
Sequence Similarity Network Analysis Provides Insight into the Temporal and Geographical Distribution of Mutations in SARS-CoV-2 Spike Protein

 , and
, and
Abstract
:1. Introduction
1.1. Spike Protein Structure
1.2. Network Analysis of Spike Protein
2. Materials and Methods
2.1. Dataset Preparation
2.2. Alignment of Spike Protein
2.3. Network Analysis
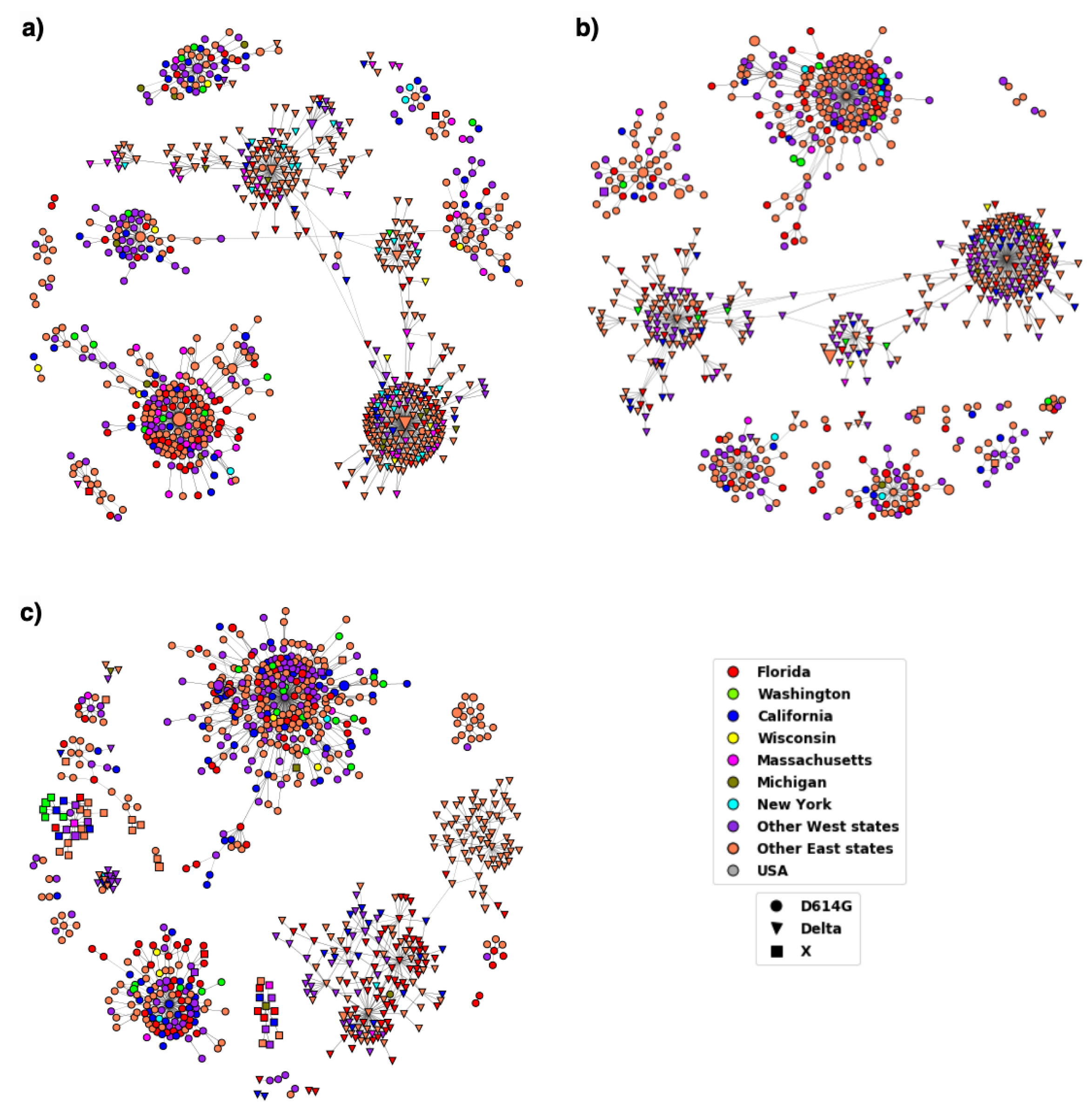
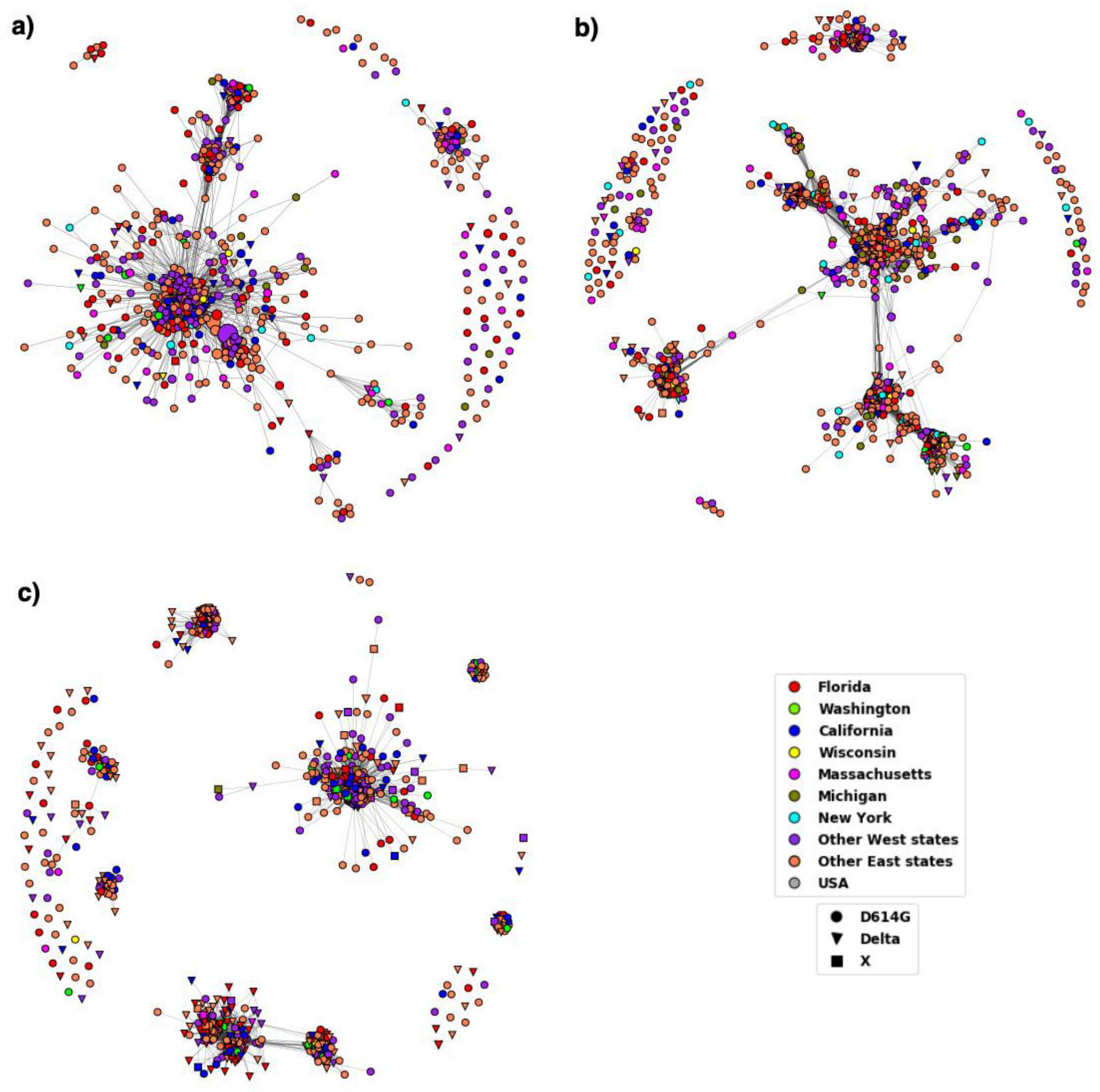
3. Results
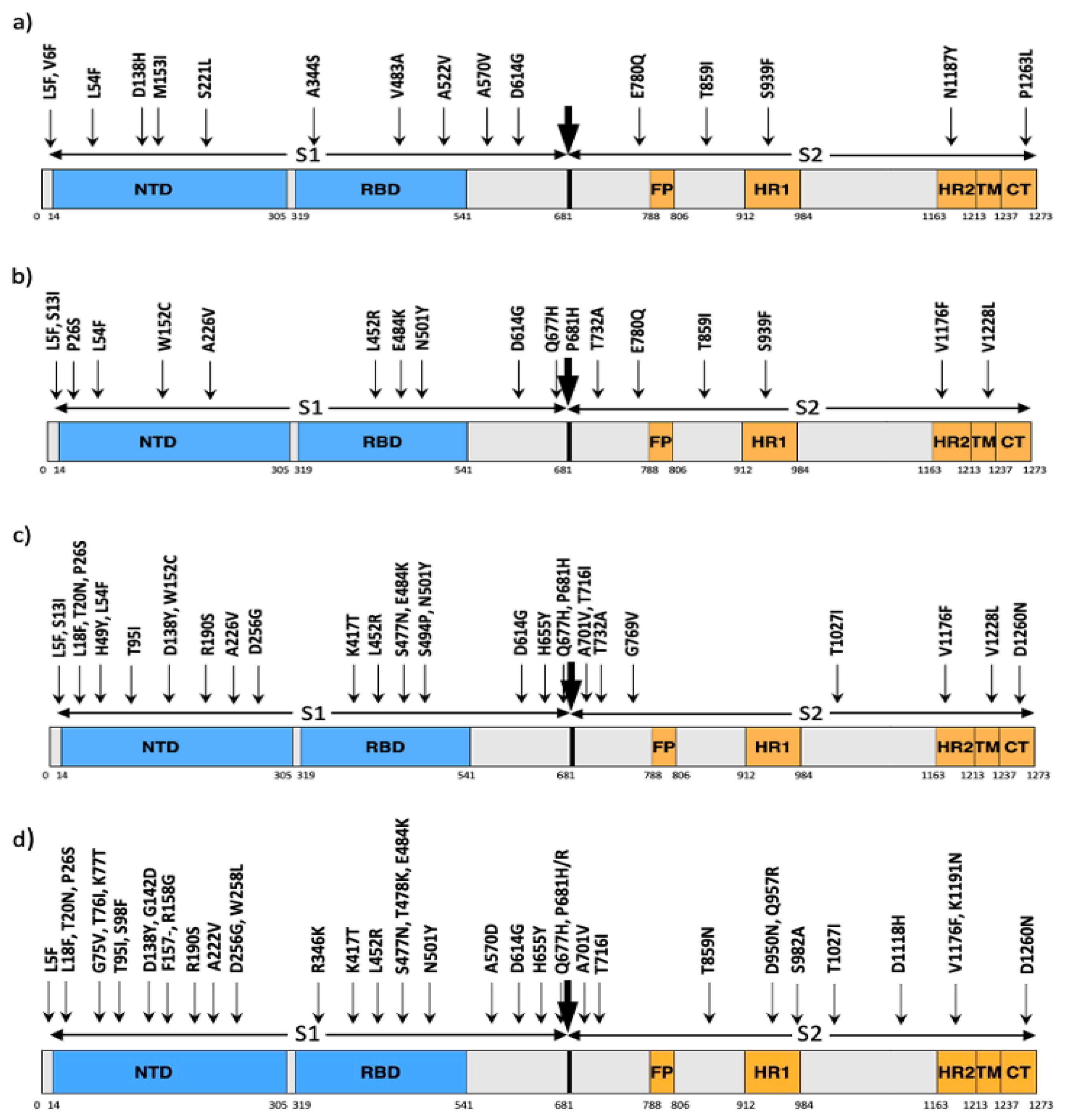
3.1. Variations in Spike Protein
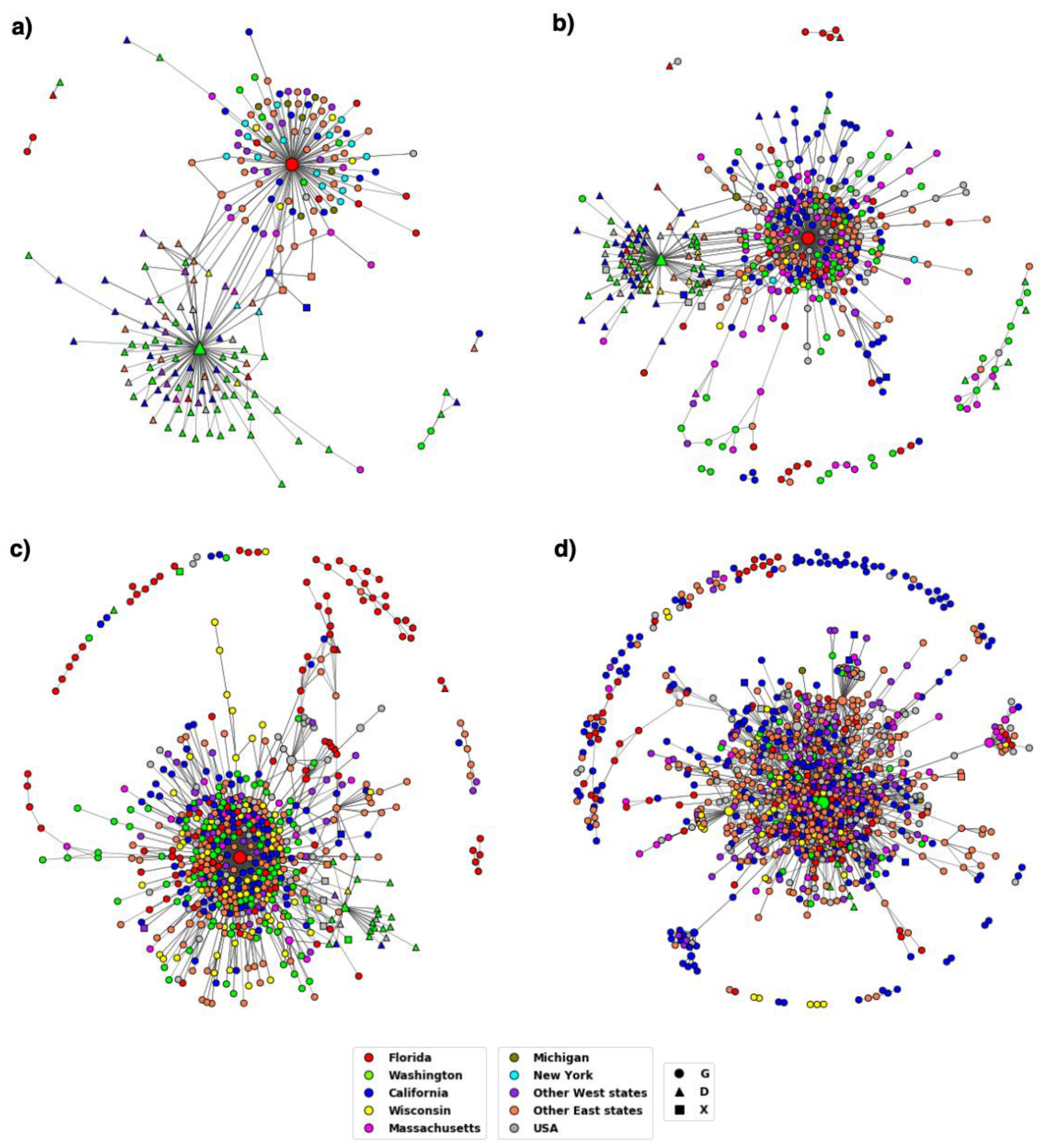
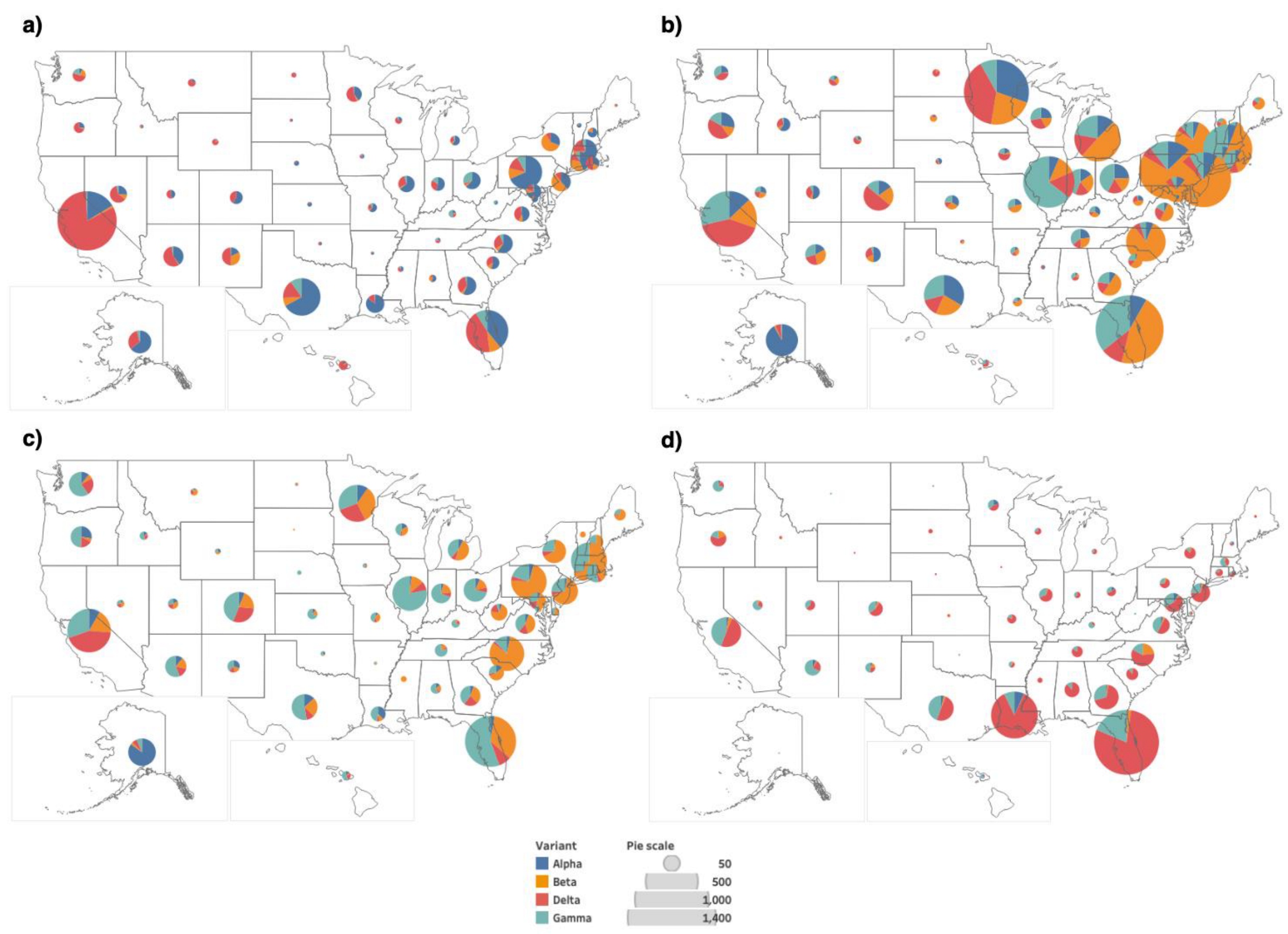
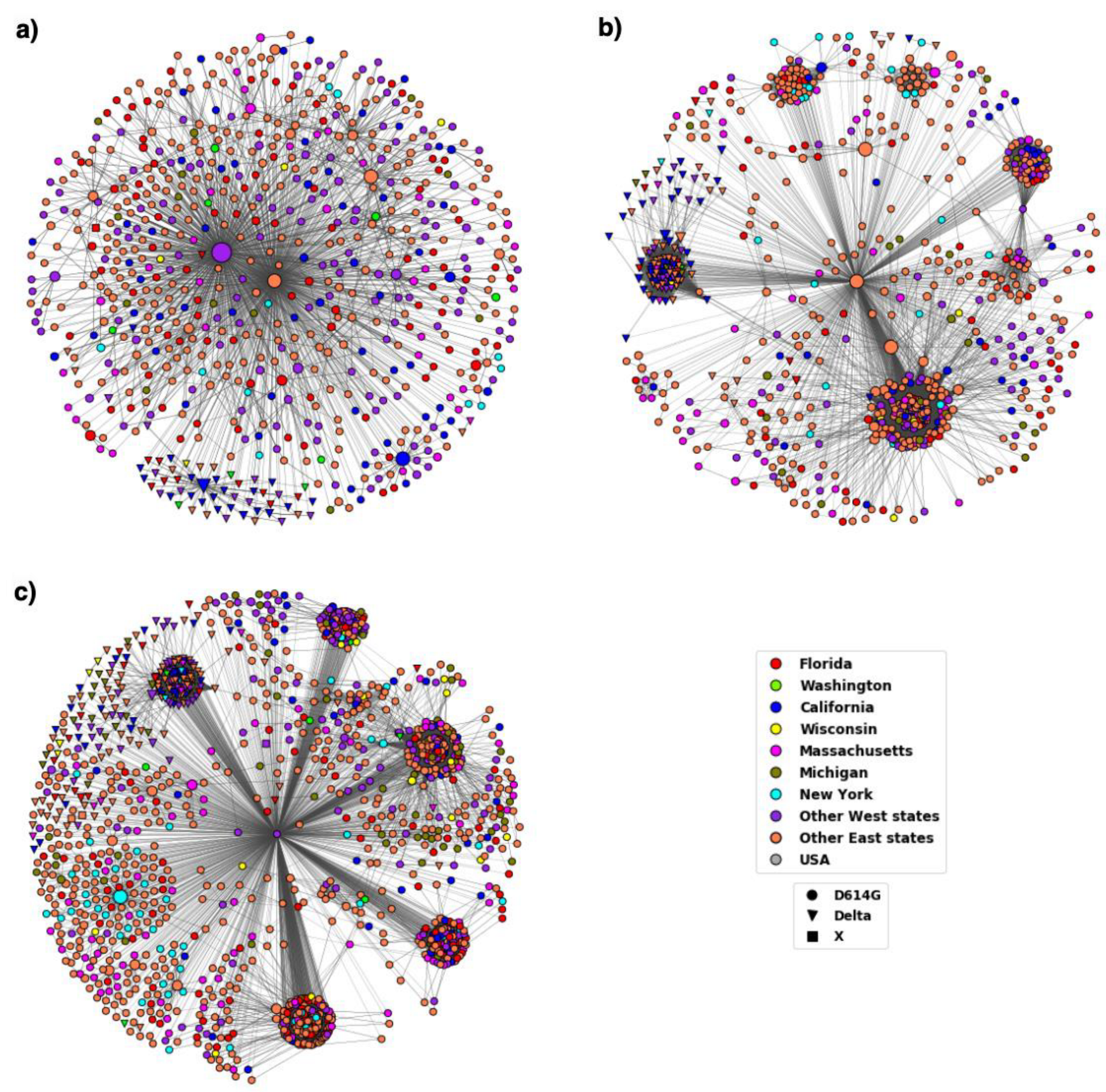
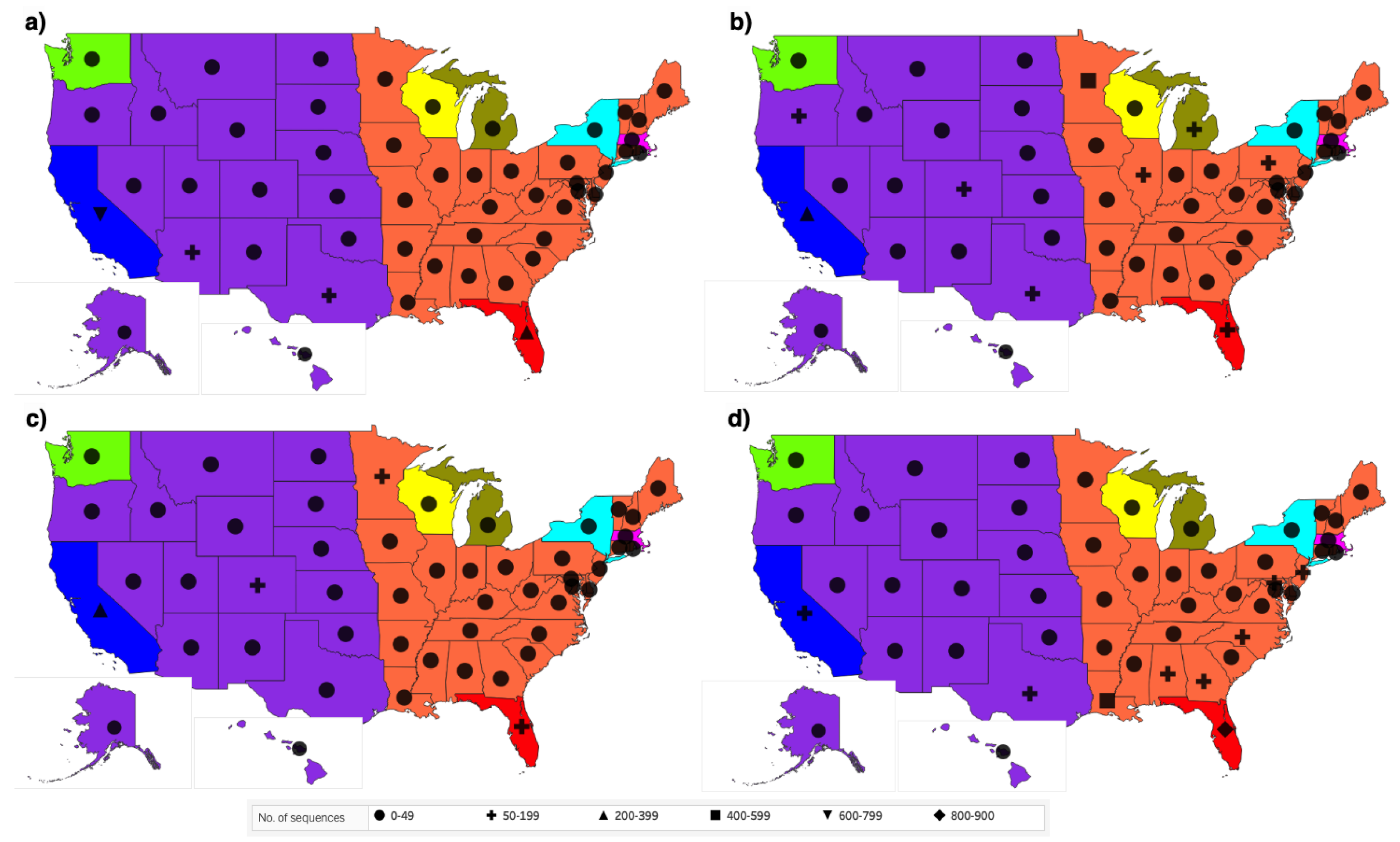
3.2. Distribution of Highly Transmissible Variants
