

Diversity and Complexity of Internally Deleted Viral Genomes in Influenza A Virus Subpopulations with Enhanced Interferon-Inducing Phenotypes
 , ,
, ,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Viruses and Cells
2.2. Genome Amplification, Deep Sequencing, and Sequence Analysis
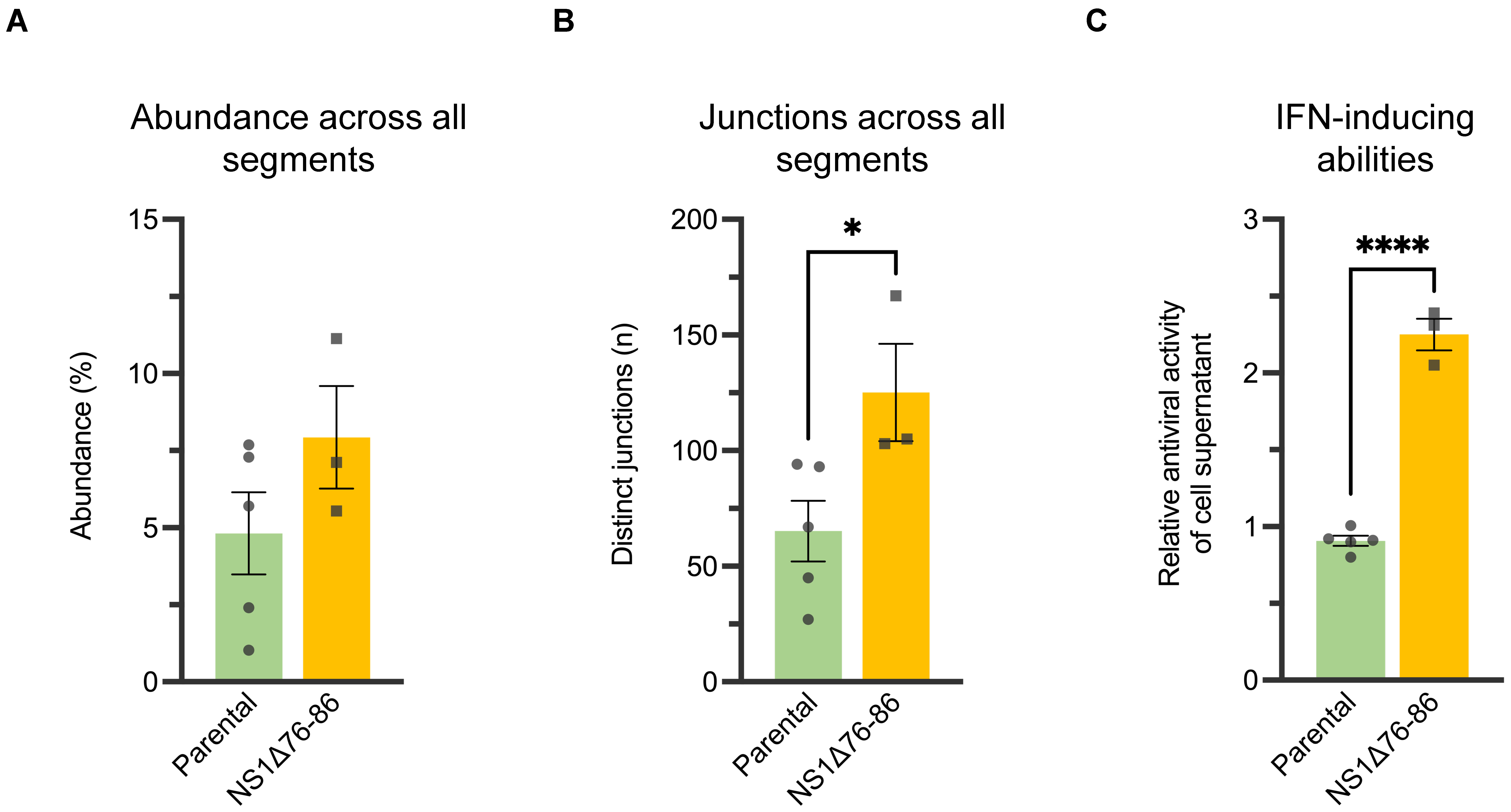
2.3. Induction and Quantification of Biologically Active IFNs
2.4. Statistical Analysis, Visualization, and Availability of Data
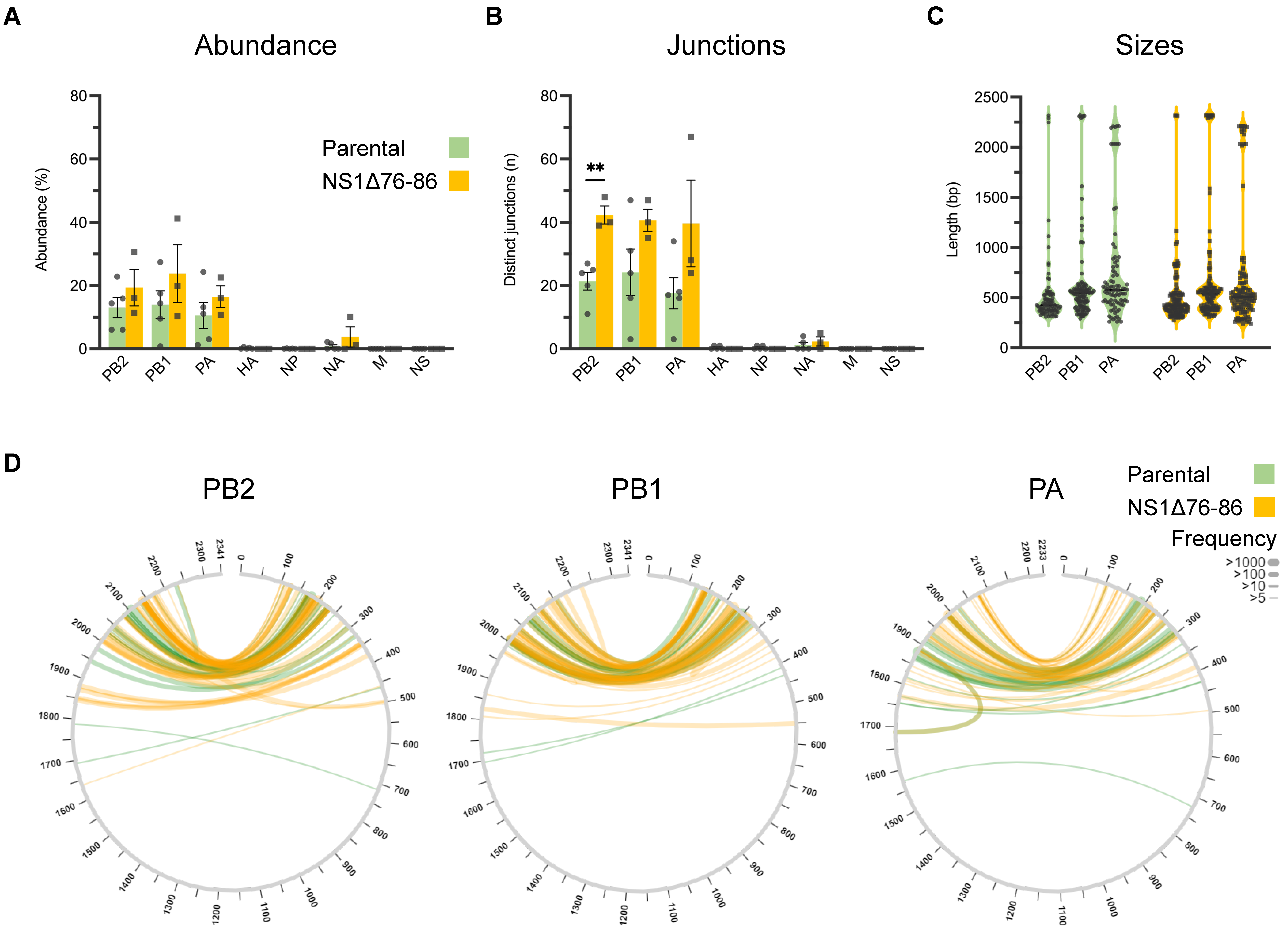
3. Results
3.1. ID vRNA Abundance and Pattern Can Be Preserved in Subcultured PC4 Virus
3.2. Physical Separation of PC4 Viral Subpopulations through Plaque Purification Significantly Reduced ID vRNA Abundance
3.3. ID vRNAs Can Encode Large Libraries of Intrinsically Disordered Polypeptides with Potential Protein Binding Sites
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Domingo, E.; Sheldon, J.; Perales, C. Viral quasispecies evolution. Microbiol. Mol. Biol. Rev. 2012, 76, 159–216. [Google Scholar] [CrossRef] [PubMed]
- Ghorbani, A.; Ngunjiri, J.M.; Lee, C.-W. Influenza A Virus Subpopulations and Their Implication in Pathogenesis and Vaccine Development. Annu. Rev. Anim. Biosci. 2020, 8, 247–267. [Google Scholar] [CrossRef] [PubMed]
- Brooke, C.B. Population diversity and collective interactions during influenza virus infection. J. Virol. 2017, 91, e01164-17. [Google Scholar] [CrossRef] [PubMed]
- von Magnus, P. Propagation of the PR8 Strain of Influenza A Virus in Chick Embryos. II. The Formation of “Incomplete” Virus following Inoculation of Large Doses of Seed Virus. Acta Pathol. Microbiol. Scand. 1951, 28, 278–293. [Google Scholar] [CrossRef] [PubMed]
- von Magnus, P. Incomplete forms of influenza virus. Adv. Virus Res. 1954, 2, 59–79. [Google Scholar] [PubMed]
- Marcus, P.I.; Ngunjiri, J.M.; Sekellick, M.J. Dynamics of biologically active subpopulations of influenza virus: Plaque-forming, noninfectious cell-killing, and defective interfering particles. J. Virol. 2009, 83, 8122–8130. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Nayak, D.; Chambers, T.; Akkina, R. Defective-interfering (DI) RNAs of influenza viruses: Origin, structure, expression, and interference. In Current Topics in Microbiology and Immunology; Springer: Berlin/Heidelberg, Germany, 1985; pp. 103–151. [Google Scholar]
- Pelz, L.; Rüdiger, D.; Dogra, T.; Alnaji, F.G.; Genzel, Y.; Brooke, C.B.; Kupke, S.Y.; Reichl, U. Semi-continuous propagation of influenza A virus and its defective interfering particles: Analyzing the dynamic competition to select candidates for antiviral therapy. J. Virol. 2021, 95, e01174-21. [Google Scholar] [CrossRef]
