. Biol. 2018, 26, 50–57. [Google Scholar] [CrossRef]
- Vercellino, I.; Sazanov, L.A. Structure and assembly of the mammalian mitochondrial supercomplex CIII2CIV. Nature 2021, 598, 364–367. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Vizarra, E.; Ugalde, C. Cooperative assembly of the mitochondrial respiratory chain. Trends Biochem. Sci. 2022, 47, 999–1008. [Google Scholar] [CrossRef]
- Acín-Pérez, R.; Fernández-Silva, P.; Peleato, M.L.; Pérez-Martos, A.; Enriquez, J.A. Respiratory Active Mitochondrial Supercomplexes. Mol. Cell 2008, 32, 529–539. [Google Scholar] [CrossRef] [PubMed]
- Muhleip, A.; Flygaard, R.K.; Baradaran, R.; Haapanen, O.; Gruhl, T.; Tobiasson, V.; Maréchal, A.; Sharma, V.; Amunts, A. Structural basis of mitochondrial membrane bending by the I-II-III(2)-IV(2) supercomplex. Nature 2023, 615, 934–938. [Google Scholar] [CrossRef]
- Azuma, K.; Ikeda, K.; Inoue, S. Functional Mechanisms of Mitochondrial Respiratory Chain Supercomplex Assembly Factors and Their Involvement in Muscle Quality. Int. J. Mol. Sci. 2020, 21, 3182. [Google Scholar] [CrossRef]
- Lobo-Jarne, T.; Nývltová, E.; Pérez-Pérez, R.; Timón-Gómez, A.; Molinié, T.; Choi, A.; Mourier, A.; Fontanesi, F.; Ugalde, C.; Barrientos, A. Human COX7A2L Regulates Complex III Biogenesis and Promotes Supercomplex Organization Remodeling without Affecting Mitochondrial Bioenergetics. Cell Rep. 2018, 25, 1786–1799.e4. [Google Scholar] [CrossRef] [PubMed]
- Kohler, A.; Barrientos, A.; Fontanesi, F.; Ott, M. The functional significance of mitochondrial respiratory chain supercomplexes. Embo Rep. 2023, 24, e57092. [Google Scholar] [CrossRef]
- Garaude, J.; Acín-Pérez, R.; Martínez-Cano, S.; Enamorado, M.; Ugolini, M.; Nistal-Villán, E.; Hervás-Stubbs, S.; Pelegrín, P.; Sander, L.E.; Enríquez, J.A.; et al. Mitochondrial respiratory-chain adaptations in macrophages contribute to antibacterial host defense. Nat. Immunol. 2016, 17, 1037–1045. [Google Scholar] [CrossRef] [PubMed]
- Hanada, Y.; Ishihara, N.; Wang, L.; Otera, H.; Ishihara, T.; Koshiba, T.; Mihara, K.; Ogawa, Y.; Nomura, M. MAVS is energized by Mff which senses mitochondrial metabolism via AMPK for acute antiviral immunity. Nat. Commun. 2020, 11, 5711. [Google Scholar] [CrossRef] [PubMed]
- Koshiba, T.; Yasukawa, K.; Yanagi, Y.; Kawabata, S.-I. Mitochondrial membrane potential is required for MAVS-mediated antiviral signaling. Sci. Signal. 2011, 4, ra7. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-J.; Ahn, D.-G.; Syed, G.H.; Siddiqui, A. The essential role of mitochondrial dynamics in antiviral immunity. Mitochondrion 2017, 41, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wu, K.; Zeng, S.; Zou, L.; Li, X.; Xu, C.; Li, B.; Liu, X.; Li, Z.; Zhu, W.; et al. The Role of Mitophagy in Viral Infection. Cells 2022, 11, 711. [Google Scholar] [CrossRef] [PubMed]
- Koonin, E.V.; Krupovic, M.; Agol, V.I. The Baltimore Classification of Viruses 50 Years Later: How Does It Stand in the Light of Virus Evolution? Microbiol. Mol. Biol. Rev. 2021, 85, e0005321. [Google Scholar] [CrossRef]
- Grimbert, S.; Johanet, C.; Bendjaballah, F.; Homberg, J.; Poupon, R.; Beaugrand, M. Antimitochondrial antibodies in patients with chronic hepatitis C. Liver Int. 1996, 16, 161–165. [Google Scholar] [CrossRef]
- Barbaro, G.; Di Lorenzo, G.; Asti, A.; Ribersani, M.; Belloni, G.; Grisorio, B.; Filice, G.; Barbarini, G. Hepatocellular mitochondrial alterations in patients with chronic hepatitis C: Ultrastructural and biochemical findings. Am. J. Gastroenterol. 1999, 94, 2198–2205. [Google Scholar] [CrossRef]
- Korenaga, M.; Wang, T.; Li, Y.; Showalter, L.A.; Chan, T.; Sun, J.; Weinman, S.A. Hepatitis C Virus Core Protein Inhibits Mitochondrial Electron Transport and Increases Reactive Oxygen Species (ROS) Production. J. Biol. Chem. 2005, 280, 37481–37488. [Google Scholar] [CrossRef] [PubMed]
- Piccoli, C.; Scrima, R.; Quarato, G.; D’Aprile, A.; Ripoli, M.; Lecce, L.; Boffoli, D.; Moradpour, D.; Capitanio, N. Hepatitis C virus protein expression causes calcium-mediated mitochondrial bioenergetic dysfunction and nitro-oxidative stress. Hepatology 2007, 46, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Quarato, G.; Scrima, R.; Ripoli, M.; Agriesti, F.; Moradpour, D.; Capitanio, N.; Piccoli, C. Protective role of amantadine in mitochondrial dysfunction and oxidative stress mediated by hepatitis C virus protein expression. Biochem. Pharmacol. 2014, 89, 545–556. [Google Scholar] [CrossRef] [PubMed]
- Cortelli, P.; Mandrioli, J.; Zeviani, M.; Lodi, R.; Prata, C.; Pecorari, M.; Orlando, G.; Guaraldi, G. Mitochondrial complex III deficiency in a case of HCV related noninflammatory myopathy. J. Neurol. 2007, 254, 1450–1452. [Google Scholar] [CrossRef] [PubMed]