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- COVID Live—Coronavirus Statistics—Worldometer. Available online: https://www.worldometers.info/coronavirus/ (accessed on 22 December 2021).
- Gorbalenya, A.E.; Baker, S.C.; Baric, R.S.; de Groot, R.J.; Drosten, C.; Gulyaeva, A.A.; Haagmans, B.L.; Lauber, C.; Leontovich, A.M.; Neuman, B.W.; et al. The species Severe acute respiratory syndrome-related coronavirus: Classifying 2019-nCoV and naming it SARS-CoV-2. Nat. Microbiol. 2020, 5, 536–544. [Google Scholar] [CrossRef] [Green Version]
- Demers-Mathieu, V.; Dung, M.; Mathijssen, G.B.; Sela, D.A.; Seppo, A.; Järvinen, K.M.; Medo, E. Difference in levels of SARS-CoV-2 S1 and S2 subunits- and nucleocapsid protein-reactive SIgM/IgM, IgG and SIgA/IgA antibodies in human milk. J. Perinatol. 2021, 41, 850–859. [Google Scholar] [CrossRef] [PubMed]
- Duan, L.; Zheng, Q.; Zhang, H.; Niu, Y.; Lou, Y.; Wang, H. The SARS-CoV-2 Spike Glycoprotein Biosynthesis, Structure, Function, and Antigenicity: Implications for the Design of Spike-Based Vaccine Immunogens. Front. Immunol. 2020, 11, 576622. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Yang, C.; Xu, X.F.; Xu, W.; Liu, S.W. Structural and functional properties of SARS-CoV-2 spike protein: Potential antivirus drug development for COVID-19. Acta Pharmacol. Sin. 2020, 41, 1141–1149. [Google Scholar] [CrossRef]
- Grant, O.C.; Montgomery, D.; Ito, K.; Woods, R.J. Analysis of the SARS-CoV-2 spike protein glycan shield reveals implications for immune recognition. Sci. Rep. 2020, 10, 14991. [Google Scholar] [CrossRef]