- Tapia, K.; Kim, W.-K.; Sun, Y.; Mercado-López, X.; Dunay, E.; Wise, M.; Adu, M.; López, C.B. Defective viral genomes arising in vivo provide critical danger signals for the triggering of lung antiviral immunity. PLoS Pathog. 2013, 9, e1003703. [Google Scholar] [CrossRef]
- Saira, K.; Lin, X.; DePasse, J.V.; Halpin, R.; Twaddle, A.; Stockwell, T.; Angus, B.; Cozzi-Lepri, A.; Delfino, M.; Dugan, V.; et al. Sequence analysis of in vivo defective interfering-like RNA of influenza A H1N1 pandemic virus. J. Virol. 2013, 87, 8064–8074. [Google Scholar] [CrossRef]
- Ngunjiri, J.M.; Ali, A.; Boyaka, P.; Marcus, P.I.; Lee, C.W. In Vivo Assessment of NS1-Truncated Influenza Virus with a Novel SLSYSINWRH Motif as a Self-Adjuvanting Live Attenuated Vaccine. PLoS ONE 2015, 10, e0118934. [Google Scholar] [CrossRef]
- Dimmock, N.J.; Rainsford, E.W.; Scott, P.D.; Marriott, A.C. Influenza virus protecting RNA: An effective prophylactic and therapeutic antiviral. J. Virol. 2008, 82, 8570–8578. [Google Scholar] [CrossRef] [PubMed]
- Easton, A.J.; Scott, P.D.; Edworthy, N.L.; Meng, B.; Marriott, A.C.; Dimmock, N.J. A novel broad-spectrum treatment for respiratory virus infections: Influenza-based defective interfering virus provides protection against pneumovirus infection in vivo. Vaccine 2011, 29, 2777–2784. [Google Scholar] [CrossRef] [PubMed]
- Wasik, M.A.; Eichwald, L.; Genzel, Y.; Reichl, U. Cell culture-based production of defective interfering particles for influenza antiviral therapy. Appl. Microbiol. Biotechnol. 2018, 102, 1167–1177. [Google Scholar] [CrossRef] [PubMed]
- Dimmock, N.J.; Easton, A.J. Defective interfering influenza virus RNAs: Time to reevaluate their clinical potential as broad-spectrum antivirals? J. Virol. 2014, 88, 5217–5227. [Google Scholar] [CrossRef] [PubMed]
- Marriott, A.C.; Dimmock, N.J. Defective interfering viruses and their potential as antiviral agents. Rev. Med. Virol. 2010, 20, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Marcus, P.I.; Ngunjiri, J.M.; Sekellick, M.J.; Wang, L.; Lee, C.W. In vitro analysis of virus particle subpopulations in candidate live-attenuated influenza vaccines distinguishes effective from ineffective vaccines. J. Virol. 2010, 84, 10974–10981. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Noble, S.; McLain, L.; Dimmock, N.J. Interfering vaccine: A novel antiviral that converts a potentially virulent infection into one that is subclinical and immunizing. Vaccine 2004, 22, 3018–3025. [Google Scholar] [CrossRef]
- Mann, A.; Marriott, A.; Balasingam, S.; Lambkin, R.; Oxford, J.; Dimmock, N. Interfering vaccine (defective interfering influenza A virus) protects ferrets from influenza, and allows them to develop solid immunity to reinfection. Vaccine 2006, 24, 4290–4296. [Google Scholar] [CrossRef]