- Jassey, A.; Liu, C.-H.; Changou, C.A.; Richardson, C.D.; Hsu, H.-Y.; Lin, L.-T. Hepatitis C Virus Non-Structural Protein 5A (NS5A) Disrupts Mitochondrial Dynamics and Induces Mitophagy. Cells 2019, 8, 290. [Google Scholar] [CrossRef]
- Gong, G.; Waris, G.; Tanveer, R.; Siddiqui, A. Human hepatitis C virus NS5A protein alters intracellular calcium levels, induces oxidative stress, and activates STAT-3 and NF-κB. Proc. Natl. Acad. Sci. USA 2001, 98, 9599–9604. [Google Scholar] [CrossRef] [PubMed]
- Gerresheim, G.K.; Bathke, J.; Michel, A.M.; Andreev, D.E.; Shalamova, L.A.; Rossbach, O.; Hu, P.; Glebe, D.; Fricke, M.; Marz, M.; et al. Cellular Gene Expression during Hepatitis C Virus Replication as Revealed by Ribosome Profiling. Int. J. Mol. Sci. 2019, 20, 1321. [Google Scholar] [CrossRef] [PubMed]
- Ramage, H.R.; Kumar, G.R.; Verschueren, E.; Johnson, J.R.; Von Dollen, J.; Johnson, T.; Newton, B.; Shah, P.; Horner, J.; Krogan, N.J.; et al. A combined proteomics/genomics approach links hepatitis C virus infection with nonsense-mediated mRNA decay. Mol. Cell 2015, 57, 329–340. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Xiu, W.; Xu, J.; Chen, X.; Wang, G.; Duan, J.; Sun, L.; Liu, B.; Xie, W.; Pu, G.; et al. Increased CHCHD2 expression promotes liver fibrosis in nonalcoholic steatohepatitis via Notch/osteopontin signaling. J. Clin. Investig. 2022, 7, e162402. [Google Scholar] [CrossRef]
- Aras, S.; Bai, M.; Lee, I.; Springett, R.; Hüttemann, M.; Grossman, L.I. MNRR1 (formerly CHCHD2) is a bi-organellar regulator of mitochondrial metabolism. Mitochondrion 2015, 20, 43–51. [Google Scholar] [CrossRef]
- Aras, S.; Purandare, N.; Gladyck, S.; Somayajulu-Nitu, M.; Zhang, K.; Wallace, D.C.; Grossman, L.I. Mitochondrial Nuclear Retrograde Regulator 1 (MNRR1) rescues the cellular phenotype of MELAS by inducing homeostatic mechanisms. Proc. Natl. Acad. Sci. USA 2020, 117, 32056–32065. [Google Scholar] [CrossRef] [PubMed]
- Beckham, J.D.; Pastula, D.M.; Massey, A.; Tyler, K.L. Zika Virus as an Emerging Global Pathogen: Neurological Complications of Zika Virus. JAMA Neurol. 2016, 73, 875–879. [Google Scholar] [CrossRef] [PubMed]
- Bhatnagar, J.; Rabeneck, D.B.; Martines, R.B.; Reagan-Steiner, S.; Ermias, Y.; Estetter, L.B.; Suzuki, T.; Ritter, J.; Keating, M.K.; Al, J.B.E.; et al. Zika Virus RNA Replication and Persistence in Brain and Placental Tissue. Emerg. Infect. Dis. 2017, 23, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.K.; Shin, O.S. Zika virus modulates mitochondrial dynamics, mitophagy, and mitochondria-derived vesicles to facilitate viral replication in trophoblast cells. Front. Immunol. 2023, 14, 1203645. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Wang, J.; Yang, Y.; Qu, S.; Wan, F.; Zhang, Z.; Wang, R.; Li, G.; Cong, H. Zika virus infection induced apoptosis by modulating the recruitment and activation of pro-apoptotic protein Bax. J. Virol. 2021, 95, 10-1128. [Google Scholar] [CrossRef] [PubMed]
- Xin, Q.L.; Deng, C.L.; Chen, X.; Wang, J.; Wang, S.B.; Wang, W.; Deng, F.; Zhang, B.; Xiao, G.; Zhang, L.K. Quantitative Proteomic Analysis of Mosquito C6/36 Cells Reveals Host Proteins Involved in Zika Virus Infection. J. Virol. 2017, 91, 10-1128. [Google Scholar] [CrossRef] [PubMed]
- Daniels, B.P.; Kofman, S.B.; Smith, J.R.; Norris, G.T.; Snyder, A.G.; Kolb, J.P.; Gao, X.; Locasale, J.W.; Martinez, J.; Gale, M.; et al. The Nucleotide Sensor ZBP1 and Kinase RIPK3 Induce the Enzyme IRG1 to Promote an Antiviral Metabolic State in Neurons. Immunity 2019, 50, 64–76.e4. [Google Scholar] [CrossRef] [PubMed]
- Yau, C.; Low, J.Z.; Gan, E.S.; Kwek, S.S.; Cui, L.; Tan, H.C.; Mok, D.Z.; Chan, C.Y.; Sessions, O.M.; Watanabe, S.; et al. Dysregulated metabolism underpins Zika-virus-infection-associated impairment in fetal development. Cell Rep. 2021, 37, 110118. [Google Scholar] [CrossRef] [PubMed]
- Airo, A.M.; Felix-Lopez, A.; Mancinelli, V.; Evseev, D.; Lopez-Orozco, J.; Shire, K.; Paszkowski, P.; Frappier, L.; Magor, K.E.; Hobman, T.C. Flavivirus Capsid Proteins Inhibit the Interferon Response. Viruses 2022, 14, 968. [Google Scholar] [CrossRef]
- He, J.; Ford, H.C.; Carroll, J.; Douglas, C.; Gonzales, E.; Ding, S.; Fearnley, I.M.; Walker, J.E. Assembly of the membrane domain of ATP synthase in human mitochondria. Proc. Natl. Acad. Sci. USA 2018, 115, 2988–2993. [Google Scholar] [CrossRef]
- Lim, P.-Y.; Behr, M.J.; Chadwick, C.M.; Shi, P.-Y.; Bernard, K.A. Keratinocytes Are Cell Targets of West Nile Virus In Vivo. J. Virol. 2011, 85, 5197–5201. [Google Scholar] [CrossRef]
- Sampson, B.A.; Ambrosi, C.; Charlot, A.; Reiber, K.; Veress, J.F.; Armbrustmacher, V. The pathology of human West Nile virus infection. Hum. Pathol. 2000, 31, 527–531. [Google Scholar] [CrossRef] [PubMed]
- Koh, W.-L.; Ng, M.-L. Molecular Mechanisms of West Nile Virus Pathogenesis in Brain Cells. Emerg. Infect. Dis. 2005, 11, 629–632. [Google Scholar] [CrossRef] [PubMed]