- Ni, W.; Yang, X.; Yang, D.; Bao, J.; Li, R.; Xiao, Y.; Hou, C.; Wang, H.; Liu, J.; Yang, D.; et al. Role of angiotensin-converting enzyme 2 (ACE2) in COVID-19. Crit. Care 2020, 24, 422. [Google Scholar] [CrossRef]
- Ou, X.; Liu, Y.; Lei, X.; Li, P.; Mi, D.; Ren, L.; Guo, L.; Guo, R.; Chen, T.; Hu, J.; et al. Characterization of spike glycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nat. Commun. 2020, 11, 1620. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Wu, J.; Nie, J.; Zhang, L.; Hao, H.; Liu, S.; Zhao, C.; Zhang, Q.; Liu, H.; Nie, L.; et al. The Impact of Mutations in SARS-CoV-2 Spike on Viral Infectivity and Antigenicity. Cell 2020, 182, 1284–1294. [Google Scholar] [CrossRef]
- Ugurel, O.M.; Ata, O.; Turgut-Balik, D. An updated analysis of variations in SARS-CoV-2 genome. Turk. J. Biol. 2020, 44, 157–167. [Google Scholar] [CrossRef]
- Zhao, L.; Abbasi, A.B.; Illingworth, C.J.R. Mutational load causes stochastic evolutionary outcomes in acute RNA viral infection. Virus Evol. 2019, 5, vez008. [Google Scholar] [CrossRef]
- Martínez-Flores, D.; Zepeda-Cervantes, J.; Cruz-Reséndiz, A.; Aguirre-Sampieri, S.; Sampieri, A.; Vaca, L. SARS-CoV-2 Vaccines Based on the Spike Glycoprotein and Implications of New Viral Variants. Front. Immunol. 2021, 12, 2774. [Google Scholar] [CrossRef]
- Heinz, F.X.; Stiasny, K. Distinguishing features of current COVID-19 vaccines: Knowns and unknowns of antigen presentation and modes of action. npj Vaccines 2021, 6, 104. [Google Scholar] [CrossRef]
- Wambani, J.; Okoth, P. Scope of SARS-CoV-2 variants, mutations, and vaccine technologies. Egypt. J. Intern. Med. 2022, 34, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Jia, Z.; Gong, W. Will Mutations in the Spike Protein of SARS-CoV-2 Lead to the Failure of COVID-19 Vaccines? J. Korean Med. Sci. 2021, 36, e124. [Google Scholar] [CrossRef]
- Zhang, L.; Jackson, C.B.; Mou, H.; Ojha, A.; Rangarajan, E.S.; Izard, T.; Farzan, M.; Choe, H. The D614G mutation in the SARS-CoV-2 spike protein reduces S1 shedding and increases infectivity. BioRxiv 2020, 148726. [Google Scholar] [CrossRef]
- SARS-CoV-2 Variant Classifications and Definitions. Available online: https://www.cdc.gov/coronavirus/2019-ncov/variants/variant-classifications.html (accessed on 17 January 2022).