- Dimmock, N.J.; Beck, S.; McLain, L. Protection of mice from lethal influenza: Evidence that defective interfering virus modulates the immune response and not virus multiplication. J. Gen. Virol. 1986, 67, 839–850. [Google Scholar] [CrossRef]
- Davis, A.R.; Hiti, A.L.; Nayak, D.P. Influenza defective interfering viral RNA is formed by internal deletion of genomic RNA. Proc. Natl. Acad. Sci. USA 1980, 77, 215–219. [Google Scholar] [CrossRef]
- Duhaut, S.D.; Dimmock, N.J. Heterologous protection of mice from a lethal human H1N1 influenza A virus infection by H3N8 equine defective interfering virus: Comparison of defective RNA sequences isolated from the DI inoculum and mouse lung. Virology 1998, 248, 241–253. [Google Scholar] [CrossRef] [PubMed]
- Jennings, P.A.; Finch, J.T.; Winter, G.; Robertson, J.S. Does the higher order structure of the influenza virus ribonucleoprotein guide sequence rearrangements in influenza viral RNA? Cell 1983, 34, 619–627. [Google Scholar] [CrossRef] [PubMed]
- Fodor, E.; Crow, M.; Mingay, L.J.; Deng, T.; Sharps, J.; Fechter, P.; Brownlee, G.G. A single amino acid mutation in the PA subunit of the influenza virus RNA polymerase inhibits endonucleolytic cleavage of capped RNAs. J. Virol. 2002, 76, 8989–9001. [Google Scholar] [CrossRef] [PubMed]
- Nayak, D.; Tobita, K.; Janda, J.; Davis, A.; De, B. Homologous interference mediated by defective interfering influenza virus derived from a temperature-sensitive mutant of influenza virus. J. Virol. 1978, 28, 375–386. [Google Scholar] [CrossRef]
- Ngunjiri, J.M.; Lee, C.-W.; Ali, A.; Marcus, P.I. Influenza virus interferon-inducing particle efficiency is reversed in avian and mammalian cells, and enhanced in cells co-infected with defective-interfering particles. J. Interferon Cytokine Res. 2012, 32, 280–285. [Google Scholar] [CrossRef]
- te Velthuis, A.J.; Long, J.C.; Bauer, D.L.; Fan, R.L.; Yen, H.-L.; Sharps, J.; Siegers, J.Y.; Killip, M.J.; French, H.; Oliva-Martín, M.J. Mini viral RNAs act as innate immune agonists during influenza virus infection. Nat. Microbiol. 2018, 3, 1234–1242. [Google Scholar] [CrossRef]
- Fodor, E.; Te Velthuis, A.J. Structure and function of the influenza virus transcription and replication machinery. Cold Spring Harb. Perspect. Med. 2020, 10, a038398. [Google Scholar] [CrossRef]
- Perez, J.T.; Varble, A.; Sachidanandam, R.; Zlatev, I.; Manoharan, M.; García-Sastre, A.; tenOever, B.R. Influenza A virus-generated small RNAs regulate the switch from transcription to replication. Proc. Natl. Acad. Sci. USA 2010, 107, 11525–11530. [Google Scholar] [CrossRef]
- Koire, A.; Gilbert, B.E.; Sucgang, R.; Zechiedrich, L. Repurposed transcriptomic data reveal small viral RNA produced by influenza virus during infection in mice. PLoS ONE 2016, 11, e0165729. [Google Scholar] [CrossRef]