- Mingo-Casas, P.; Blázquez, A.-B.; de Cedrón, M.G.; San-Félix, A.; Molina, S.; Escribano-Romero, E.; Calvo-Pinilla, E.; de Oya, N.J.; de Molina, A.R.; Saiz, J.-C.; et al. Glycolytic shift during West Nile virus infection provides new therapeutic opportunities. J. Neuroinflamm. 2023, 20, 217. [Google Scholar] [CrossRef] [PubMed]
- Peng, B.-H.; Wang, T. West Nile Virus Induced Cell Death in the Central Nervous System. Pathogens 2019, 8, 215. [Google Scholar] [CrossRef] [PubMed]
- Petersen, L.R.; Brault, A.C.; Nasci, R.S. West Nile Virus: Review of the Literature. JAMA 2013, 310, 308–315. [Google Scholar] [CrossRef] [PubMed]
- Miller, B.; Silverstein, A.; Flores, M.; Cao, K.; Kumagai, H.; Mehta, H.H.; Yen, K.; Kim, S.J.; Cohen, P. Host mitochondrial transcriptome response to SARS-CoV-2 in multiple cell models and clinical samples. Sci. Rep. 2021, 11, 3. [Google Scholar] [CrossRef] [PubMed]
- Guarnieri, J.W.; Dybas, J.M.; Fazelinia, H.; Kim, M.S.; Frere, J.; Zhang, Y.; Soto Albrecht, Y.; Murdock, D.G.; Angelin, A.; Singh, L.N.; et al. Core mitochondrial genes are down-regulated during SARS-CoV-2 infection of rodent and human hosts. Sci. Transl. Med. 2023, 15, eabq1533. [Google Scholar] [CrossRef] [PubMed]
- Archer, S.L.; Dasgupta, A.; Chen, K.H.; Wu, D.; Baid, K.; Mamatis, J.E.; Gonzalez, V.; Read, A.; Bentley, R.E.; Martin, A.Y.; et al. SARS-CoV-2 mitochondriopathy in COVID-19 pneumonia exacerbates hypoxemia. Redox Biol. 2022, 58, 102508. [Google Scholar] [CrossRef] [PubMed]
- Codo, A.C.; Davanzo, G.G.; de Brito Monteiro, L.; de Souza, G.F.; Muraro, S.P.; Virgilio-da-Silva, J.V.; Prodonoff, J.S.; Carregari, V.C.; de Biagi Junior, C.A.O.; Crunfli, F.; et al. Elevated Glucose Levels Favor SARS-CoV-2 Infection and Monocyte Response through a HIF-1alpha/Glycolysis-Dependent Axis. Cell Metab. 2020, 32, 437–446.e5. [Google Scholar] [CrossRef]
- Ajaz, S.; McPhail, M.J.; Singh, K.K.; Mujib, S.; Trovato, F.M.; Napoli, S.; Agarwal, K. Mitochondrial metabolic manipulation by SARS-CoV-2 in peripheral blood mononuclear cells of patients with COVID-19. Am. J. Physiol. Cell Physiol. 2021, 320, C57–C65. [Google Scholar] [CrossRef] [PubMed]
- Mozzi, A.; Oldani, M.; Forcella, M.E.; Vantaggiato, C.; Cappelletti, G.; Pontremoli, C.; Valenti, F.; Forni, D.; Saresella, M.; Biasin, M.; et al. SARS-CoV-2 ORF3c impairs mitochondrial respiratory metabolism, oxidative stress, and autophagic flux. iScience 2023, 26, 107118. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Wang, L.; Dong, C.; Che, Y.; Jiang, L.; Liu, L.; Zhao, H.; Liao, Y.; Sheng, Y.; Dong, S.; et al. The interaction of the SARS coronavirus non-structural protein 10 with the cellular oxido-reductase system causes an extensive cytopathic effect. J. Clin. Virol. 2005, 34, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Kakavandi, S.; Zare, I.; VaezJalali, M.; Dadashi, M.; Azarian, M.; Akbari, A.; Ramezani Farani, M.; Zalpoor, H.; Hajikhani, B. Structural and non-structural proteins in SARS-CoV-2: Potential aspects to COVID-19 treatment or prevention of progression of related diseases. Cell Commun. Signal. 2023, 21, 110. [Google Scholar] [CrossRef] [PubMed]
- Claus, C.; Schönefeld, K.; Hübner, D.; Chey, S.; Reibetanz, U.; Liebert, U.G. Activity Increase in Respiratory Chain Complexes by Rubella Virus with Marginal Induction of Oxidative Stress. J. Virol. 2013, 87, 8481–8492. [Google Scholar] [CrossRef] [PubMed]
- Bilz, N.C.; Jahn, K.; Lorenz, M.; Lüdtke, A.; Hübschen, J.M.; Geyer, H.; Mankertz, A.; Hübner, D.; Liebert, U.G.; Claus, C. Rubella Viruses Shift Cellular Bioenergetics to a More Oxidative and Glycolytic Phenotype with a Strain-Specific Requirement for Glutamine. J. Virol. 2018, 92, 10-1128. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Bowden, D.S.; Marshall, J.A. Membrane junctions associated with rubella virus infected cells. J. Submicrosc. Cytol. Pathol. 1996, 28, 101–108. [Google Scholar] [PubMed]
- Beatch, M.D.; Hobman, T.C. Rubella Virus Capsid Associates with Host Cell Protein p32 and Localizes to Mitochondria. J. Virol. 2000, 74, 5569–5576. [Google Scholar] [CrossRef] [PubMed]
- Ebermann, L.; Wika, S.; Klumpe, I.; Hammer, E.; Klingel, K.; Lassner, D.; Völker, U.; Erben, U.; Zeichhardt, H.; Schultheiss, H.-P.; et al. The mitochondrial respiratory chain has a critical role in the antiviral process in Coxsackievirus B3-induced myocarditis. Lab. Investig. 2012, 92, 125–134. [Google Scholar] [CrossRef]
- Qu, C.; Zhang, S.; Wang, W.; Li, M.; Wang, Y.; Heijde-Mulder, M.; Shokrollahi, E.; Hakim, M.S.; Raat, N.J.H.; Peppelenbosch, M.P.; et al. Mitochondrial electron transport chain complex III sustains hepatitis E virus replication and represents an antiviral target. FASEB J. 2018, 33, 1008–1019. [Google Scholar] [CrossRef]
- Tian, J.; Shi, R.; Xiao, P.; Liu, T.; She, R.; Wu, Q.; An, J.; Hao, W.; Soomro, M. Hepatitis E Virus Induces Brain Injury Probably Associated With Mitochondrial Apoptosis. Front. Cell. Infect. Microbiol. 2019, 9, 433. [Google Scholar] [CrossRef] [PubMed]