- Ricci, G.; Salpini, R.; Svicher, V.; Alkhatib, M.; Parra-Lucares, A.; Segura, P.; Rojas, V.; Pumarino, C.; Saint-Pierre, G.; Toro, L. Emergence of SARS-CoV-2 Variants in the World: How Could This Happen? Life 2022, 12, 194. [Google Scholar] [CrossRef]
- Araf, Y.; Akter, F.; Tang, Y.D.; Fatemi, R.; Parvez, M.S.A.; Zheng, C.; Hossain, M.G. Omicron variant of SARS-CoV-2: Genomics, transmissibility, and responses to current COVID-19 vaccines. J. Med. Virol. 2022, 94, 1825–1832. [Google Scholar] [CrossRef]
- Rambaut, A.; Holmes, E.C.; O’Toole, Á.; Hill, V.; McCrone, J.T.; Ruis, C.; du Plessis, L.; Pybus, O.G. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat. Microbiol. 2020, 5, 1403. [Google Scholar] [CrossRef]
- Hadfield, J.; Megill, C.; Bell, S.M.; Huddleston, J.; Potter, B.; Callender, C.; Sagulenko, P.; Bedford, T.; Neher, R.A. Nextstrain: Real-time tracking of pathogen evolution. Bioinformatics 2018, 34, 4121–4123. [Google Scholar] [CrossRef]
- Paudel, S.; Dahal, A.; Kumar Bhattarai, H.; Yavropoulou, M.; Paraskevis, D.; Tsiodras, S. Temporal Analysis of SARS-CoV-2 Variants during the COVID-19 Pandemic in Nepal. COVID 2021, 1, 423–434. [Google Scholar] [CrossRef]
- Atkinson, H.J.; Morris, J.H.; Ferrin, T.E.; Babbitt, P.C. Using sequence similarity networks for visualization of relationships across diverse protein superfamilies. PLoS ONE 2009, 4, e4345. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.; Karkar, S.; Bapteste, E.; Yee, N.; Falkowski, P.; Bhattacharya, D. Sequence similarity network reveals the imprints of major diversification events in the evolution of microbial life. Front. Ecol. Evol. 2014, 2, 72. [Google Scholar] [CrossRef] [Green Version]
- Copp, J.N.; Akiva, E.; Babbitt, P.C.; Tokuriki, N. Revealing Unexplored Sequence-Function Space Using Sequence Similarity Networks. Biochemistry 2018, 57, 4651–4662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- González, J.M. Visualizing the superfamily of metallo-β-lactamases through sequence similarity network neighborhood connectivity analysis. Heliyon 2021, 7, e05867. [Google Scholar] [CrossRef]
- Cheang, Q.W.; Sheng, S.; Xu, L.; Liang, Z.-X. Large-scale sequence similarity analysis reveals the scope of sequence and function divergence in PilZ domain proteins. bioRxiv 2020, 2, 943704. [Google Scholar] [CrossRef]
- Padhan, K.; Parvez, M.K.; Al-Dosari, M.S. Comparative sequence analysis of SARS-CoV-2 suggests its high transmissibility and pathogenicity. Future Virol. 2021, 16, 245–254. [Google Scholar] [CrossRef]
- Ahmadi, E.; Zabihi, M.R.; Hosseinzadeh, R.; Khosroshahi, L.M.; Noorbakhsh, F. SARS-CoV-2 spike protein displays sequence similarities with paramyxovirus surface proteins; a bioinformatics study. PLoS ONE 2021, 16, e0260360. [Google Scholar] [CrossRef]
- Khaledian, E.; Ulusan, S.; Erickson, J.; Fawcett, S.; Letko, M.C.; Broschat, S.L. Sequence determinants of human-cell entry identified in ACE2-independent bat sarbecoviruses: A combined laboratory and computational network science approach. EBioMedicine 2022, 79, 103990. [Google Scholar] [CrossRef]
- NCBI Virus. Available online: https://www.ncbi.nlm.nih.gov/labs/virus/vssi/#/ (accessed on 22 October 2020).