- Baum, A.; Sachidanandam, R.; García-Sastre, A. Preference of RIG-I for short viral RNA molecules in infected cells revealed by next-generation sequencing. Proc. Natl. Acad. Sci. USA 2010, 107, 16303–16308. [Google Scholar] [CrossRef]
- Dimmock, N.J.; Easton, A.J. Cloned defective interfering influenza RNA and a possible pan-specific treatment of respiratory virus diseases. Viruses 2015, 7, 3768–3788. [Google Scholar] [CrossRef] [PubMed]
- Russell, A.B.; Elshina, E.; Kowalsky, J.R.; te Velthuis, A.J.; Bloom, J.D. Single-cell virus sequencing of influenza infections that trigger innate immunity. J. Virol. 2019, 93, e00500-19. [Google Scholar] [CrossRef] [PubMed]
- Bracci, L.; Canini, I.; Venditti, M.; Spada, M.; Puzelli, S.; Donatelli, I.; Belardelli, F.; Proietti, E. Type I IFN as a vaccine adjuvant for both systemic and mucosal vaccination against influenza virus. Vaccine 2006, 24 (Suppl. 2), S56–S57. [Google Scholar] [CrossRef] [PubMed]
- Bracci, L.; Canini, I.; Puzelli, S.; Sestili, P.; Venditti, M.; Spada, M.; Donatelli, I.; Belardelli, F.; Proietti, E. Type I IFN is a powerful mucosal adjuvant for a selective intranasal vaccination against influenza virus in mice and affects antigen capture at mucosal level. Vaccine 2005, 23, 2994–3004. [Google Scholar] [CrossRef]
- Proietti, E.; Bracci, L.; Puzelli, S.; Di Pucchio, T.; Sestili, P.; De Vincenzi, E.; Venditti, M.; Capone, I.; Seif, I.; De Maeyer, E. Type I IFN as a natural adjuvant for a protective immune response: Lessons from the influenza vaccine model. J. Immunol. 2002, 169, 375–383. [Google Scholar] [CrossRef]
- Marcus, P.I.; Girshick, T.; van der Heide, L.; Sekellick, M.J. Super-sentinel chickens and detection of low-pathogenicity influenza virus. Emerg. Infect. Dis. 2007, 13, 1608–1610. [Google Scholar] [CrossRef]
- Jang, H.; Ngunjiri, J.M.; Lee, C.-W. Association between interferon response and protective efficacy of NS1-truncated mutants as influenza vaccine candidates in chickens. PLoS ONE 2016, 11, e0156603. [Google Scholar] [CrossRef]
- Rosário-Ferreira, N.; Preto, A.J.; Melo, R.; Moreira, I.S.; Brito, R.M. The Central Role of Non-Structural Protein 1 (NS1) in Influenza Biology and Infection. Int. J. Mol. Sci. 2020, 21, 1511. [Google Scholar] [CrossRef]
- Richt, J.A.; García-Sastre, A. Attenuated influenza virus vaccines with modified NS1 proteins. In Vaccines for Pandemic Influenza; Compans, R.W., Orenstein, W.A., Eds.; Springer: Berlin/Heidelberg, Germany, 2009; pp. 177–195. [Google Scholar]
- Wang, L.; Suarez, D.; Pantin-Jackwood, M.; Mibayashi, M.; Garcia-Sastre, A.; Saif, Y.; Lee, C.-W. Characterization of influenza virus variants with different sizes of the non-structural (NS) genes and their potential as a live influenza vaccine in poultry. Vaccine 2008, 26, 3580–3586. [Google Scholar] [CrossRef]