- Kuiken, T.; Taubenberger, J.K. Pathology of human influenza revisited. Vaccine 2008, 26, D59–D66. [Google Scholar] [CrossRef] [PubMed]
- Derakhshan, M.; Willcocks, M.M.; Salako, M.A.; Kass, G.E.N.; Carter, M.J. Human herpesvirus 1 protein US3 induces an inhibition of mitochondrial electron transport. J. Gen. Virol. 2006, 87, 2155–2159. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Wang, H.; Bai, L.; Yu, Y.; Sun, Z.; Yan, Y.; Zhou, J. Mitochondrial proteomic analysis of human host cells infected with H3N2 swine influenza virus. J. Proteom. 2013, 91, 136–150. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Huang, H.; Li, B.; Li, H.; Zhao, Y.; Li, Y.; Ye, W.; Qi, W.; Tang, W.; Wang, L. Identification of cytochrome c oxidase subunit 4 isoform 1 as a positive regulator of influenza virus replication. Front. Microbiol. 2022, 13, 862205. [Google Scholar] [CrossRef] [PubMed]
- Othumpangat, S.; Noti, J.D.; Beezhold, D.H. Lung epithelial cells resist influenza A infection by inducing the expression of cytochrome c oxidase VIc which is modulated by miRNA 4276. Virology 2014, 468–470, 256–264. [Google Scholar] [CrossRef] [PubMed]
- Oleynikov, I.P.; Sudakov, R.V.; Radyukhin, V.A.; Arutyunyan, A.M.; Azarkina, N.V.; Vygodina, T.V. Interaction of Amphipathic Peptide from Influenza Virus M1 Protein with Mitochondrial Cytochrome Oxidase. Int. J. Mol. Sci. 2023, 24, 4119. [Google Scholar] [CrossRef] [PubMed]
- Keshavarz, M.; Solaymani-Mohammadi, F.; Namdari, H.; Arjeini, Y.; Mousavi, M.J.; Rezaei, F. Metabolic host response and therapeutic approaches to influenza infection. Cell. Mol. Biol. Lett. 2020, 25, 15. [Google Scholar] [CrossRef] [PubMed]
- Alandijany, T.; Kammouni, W.; Chowdhury, S.K.R.; Fernyhough, P.; Jackson, A.C. Mitochondrial dysfunction in rabies virus infection of neurons. J. Neuro Virol. 2013, 19, 537–549. [Google Scholar] [CrossRef]
- Kammouni, W.; Wood, H.; Saleh, A.; Appolinario, C.M.; Fernyhough, P.; Jackson, A.C. Rabies virus phosphoprotein interacts with mitochondrial Complex I and induces mitochondrial dysfunction and oxidative stress. J. Neuro Virol. 2015, 21, 370–382. [Google Scholar] [CrossRef]
- Harsha, P.K.; Ranganayaki, S.; Yale, G.; Dey, G.; Mangalaparthi, K.K.; Yarlagadda, A.; Sagar, B.K.C.; Mahadevan, A.; Bharath, M.M.S.; Mani, R.S. Mitochondrial Dysfunction in Rabies Virus-Infected Human and Canine Brains. Neurochem. Res. 2022, 47, 1610–1636. [Google Scholar] [CrossRef]
- Hu, M.; Schulze, K.E.; Ghildyal, R.; Henstridge, D.C.; Kolanowski, J.L.; New, E.J.; Hong, Y.; Hsu, A.C.; Hansbro, P.M.; Wark, P.A.; et al. Respiratory syncytial virus co-opts host mitochondrial function to favour infectious virus production. Elife 2019, 8, e42448. [Google Scholar] [CrossRef]
- Hu, M.; Bogoyevitch, M.A.; Jans, D.A. Respiratory Syncytial Virus Matrix Protein Is Sufficient and Necessary to Remodel Host Mitochondria in Infection. Cells 2023, 12, 1311. [Google Scholar] [CrossRef] [PubMed]
- Sato, H.; Hoshi, M.; Ikeda, F.; Fujiyuki, T.; Yoneda, M.; Kai, C. Downregulation of mitochondrial biogenesis by virus infection triggers antiviral responses by cyclic GMP-AMP synthase. PLoS Pathog. 2021, 17, e1009841. [Google Scholar] [CrossRef] [PubMed]
- Somasundaran, M.; Zapp, M.L.; Beattie, L.K.; Pang, L.; Byron, K.S.; Bassell, G.J.; Sullivan, J.L.; Singer, R.H. Localization of HIV RNA in mitochondria of infected cells: Potential role in cytopathogenicity. J. Cell Biol. 1994, 126, 1353–1360. [Google Scholar] [CrossRef] [PubMed]
- Macreadie, I.G.; Thorburn, D.R.; Kirby, D.M.; Castelli, L.A.; de Rozario, N.L.; Azad, A.A. HIV-1 protein Vpr causes gross mitochondrial dysfunction in the yeast Saccharomyces cerevisiae. FEBS Lett. 1997, 410, 145–149. [Google Scholar] [CrossRef]
- Tripathy, M.K.; Mitra, D. Differential modulation of mitochondrial OXPHOS system during HIV-1 induced T-cell apoptosis: Up regulation of Complex-IV subunit COX-II and its possible implications. Apoptosis 2010, 15, 28–40. [Google Scholar] [CrossRef] [PubMed]
- Ladha, J.S.; Tripathy, M.K.; Mitra, D. Mitochondrial complex I activity is impaired during HIV-1-induced T-cell apoptosis. Cell Death Differ. 2005, 12, 1417–1428. [Google Scholar] [CrossRef]
- Miró, O.; López, S.; Martínez, E.; Pedrol, E.; Milinkovic, A.; Deig, E.; Garrabou, G.; Casademont, J.; Gatell, J.M.; Cardellach, F. Mitochondrial Effects of HIV Infection on the Peripheral Blood Mononuclear Cells of HIV-Infected Patients Who Were Never Treated with Antiretrovirals. Clin. Infect. Dis. 2004, 39, 710–716. [Google Scholar] [CrossRef]
- Kaur, H.; Minchella, P.; Alvarez-Carbonell, D.; Purandare, N.; Nagampalli, V.K.; Blankenberg, D.; Hulgan, T.; Gerschenson, M.; Karn, J.; Aras, S.; et al. Contemporary Antiretroviral Therapy Dysregulates Iron Transport and Augments Mitochondrial Dysfunction in HIV-Infected Human Microglia and Neural-Lineage Cells. Int. J. Mol. Sci. 2023, 24, 12242. [Google Scholar] [CrossRef]
- Ogawa, M.; Takemoto, Y.; Sumi, S.; Inoue, D.; Kishimoto, N.; Takamune, N.; Shoji, S.; Suzu, S.; Misumi, S. ATP generation in a host cell in early-phase infection is increased by upregulation of cytochrome c oxidase activity via the p2 peptide from human immunodeficiency virus type 1 Gag. Retrovirology 2015, 12, 97. [Google Scholar] [CrossRef] [PubMed]
- Lecoeur, H.; Borgne-Sanchez, A.; Chaloin, O.; El-Khoury, R.; Brabant, M.; Langonné, A.; Porceddu, M.; Brière, J.-J.; Buron, N.; Rebouillat, D.; et al. HIV-1 Tat protein directly induces mitochondrial membrane permeabilization and inactivates cytochrome c oxidase. Cell Death Dis. 2012, 3, e282. [Google Scholar] [CrossRef] [PubMed]
- Yavlovich, A.; Viard, M.; Zhou, M.; Veenstra, T.D.; Wang, J.M.; Gong, W.; Heldman, E.; Blumenthal, R.; Raviv, Y. Ectopic ATP synthase facilitates transfer of HIV-1 from antigen-presenting cells to CD4+ target cells. Blood 2012, 120, 1246–1253. [Google Scholar] [CrossRef] [PubMed]
- Castellano, P.; Prevedel, L.; Valdebenito, S.; Eugenin, E.A. HIV infection and latency induce a unique metabolic signature in human macrophages. Sci. Rep. 2019, 9, 3941. [Google Scholar] [CrossRef] [PubMed]
- Schank, M.; Zhao, J.; Moorman, J.P.; Yao, Z.Q. The Impact of HIV- and ART-Induced Mitochondrial Dysfunction in Cellular Senescence and Aging. Cells 2021, 10, 174. [Google Scholar] [CrossRef] [PubMed]
- Takada, S.; Shirakata, Y.; Kaneniwa, N.; Koike, K. Association of hepatitis B virus X protein with mitochondria causes mitochondrial aggregation at the nuclear periphery, leading to cell death. Oncogene 1999, 18, 6965–6973. [Google Scholar] [CrossRef] [PubMed]
- Rahmani, Z.; Huh, K.-W.; Lasher, R.; Siddiqui, A. Hepatitis B Virus X Protein Colocalizes to Mitochondria with a Human Voltage-Dependent Anion Channel, HVDAC3, and Alters Its Transmembrane Potential. J. Virol. 2000, 74, 2840–2846. [Google Scholar] [CrossRef]
- Li, D.; Wang, X.Z.; Yu, J.P.; Chen, Z.X.; Huang, Y.H.; Tao, Q.M. Cytochrome C oxidase III interacts with hepatitis B virus X protein in vivo by yeast two-hybrid system. World J. Gastroenterol. 2004, 10, 2805–2808. [Google Scholar] [CrossRef]
- Zou, L.-Y.; Zheng, B.-Y.; Fang, X.-F.; Li, D.; Huang, Y.-H.; Chen, Z.-X.; Zhou, L.-Y.; Wang, X.-Z. HBx co-localizes with COXIII in HL-7702 cells to upregulate mitochondrial function and ROS generation. Oncol. Rep. 2015, 33, 2461–2467. [Google Scholar] [CrossRef]
- Lee, Y.I.; Hwang, J.M.; Im, J.H.; Lee, Y.I.; Kim, N.S.; Kim, D.G.; Yu, D.Y.; Moon, H.B.; Park, S.K. Human Hepatitis B Virus-X Protein Alters Mitochondrial Function and Physiology in Human Liver Cells. J. Biol. Chem. 2004, 279, 15460–15471. [Google Scholar] [CrossRef]
- Honkoop, P.; De Man, R.A.; Scholte, H.R.; Zondervan, P.E.; Van Den Berg, J.W.; Rademakers, L.H.; Schalm, S.W. Effect of lamivudine on morphology and function of mitochondria in patients with chronic hepatitis B. Hepatology 1997, 26, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhu, Y.; Feng, S.; Ishida, Y.; Chiu, T.P.; Saito, T.; Wang, S.; Ann, D.K.; Ou, J.H.J. Macrophages activated by hepatitis B virus have distinct metabolic profiles and suppress the virus via IL-1beta to downregulate PPARalpha and FOXO3. Cell Rep. 2022, 38, 110284. [Google Scholar] [CrossRef] [PubMed]
- Giosa, D.; Lombardo, D.; Musolino, C.; Chines, V.; Raffa, G.; di Tocco, F.C.; D’aliberti, D.; Caminiti, G.; Saitta, C.; Alibrandi, A.; et al. Mitochondrial DNA is a target of HBV integration. Commun. Biol. 2023, 6, 684. [Google Scholar] [CrossRef] [PubMed]
- Fisicaro, P.; Barili, V.; Montanini, B.; Acerbi, G.; Ferracin, M.; Guerrieri, F.; Salerno, D.; Boni, C.; Massari, M.; Cavallo, M.C.; et al. Targeting mitochondrial dysfunction can restore antiviral activity of exhausted HBV-specific CD8 T cells in chronic hepatitis B. Nat. Med. 2017, 23, 327–336. [Google Scholar] [CrossRef]
- Britt, W.J. Maternal Immunity and the Natural History of Congenital Human Cytomegalovirus Infection. Viruses 2018, 10, 405. [Google Scholar] [CrossRef] [PubMed]
- Combs, J.A.; Norton, E.B.; Saifudeen, Z.R.; Bentrup, K.H.Z.; Katakam, P.V.; Morris, C.A.; Myers, L.; Kaur, A.; Sullivan, D.E.; Zwezdaryk, K.J. Human Cytomegalovirus Alters Host Cell Mitochondrial Function during Acute Infection. J. Virol. 2020, 94, 10-1128. [Google Scholar] [CrossRef] [PubMed]
- Munger, J.; Bajad, S.U.; Coller, H.A.; Shenk, T.; Rabinowitz, J.D. Dynamics of the Cellular Metabolome during Human Cytomegalovirus Infection. PLoS Pathog. 2006, 2, e132. [Google Scholar] [CrossRef]
- Betsinger, C.N.; Jankowski, C.S.; Hofstadter, W.A.; Federspiel, J.D.; Otter, C.J.; Beltran, P.M.J.; Cristea, I.M. The human cytomegalovirus protein pUL13 targets mitochondrial cristae architecture to increase cellular respiration during infection. Proc. Natl. Acad. Sci. USA 2021, 118, e2101675118. [Google Scholar] [CrossRef]