- Jang, H.; Elaish, M.; KC, M.; Abundo, M.C.; Ghorbani, A.; Ngunjiri, J.M.; Lee, C.W. Efficacy and synergy of live-attenuated and inactivated influenza vaccines in young chickens. PLoS ONE 2018, 13, e0195285. [Google Scholar] [CrossRef]
- Ghorbani, A.; Ngunjiri, J.M.; Xia, M.; Elaish, M.; Jang, H.; Mahesh, K.C.; Abundo, M.C.; Jiang, X.; Lee, C.W. Heterosubtypic protection against avian influenza virus by live attenuated and chimeric norovirus P-particle-M2e vaccines in chickens. Vaccine 2019, 37, 1356–1364. [Google Scholar] [CrossRef] [PubMed]
- Ghorbani, A.; Abundo, M.C.; Ji, H.; Taylor, K.J.; Ngunjiri, J.M.; Lee, C.-W. Viral subpopulation screening guides in designing a high interferon-inducing live attenuated influenza vaccine by targeting rare mutations in NS1 and PB2 proteins. J. Virol. 2020, 95, e01722-20. [Google Scholar] [CrossRef] [PubMed]
- Ghorbani, A.; Ngunjiri, J.M.; Abundo, M.E.C.; Pantin-Jackwood, M.; Kenney, S.P.; Lee, C.-W. Development of in ovo-compatible NS1-truncated live attenuated influenza vaccines by modulation of hemagglutinin cleavage and polymerase acidic X frameshifting sites. Vaccine 2023, 41, 1848–1858. [Google Scholar] [CrossRef] [PubMed]
- Alnaji, F.G.; Holmes, J.R.; Rendon, G.; Vera, J.C.; Fields, C.J.; Martin, B.E.; Brooke, C.B. Sequencing Framework for the Sensitive Detection and Precise Mapping of Defective Interfering Particle-Associated Deletions across Influenza A and B Viruses. J. Virol. 2019, 93, e00354-19. [Google Scholar] [CrossRef]
- Schat, K.; Sellers, H. Cell-culture methods. In A laboratory Manual for the Identification and Characterization of Avian Pathogens, 5th ed.; Dufour-Zavala, L., Ed.; American Association of Avian Pathologists: Jacksonville, FL, USA, 2008; pp. 195–203. [Google Scholar]
- Carver, D.H.; Marcus, P.I. Enhanced interferon production from chick embryo cells aged in in vitro. Virology 1967, 32, 247–257. [Google Scholar] [CrossRef]
- Sekellick, M.J.; Marcus, P.I. [18] Induction of high titer chicken interferon. In Methods in Enzymology; Pestka, S., Ed.; Elsevier: Cambridge, MA, USA, 1986; Volume 119, pp. 115–125. [Google Scholar]
- Sekellick, M.J.; Biggers, W.J.; Marcus, P.I. Development of the interferon system. I. In chicken cells development in ovo continues on time in vitro. Vitr. Cell. Dev. Biol. 1990, 26, 997–1003. [Google Scholar] [CrossRef]
- Zhou, B.; Donnelly, M.E.; Scholes, D.T.; St. George, K.; Hatta, M.; Kawaoka, Y.; Wentworth, D.E. Single-reaction genomic amplification accelerates sequencing and vaccine production for classical and Swine origin human influenza a viruses. J. Virol. 2009, 83, 10309–10313. [Google Scholar] [CrossRef]
- Borges, V.; Pinheiro, M.; Pechirra, P.; Guiomar, R.; Gomes, J.P. INSaFLU: An automated open web-based bioinformatics suite “from-reads” for influenza whole-genome-sequencing-based surveillance. Genome Med. 2018, 10, 46. [Google Scholar] [CrossRef]