- Kaarbø, M.; Ager-Wick, E.; Osenbroch, P.; Kilander, A.; Skinnes, R.; Müller, F.; Eide, L. Human cytomegalovirus infection increases mitochondrial biogenesis. Mitochondrion 2011, 11, 935–945. [Google Scholar] [CrossRef]
- Karniely, S.; Weekes, M.P.; Antrobus, R.; Rorbach, J.; van Haute, L.; Umrania, Y.; Smith, D.L.; Stanton, R.J.; Minczuk, M.; Lehner, P.J.; et al. Human Cytomegalovirus Infection Upregulates the Mitochondrial Transcription and Translation Machineries. mBio 2016, 7, e00029. [Google Scholar] [CrossRef]
- Huang, G.; Lu, H.; Hao, A.; Ng, D.C.H.; Ponniah, S.; Guo, K.; Lufei, C.; Zeng, Q.; Cao, X. GRIM-19, a Cell Death Regulatory Protein, Is Essential for Assembly and Function of Mitochondrial Complex, I. Mol. Cell. Biol. 2004, 24, 8447–8456. [Google Scholar] [CrossRef] [PubMed]
- Reeves, M.B.; Davies, A.A.; McSharry, B.P.; Wilkinson, G.W.; Sinclair, J.H. Complex I Binding by a Virally Encoded RNA Regulates Mitochondria-Induced Cell Death. Science 2007, 316, 1345–1348. [Google Scholar] [CrossRef]
- Damania, B.; Kenney, S.C.; Raab-Traub, N. Epstein-Barr virus: Biology and clinical disease. Cell 2022, 185, 3652–3670. [Google Scholar] [CrossRef] [PubMed]
- Mrozek-Gorska, P.; Buschle, A.; Pich, D.; Schwarzmayr, T.; Fechtner, R.; Scialdone, A.; Hammerschmidt, W. Epstein–Barr virus reprograms human B lymphocytes immediately in the prelatent phase of infection. Proc. Natl. Acad. Sci. USA 2019, 116, 16046–16055. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.W.; Shen, H.; Nobre, L.; Ersing, I.; Paulo, J.A.; Trudeau, S.; Wang, Z.; Smith, N.A.; Ma, Y.; Reinstadler, B.; et al. Epstein-Barr-Virus-Induced One-Carbon Metabolism Drives B Cell Transformation. Cell Metab. 2019, 30, 539–555.e11. [Google Scholar] [CrossRef] [PubMed]
- Schober, F.A.; Moore, D.; Atanassov, I.; Moedas, M.F.; Clemente, P.; Végvári, A.; El Fissi, N.; Filograna, R.; Bucher, A.-L.; Hinze, Y.; et al. The one-carbon pool controls mitochondrial energy metabolism via complex I and iron-sulfur clusters. Sci. Adv. 2021, 7, eabf0717. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Maklashina, E.; Cecchini, G.; Iverson, T.M. The roles of SDHAF2 and dicarboxylate in covalent flavinylation of SDHA, the human complex II flavoprotein. Proc. Natl. Acad. Sci. USA 2020, 117, 23548–23556. [Google Scholar] [CrossRef]
- Van Vranken, J.G.; Bricker, D.K.; Dephoure, N.; Gygi, S.P.; Cox, J.E.; Thummel, C.S.; Rutter, J. SDHAF4 Promotes Mitochondrial Succinate Dehydrogenase Activity and Prevents Neurodegeneration. Cell Metab. 2014, 20, 241–252. [Google Scholar] [CrossRef]
- Na, U.; Yu, W.; Cox, J.; Bricker, D.K.; Brockmann, K.; Rutter, J.; Thummel, C.S.; Winge, D.R. The LYR Factors SDHAF1 and SDHAF3 Mediate Maturation of the Iron-Sulfur Subunit of Succinate Dehydrogenase. Cell Metab. 2014, 20, 253–266. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, K.; Thorne, R.F.; Shi, R.; Zhang, Q.; Wu, M.; Liu, L. Mitochondrial SENP2 regulates the assembly of SDH complex under metabolic stress. Cell Rep. 2023, 42, 112041. [Google Scholar] [CrossRef]
- Selby, T.L.; Biel, N.; Varn, M.; Patel, S.; Patel, A.; Hilding, L.; Ray, A.; Ross, T.; Cramblet, W.T.; Moss, C.R.; et al. The Epstein-Barr Virus Oncoprotein, LMP1, Regulates the Function of SENP2, a SUMO-protease. Sci. Rep. 2019, 9, 9523. [Google Scholar] [CrossRef]
- McBride, A.A. The papillomavirus E2 proteins. Virology 2013, 445, 57–79. [Google Scholar] [CrossRef] [PubMed]
- Lai, D.; Tan, C.L.; Gunaratne, J.; Quek, L.S.; Nei, W.; Thierry, F.; Bellanger, S. Localization of HPV-18 E2 at Mitochondrial Membranes Induces ROS Release and Modulates Host Cell Metabolism. PLoS ONE 2013, 8, e75625. [Google Scholar] [CrossRef] [PubMed]
- Kirschberg, M.; Heuser, S.; Marcuzzi, G.P.; Hufbauer, M.; Seeger, J.M.; Đukić, A.; Tomaić, V.; Majewski, S.; Wagner, S.; Wittekindt, C.; et al. ATP synthase modulation leads to an increase of spare respiratory capacity in HPV associated cancers. Sci. Rep. 2020, 10, 17339. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.-J.; Gu, P.-Q.; Zhao, W.; Ding, W.-Y.; Zhao, X.-Q.; Guo, S.-Y.; Zhong, T.-Y. The role of globular heads of the C1q receptor in HPV 16 E2-induced human cervical squamous carcinoma cell apoptosis is associated with p38 MAPK/JNK activation. J. Transl. Med. 2013, 11, 118. [Google Scholar] [CrossRef]
- Kanki, T.; Ohgaki, K.; Gaspari, M.; Gustafsson, C.M.; Fukuoh, A.; Sasaki, N.; Hamasaki, N.; Kang, D. Architectural Role of Mitochondrial Transcription Factor A in Maintenance of Human Mitochondrial DNA. Mol. Cell. Biol. 2004, 24, 9823–9834. [Google Scholar] [CrossRef] [PubMed]
- Boczonadi, V.; Ricci, G.; Horvath, R. Mitochondrial DNA transcription and translation: Clinical syndromes. Essays Biochem. 2018, 62, 321–340. [Google Scholar] [CrossRef] [PubMed]