- Sneath, P.H.; Sokal, R.R. Numerical Taxonomy: The Principles and Practice of Numerical Classification; W H Freeman & Co.: San Francisco, CA, USA, 1973. [Google Scholar]
- Zuckerkandl, E.; Pauling, L. Evolutionary divergence and convergence in proteins. In Evolving Genes and Proteins; Elsevier: Amsterdam, The Netherlands, 1965; pp. 97–166. [Google Scholar]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef]
- Huerta-Cepas, J.; Serra, F.; Bork, P. ETE 3: Reconstruction, analysis, and visualization of phylogenomic data. Mol. Biol. Evol. 2016, 33, 1635–1638. [Google Scholar] [CrossRef]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—Approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef] [PubMed]
- Erdős, G.; Pajkos, M.; Dosztányi, Z. IUPred3: Prediction of protein disorder enhanced with unambiguous experimental annotation and visualization of evolutionary conservation. Nucleic Acids Res. 2021, 49, W297–W303. [Google Scholar] [CrossRef] [PubMed]
- Dosztányi, Z.; Mészáros, B.; Simon, I. ANCHOR: Web server for predicting protein binding regions in disordered proteins. Bioinformatics 2009, 25, 2745–2746. [Google Scholar] [CrossRef]
- Mészáros, B.; Simon, I.; Dosztányi, Z. Prediction of protein binding regions in disordered proteins. PLoS Comput. Biol. 2009, 5, e1000376. [Google Scholar] [CrossRef]
- Marcus, P.I.; Rojek, J.M.; Sekellick, M.J. Interferon induction and/or production and its suppression by influenza A viruses. J. Virol. 2005, 79, 2880–2890. [Google Scholar] [CrossRef] [PubMed]
- Beauclair, G.; Mura, M.; Combredet, C.; Tangy, F.; Jouvenet, N.; Komarova, A.V. DI-tector: Defective interfering viral genomes’ detector for next-generation sequencing data. Rna 2018, 24, 1285–1296. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef]
- Dyson, H.J. Vital for Viruses: Intrinsically Disordered Proteins. J. Mol. Biol. 2023, 435, 167860. [Google Scholar] [CrossRef]
- Yewdell, J.W.; Hollý, J. DRiPs get molecular. Curr. Opin. Immunol. 2020, 64, 130–136. [Google Scholar] [CrossRef]
- Wei, J.; Yewdell, J.W. Flu DRiPs in MHC class I immunosurveillance. Virol. Sin. 2019, 34, 162–167. [Google Scholar] [CrossRef]
- Ngunjiri, J.M.; Buchek, G.M.; Mohni, K.N.; Sekellick, M.J.; Marcus, P.I. Influenza virus subpopulations: Exchange of lethal H5N1 virus NS for H1N1 virus NS triggers de novo generation of defective-interfering particles and enhances interferon-inducing particle efficiency. J. Interferon Cytokine Res. 2013, 33, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Frensing, T.; Heldt, F.S.; Pflugmacher, A.; Behrendt, I.; Jordan, I.; Flockerzi, D.; Genzel, Y.; Reichl, U. Continuous influenza virus production in cell culture shows a periodic accumulation of defective interfering particles. PLoS ONE 2013, 8, e72288. [Google Scholar] [CrossRef] [PubMed]
- Frensing, T.; Pflugmacher, A.; Bachmann, M.; Peschel, B.; Reichl, U. Impact of defective interfering particles on virus replication and antiviral host response in cell culture-based influenza vaccine production. Appl. Microbiol. Biotechnol. 2014, 98, 8999–9008. [Google Scholar] [CrossRef] [PubMed]
- Frensing, T.; Kupke, S.Y.; Bachmann, M.; Fritzsche, S.; Gallo-Ramirez, L.E.; Reichl, U. Influenza virus intracellular replication dynamics, release kinetics, and particle morphology during propagation in MDCK cells. Appl. Microbiol. Biotechnol. 2016, 100, 7181–7192. [Google Scholar] [CrossRef]
- Vasilijevic, J.; Zamarreño, N.; Oliveros, J.C.; Rodriguez-Frandsen, A.; Gómez, G.; Rodriguez, G.; Pérez-Ruiz, M.; Rey, S.; Barba, I.; Pozo, F. Reduced accumulation of defective viral genomes contributes to severe outcome in influenza virus infected patients. PLoS Pathog. 2017, 13, e1006650. [Google Scholar] [CrossRef]
- Odagiri, T.; Tobita, K. Mutation in NS2, a nonstructural protein of influenza A virus, extragenically causes aberrant replication and expression of the PA gene and leads to generation of defective interfering particles. Proc. Natl. Acad. Sci. USA 1990, 87, 5988–5992. [Google Scholar] [CrossRef]
- Pérez-Cidoncha, M.; Killip, M.J.; Oliveros, J.C.; Asensio, V.J.; Fernández, Y.; Bengoechea, J.A.; Randall, R.E.; Ortín, J. An unbiased genetic screen reveals the polygenic nature of the influenza virus anti-interferon response. J. Virol. 2014, 88, 4632–4646. [Google Scholar] [CrossRef]
- Wang, Z.; Robb, N.C.; Lenz, E.; Wolff, T.; Fodor, E.; Pleschka, S. NS reassortment of an H7-type highly pathogenic avian influenza virus affects its propagation by altering the regulation of viral RNA production and antiviral host response. J. Virol. 2010, 84, 11323–11335. [Google Scholar] [CrossRef]
- Pflug, A.; Guilligay, D.; Reich, S.; Cusack, S. Structure of influenza A polymerase bound to the viral RNA promoter. Nature 2014, 516, 355–360. [Google Scholar] [CrossRef]
- Jorba, N.; Coloma, R.; Ortín, J. Genetic trans-complementation establishes a new model for influenza virus RNA transcription and replication. PLoS Pathog. 2009, 5, e1000462. [Google Scholar] [CrossRef]
- Chang, S.; Sun, D.; Liang, H.; Wang, J.; Li, J.; Guo, L.; Wang, X.; Guan, C.; Boruah, B.M.; Yuan, L. Cryo-EM structure of influenza virus RNA polymerase complex at 4.3 Å resolution. Mol. Cell 2015, 57, 925–935. [Google Scholar] [CrossRef] [PubMed]
- Mari, R.; de la Luna, S.; Ort, J. Influenza virus NS1 protein interacts with viral transcription-replication complexes in vivo. J. Gen. Virol. 1997, 78, 2447–2451. [Google Scholar]