- Guarnieri, J.W.; Dybas, J.M.; Fazelinia, H.; Kim, M.S.; Frere, J.; Zhang, Y.; Albrecht, Y.S.; Murdock, D.G.; Angelin, A.; Singh, L.N.; et al. Targeted down Regulation of Core Mitochondrial Genes during SARS-CoV-2 Infection. bioRxiv 2022. Available online: https://www.biorxiv.org/content/10.1101/2022.02.19.481089v1 (accessed on 1 October 2023).
- Too, I.H.K.; Bonne, I.; Tan, E.L.; Chu, J.J.H.; Alonso, S. Prohibitin plays a critical role in Enterovirus 71 neuropathogenesis. PLoS Pathog. 2018, 14, e1006778. [Google Scholar] [CrossRef]
- Lee, I.-K.; Lee, S.-A.; Kim, H.; Won, Y.-S.; Kim, B.-J. Induction of endoplasmic reticulum-derived oxidative stress by an occult infection related S surface antigen variant. World J. Gastroenterol. 2015, 21, 6872–6883. [Google Scholar] [CrossRef]
- Krylatov, A.V.; Maslov, L.N.; Voronkov, N.S.; Boshchenko, A.A.; Popov, S.V.; Gomez, L.; Wang, H.; Jaggi, A.S.; Downey, J.M. Reactive Oxygen Species as Intracellular Signaling Molecules in the Cardiovascular System. Curr. Cardiol. Rev. 2018, 14, 290–300. [Google Scholar] [CrossRef] [PubMed]
- Thannickal, V.J.; Fanburg, B.L.; Yasuoka, H.; Garrett, S.M.; Nguyen, X.-X.; Artlett, C.M.; Feghali-Bostwick, C.A.; Zimmer, A.; Bagchi, A.K.; Vinayak, K.; et al. Reactive oxygen species in cell signaling. Am. J. Physiol. Cell. Mol. Physiol. 2000, 279, L1005–L1028. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.C.; Chio, C.C.; Chi, K.H.; Wu, H.M.; Lin, W.W. Superoxide anion-dependent Raf/MEK/ERK activation by peroxisome proliferator activated receptor gamma agonists 15-deoxy-delta(12,14)-prostaglandin J(2), ciglitazone, and GW1929. Exp. Cell Res. 2002, 277, 192–200. [Google Scholar] [CrossRef] [PubMed]
- Ghasemnejad-Berenji, M.; Pashapour, S. SARS-CoV-2 and the Possible Role of Raf/MEK/ERK Pathway in Viral Survival: Is This a Potential Therapeutic Strategy for COVID-19? Pharmacology 2021, 106, 119–122. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Deng, J.; Lv, L.; Kang, Q.; Ma, P.; Yan, F.; Song, X.; Gao, B.; Zhang, Y.; Xu, J. Hydrogen Peroxide Induce Human Cytomegalovirus Replication through the Activation of p38-MAPK Signaling Pathway. Viruses 2015, 7, 2816–2833. [Google Scholar] [CrossRef] [PubMed]
- Luong, T.T.D.; Tran, G.V.Q.; Shin, D.-J.; Lim, Y.-S.; Hwang, S.B. Hepatitis C Virus Exploits Death Receptor 6-mediated Signaling Pathway to Facilitate Viral Propagation. Sci. Rep. 2017, 7, 6445. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.-N.; Yang, W.K.; Kim, J.; Kim, H.S.; Kim, E.J.; Yun, H.; Park, H.; Kim, S.S.; Choe, W.; Kang, I.; et al. Reactive oxygen species stabilize hypoxia-inducible factor-1 alpha protein and stimulate transcriptional activity via AMP-activated protein kinase in DU145 human prostate cancer cells. Carcinogenesis 2008, 29, 713–721. [Google Scholar] [CrossRef]
- Willson, J.A.; Arienti, S.; Sadiku, P.; Reyes, L.; Coelho, P.; Morrison, T.; Rinaldi, G.; Dockrell, D.H.; Whyte, M.K.B.; Walmsley, S.R. Neutrophil HIF-1α stabilization is augmented by mitochondrial ROS produced via the glycerol 3-phosphate shuttle. Blood 2022, 139, 281–286. [Google Scholar] [CrossRef]
- Duette, G.; Gerber, P.P.; Rubione, J.; Perez, P.S.; Landay, A.L.; Crowe, S.M.; Liao, Z.; Witwer, K.W.; Holgado, M.P.; Salido, J.; et al. Induction of HIF-1α by HIV-1 Infection in CD4 + T Cells Promotes Viral Replication and Drives Extracellular Vesicle-Mediated Inflammation. mBio 2018, 9, e00757-18. [Google Scholar] [CrossRef]
- Lan, X.; Cheng, K.; Chandel, N.; Lederman, R.; Jhaveri, A.; Husain, M.; Malhotra, A.; Singhal, P.C. High glucose enhances HIV entry into T cells through upregulation of CXCR4. J. Leukoc. Biol. 2013, 94, 769–777. [Google Scholar] [CrossRef]
- Gullberg, R.C.; Steel, J.J.; Moon, S.; Soltani, E.; Geiss, B.J. Oxidative stress influences positive strand RNA virus genome synthesis and capping. Virology 2015, 475, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Bai, L.; Dong, J.; Liu, Z.; Rao, Y.; Feng, P.; Lan, K. Viperin catalyzes methionine oxidation to promote protein expression and function of helicases. Sci. Adv. 2019, 5, eaax1031. [Google Scholar] [CrossRef] [PubMed]
- Shim, J.-H.; Kim, K.-H.; Cho, Y.-S.; Choi, H.-S.; Song, E.Y.; Myung, P.-K.; Kang, J.S.; Suh, S.-K.; Park, S.N.; Yoon, D.-Y. Protective effect of oxidative stress in HaCaT keratinocytes expressing E7 oncogene. Amino Acids 2007, 34, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Schaedler, S.; Krause, J.; Himmelsbach, K.; Carvajal-Yepes, M.; Lieder, F.; Klingel, K.; Nassal, M.; Weiss, T.S.; Werner, S.; Hildt, E. Hepatitis B Virus Induces Expression of Antioxidant Response Element-regulated Genes by Activation of Nrf2. J. Biol. Chem. 2010, 285, 41074–41086. [Google Scholar] [CrossRef] [PubMed]