- De, B.K.; Nayak, D. Defective interfering influenza viruses and host cells: Establishment and maintenance of persistent influenza virus infection in MDBK and HeLa cells. J. Virol. 1980, 36, 847–859. [Google Scholar] [CrossRef]
- Kulkarni, P.; Bhattacharya, S.; Achuthan, S.; Behal, A.; Jolly, M.K.; Kotnala, S.; Mohanty, A.; Rangarajan, G.; Salgia, R.; Uversky, V. Intrinsically disordered proteins: Critical components of the wetware. Chem. Rev. 2022, 122, 6614–6633. [Google Scholar] [CrossRef] [PubMed]
- Wright, P.E.; Dyson, H.J. Intrinsically disordered proteins in cellular signalling and regulation. Nat. Rev. Mol. Cell Biol. 2015, 16, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Cuevas, M.V.R.; Hardy, M.-P.; Hollý, J.; Bonneil, É.; Durette, C.; Courcelles, M.; Lanoix, J.; Côté, C.; Staudt, L.M.; Lemieux, S. Most non-canonical proteins uniquely populate the proteome or immunopeptidome. Cell Rep. 2021, 34, 108815. [Google Scholar] [CrossRef]
- Specht, H.; Emmott, E.; Petelski, A.A.; Huffman, R.G.; Perlman, D.H.; Serra, M.; Kharchenko, P.; Koller, A.; Slavov, N. Single-cell proteomic and transcriptomic analysis of macrophage heterogeneity using SCoPE2. Genome Biol. 2021, 22, 1–27. [Google Scholar] [CrossRef]
- Zanker, D.J.; Oveissi, S.; Tscharke, D.C.; Duan, M.; Wan, S.; Zhang, X.; Xiao, K.; Mifsud, N.A.; Gibbs, J.; Izzard, L. Influenza A virus infection induces viral and cellular defective ribosomal products encoded by alternative reading frames. J. Immunol. 2019, 202, 3370–3380. [Google Scholar] [CrossRef]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ghorbani, A.; Ngunjiri, J.M.; Rendon, G.; Brooke, C.B.; Kenney, S.P.; Lee, C.-W. Diversity and Complexity of Internally Deleted Viral Genomes in Influenza A Virus Subpopulations with Enhanced Interferon-Inducing Phenotypes. Viruses 2023, 15, 2107. https://doi.org/10.3390/v15102107
Ghorbani A, Ngunjiri JM, Rendon G, Brooke CB, Kenney SP, Lee C-W. Diversity and Complexity of Internally Deleted Viral Genomes in Influenza A Virus Subpopulations with Enhanced Interferon-Inducing Phenotypes. Viruses. 2023; 15(10):2107. https://doi.org/10.3390/v15102107
Chicago/Turabian StyleGhorbani, Amir, John M. Ngunjiri, Gloria Rendon, Christopher B. Brooke, Scott P. Kenney, and Chang-Won Lee. 2023. "Diversity and Complexity of Internally Deleted Viral Genomes in Influenza A Virus Subpopulations with Enhanced Interferon-Inducing Phenotypes" Viruses 15, no. 10: 2107. https://doi.org/10.3390/v15102107
APA StyleGhorbani, A., Ngunjiri, J. M., Rendon, G., Brooke, C. B., Kenney, S. P., & Lee, C.-W. (2023). Diversity and Complexity of Internally Deleted Viral Genomes in Influenza A Virus Subpopulations with Enhanced Interferon-Inducing Phenotypes. Viruses, 15(10), 2107. https://doi.org/10.3390/v15102107