- Brault, C.; Lévy, P.; Duponchel, S.; Michelet, M.; Sallé, A.; Pécheur, E.-I.; Plissonnier, M.-L.; Parent, R.; Véricel, E.; Ivanov, A.V.; et al. Glutathione peroxidase 4 is reversibly induced by HCV to control lipid peroxidation and to increase virion infectivity. Gut 2014, 65, 144–154. [Google Scholar] [CrossRef]
- Speir, E.; Shibutani, T.; Yu, Z.-X.; Ferrans, V.; Epstein, S.E. Role of Reactive Oxygen Intermediates in Cytomegalovirus Gene Expression and in the Response of Human Smooth Muscle Cells to Viral Infection. Circ. Res. 1996, 79, 1143–1152. [Google Scholar] [CrossRef] [PubMed]
- Tilton, C.; Clippinger, A.J.; Maguire, T.; Alwine, J.C. Human Cytomegalovirus Induces Multiple Means To Combat Reactive Oxygen Species. J. Virol. 2011, 85, 12585–12593. [Google Scholar] [CrossRef]
- Chen, K.-K.; Minakuchi, M.; Wuputra, K.; Ku, C.-C.; Pan, J.-B.; Kuo, K.-K.; Lin, Y.-C.; Saito, S.; Lin, C.-S.; Yokoyama, K.K. Redox control in the pathophysiology of influenza virus infection. BMC Microbiol. 2020, 20, 214. [Google Scholar] [CrossRef]
- Anderton, E.; Yee, J.; Smith, P.; Crook, T.; White, R.E.; Allday, M.J. Two Epstein-Barr virus (EBV) oncoproteins cooperate to repress expression of the proapoptotic tumour-suppressor Bim: Clues to the pathogenesis of Burkitt’s lymphoma. Oncogene 2008, 27, 421–433. [Google Scholar] [CrossRef]
- You, Y.; Cheng, A.-C.; Wang, M.-S.; Jia, R.-Y.; Sun, K.-F.; Yang, Q.; Wu, Y.; Zhu, D.; Chen, S.; Liu, M.-F.; et al. The suppression of apoptosis by α-herpesvirus. Cell Death Dis. 2017, 8, e2749. [Google Scholar] [CrossRef]
- Peischard, S.; Ho, H.T.; Theiss, C.; Strutz-Seebohm, N.; Seebohm, G. A Kidnapping Story: How Coxsackievirus B3 and Its Host Cell Interact. Cell. Physiol. Biochem. 2019, 53, 121–140. [Google Scholar] [CrossRef]
- Fislová, T.; Thomas, B.; Graef, K.M.; Fodor, E. Association of the Influenza Virus RNA Polymerase Subunit PB2 with the Host Chaperonin CCT. J. Virol. 2010, 84, 8691–8699. [Google Scholar] [CrossRef]
- Arakawa, C.; Endo, A.; Kohira, R.; Fujita, Y.; Fuchigami, T.; Mugishima, H.; Ohtake, A.; Murayama, K.; Mori, M.; Miyata, R.; et al. Liver-specific mitochondrial respiratory chain complex I deficiency in fatal influenza encephalopathy. Brain Dev. 2012, 34, 115–117. [Google Scholar] [CrossRef]
- Gorai, T.; Goto, H.; Noda, T.; Watanabe, T.; Kozuka-Hata, H.; Oyama, M.; Takano, R.; Neumann, G.; Watanabe, S.; Kawaoka, Y. F1Fo-ATPase, F-type proton-translocating ATPase, at the plasma membrane is critical for efficient influenza virus budding. Proc. Natl. Acad. Sci. USA 2012, 109, 4615–4620. [Google Scholar] [CrossRef]
- Hu, M.; Bogoyevitch, M.A.; Jans, D.A. Subversion of Host Cell Mitochondria by RSV to Favor Virus Production is Dependent on Inhibition of Mitochondrial Complex I and ROS Generation. Cells 2019, 8, 1417. [Google Scholar] [CrossRef]
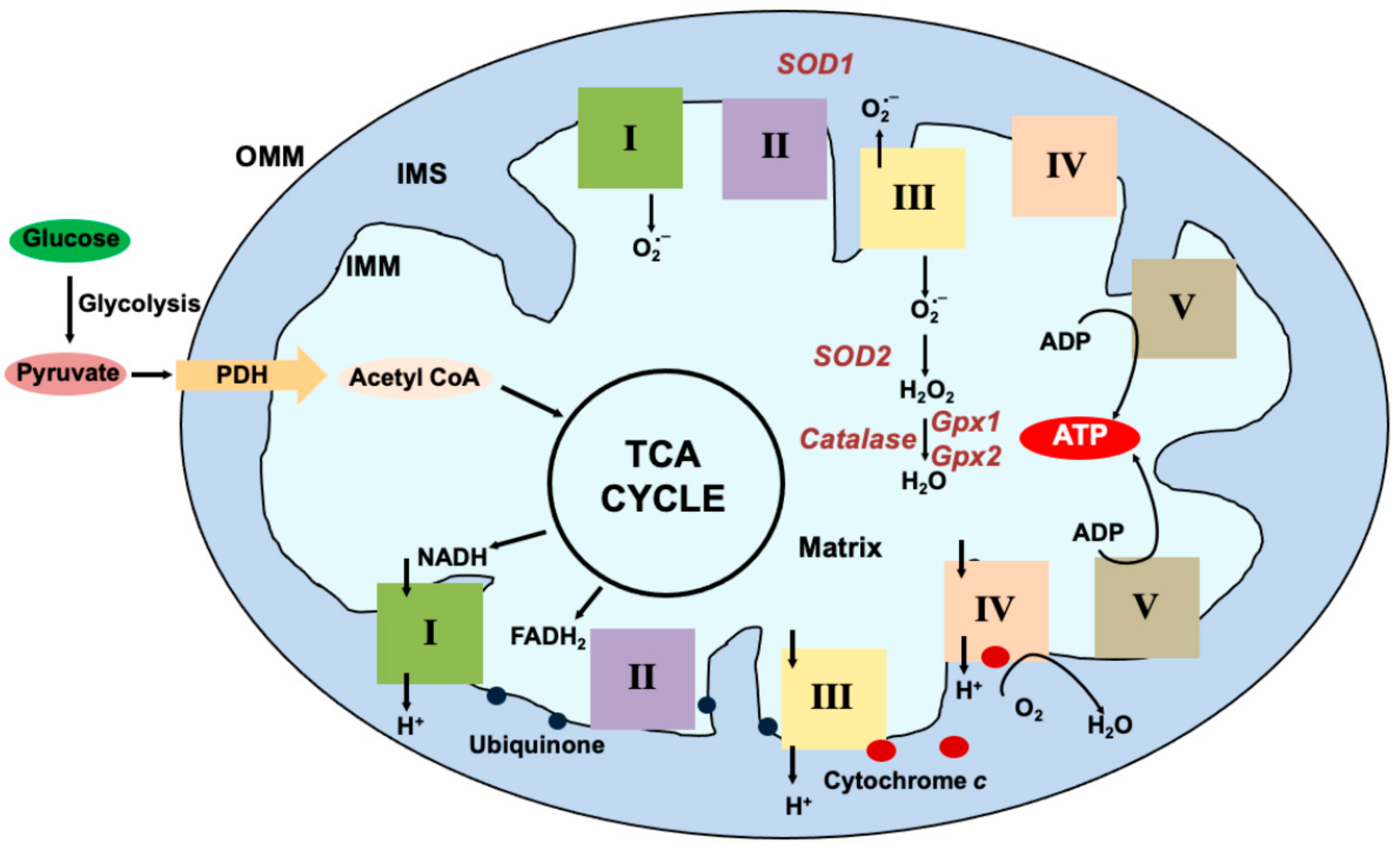

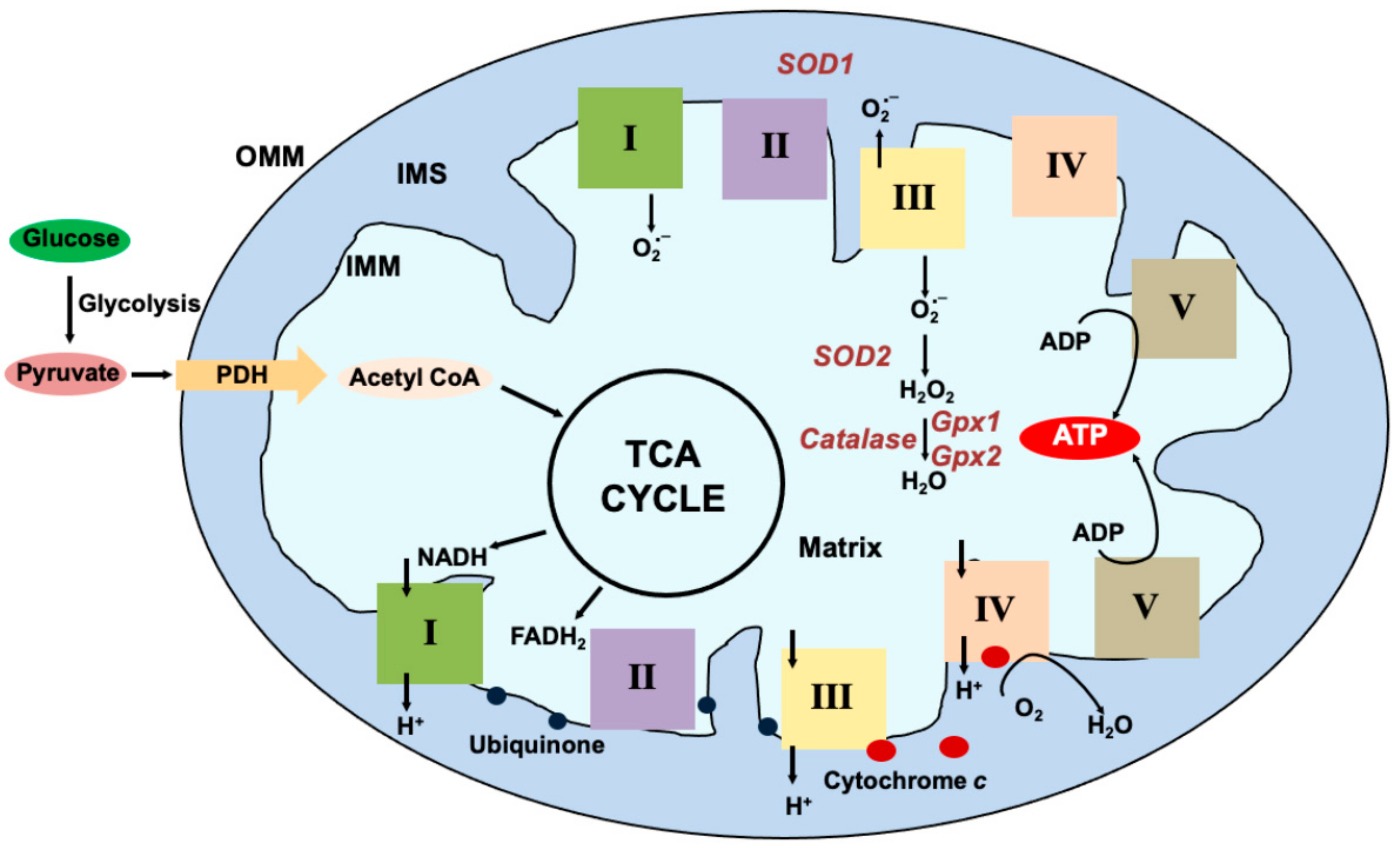{kind=link}
| Group | Virus | Complex | Effect | Reference |
|---|---|---|---|---|
| (+) ssRNA | CVB3 | I | Induces | [161] |
| CVB3 | III | Inhibits | [161] | |
| HCV | I | Inhibits | [42] | |
| HCV | III | Inhibits | [44] | |
| HEV | III | Induces | [80] | |
| Rubella Virus | II, III | Induces | [75] | |
| Rubella Virus | IV | Inhibits | [75] | |
| SARS-CoV-2 | I | Inhibits | [69] | |
| SARS-CoV-2 | III | Inhibits | [70] | |
| ZIKV | II | Inhibits | [57] | |
| ZIKV | V | Induces | [59] | |
| West Nile Virus | II | Inhibits | [63] | |
| (-) ssRNA | H5N1 Virus | IV | Induces | [69] |
| Influenza Virus | II | Induces | [162] | |
| Influenza Virus | III | Induces | [163] | |
| Influenza Virus | V | Induces | [164] | |
| Rabies Virus | I | Induces | [90] | |
| Rabies Virus | IV | Induces | [89] | |
| RSV | I | Inhibits | [165] | |
| ssRNA-RT | HIV | I | Inhibits | [98] |
| HIV | III | Inhibits | [97] | |
| HIV | IV | Induces | [101] | |
| HIV | IV | Inhibits | [102] | |
| HIV | V | Induces | [103] | |
| dsRNA-RT | HBV | I, III, IV, V | Inhibits | [110] |
| HBV | II | Inhibits | [111] | |
| dsDNA | HCMV | I, II, III, IV, V | Induces | [116] |
| EBV | II | Inhibits | [131] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Purandare, N.; Ghosalkar, E.; Grossman, L.I.; Aras, S. Mitochondrial Oxidative Phosphorylation in Viral Infections. Viruses 2023, 15, 2380. https://doi.org/10.3390/v15122380
Purandare N, Ghosalkar E, Grossman LI, Aras S. Mitochondrial Oxidative Phosphorylation in Viral Infections. Viruses. 2023; 15(12):2380. https://doi.org/10.3390/v15122380
Chicago/Turabian StylePurandare, Neeraja, Esha Ghosalkar, Lawrence I. Grossman, and Siddhesh Aras. 2023. "Mitochondrial Oxidative Phosphorylation in Viral Infections" Viruses 15, no. 12: 2380. https://doi.org/10.3390/v15122380
APA StylePurandare, N., Ghosalkar, E., Grossman, L. I., & Aras, S. (2023). Mitochondrial Oxidative Phosphorylation in Viral Infections. Viruses, 15(12), 2380. https://doi.org/10.3390/v15122380