- Catanese, H.N.; Brayton, K.A.; Gebremedhin, A.H. A nearest-neighbors network model for sequence data reveals new insight into genotype distribution of a pathogen. BMC Bioinform. 2018, 19, 475. [Google Scholar] [CrossRef]
- Ortega, J.T.; Serrano, M.L.; Pujol, F.H.; Rangel, H.R. Role of changes in SARS-CoV-2 spike protein in the interaction with the human ACE2 receptor: An in silico analysis. EXCLI J. 2020, 19, 410–417. [Google Scholar] [CrossRef]
- Wang, Q.; Zhang, Y.; Wu, L.; Zhou, H.; Yan, J.; Correspondence, J.Q. Structural and Functional Basis of SARS-CoV-2 Entry by Using Human ACE2. Cell 2020, 181, 894–904. [Google Scholar] [CrossRef]
- Jaimes, J.A.; André, N.M.; Chappie, J.S.; Millet, J.K.; Whittaker, G.R. Phylogenetic Analysis and Structural Modeling of SARS-CoV-2 Spike Protein Reveals an Evolutionary Distinct and Proteolytically Sensitive Activation Loop. J. Mol. Biol. 2020, 432, 3309–3325. [Google Scholar] [CrossRef]
- Tai, W.; He, L.; Zhang, X.; Pu, J.; Voronin, D.; Jiang, S.; Zhou, Y.; Du, L. Characterization of the receptor-binding domain (RBD) of 2019 novel coronavirus: Implication for development of RBD protein as a viral attachment inhibitor and vaccine. Cell. Mol. Immunol. 2020, 17, 613–620. [Google Scholar] [CrossRef] [Green Version]
- Cormode, G.; Muthukrishnan, S. The string edit distance matching problem with moves. In ACM Transactions on Algorithms; ACM PUB27: New York, NY, USA, 2007; Volume 3, pp. 1–19. [Google Scholar]
- Zhang, H.; Zhang, Q. MinSearch: An Efficient Algorithm for Similarity Search under Edit Distance. In ACM SIGKDD International Conference on Knowledge Discovery and Data Mining; Association for Computing Machinery: New York, NY, USA, 2020; pp. 566–576. [Google Scholar]
- Bookstein, A.; Kulyukin, V.A.; Raita, T. Generalized hamming distance. Inf. Retr. Boston. 2002, 5, 353–375. [Google Scholar] [CrossRef]
- Chan, T.M.; Golan, S.; Kociumaka, T.; Kopelowitz, T.; Porat, E. Approximating text-to-pattern Hamming distances. In Annual ACM Symposium on Theory of Computing; Association for Computing Machinery: New York, NY, USA, 2020; pp. 643–656. [Google Scholar]
- Eger, S. Sequence alignment with arbitrary steps and further generalizations, with applications to alignments in linguistics. Inf. Sci. 2013, 237, 287–304. [Google Scholar] [CrossRef]
- Muhamad, F.N.; Ahmad, R.B.; Asi, S.M.; Murad, M.N. Performance Analysis of Needleman-Wunsch Algorithm (Global) and Smith-Waterman Algorithm (Local) in Reducing Search Space and Time for Dna Sequence Alignment. J. Phys. Conf. Ser. 2018, 1019, 012085. [Google Scholar] [CrossRef]
- Lugo, W.; Seguel, J. A fast and accurate parallel algorithm for genome mapping assembly aimed at massively parallel sequencers. In Proceedings of the BCB 2015—6th ACM Conference on Bioinformatics, Computational Biology, and Health Informatics, Atlanta, GA, USA, 9–12 September 2015; Association for Computing Machinery: New York, NY, USA, 2015; pp. 574–581. [Google Scholar]
- Yin, R.; Tan, J.; Akhila, D.; Zhou, X.; Kwoh, C.K. Inference of Sequence Homology by BLAST visualization of Influenza Genome set. In ACM International Conference Proceeding Series; Association for Computing Machinery: New York, NY, USA, 2018; pp. 1–6. [Google Scholar]
- Cameron, M.; Williams, H.E. Comparing compressed sequences for faster nucleotide BLAST searches. IEEE/ACM Trans. Comput. Biol. Bioinform. 2007, 4, 349–364. [Google Scholar] [CrossRef]
- Pearson, W.R. An introduction to sequence similarity (“homology”) searching. Curr. Protoc. Bioinform. 2013, 42, 3. [Google Scholar] [CrossRef]
- Bilu, Y.; Linial, M. On the predictive power of sequence similarity in yeast. In Proceedings of the Fifth Annual International Conference on Computational Molecular Biology, Montreal, QC, Canada, 22–25 April 2001; RECOMB. Association for Computing Machinery (ACM): New York, NY, USA, 2001; pp. 39–48. [Google Scholar]
- Joshi, T.; Xu, D. Quantitative assessment of relationship between sequence similarity and function similarity. BMC Genom. 2007, 8, 222. [Google Scholar] [CrossRef] [Green Version]
- Clustal Omega < Multiple Sequence Alignment < EMBL-EBI. Available online: https://www.ebi.ac.uk/Tools/msa/clustalo/ (accessed on 1 March 2021).
- MView < Multiple Sequence Alignment < EMBL-EBI. Available online: https://www.ebi.ac.uk/Tools/msa/mview/ (accessed on 1 March 2021).
- Csardi, G.; Nepusz, T. The igraph software package for complex network research. Inter J. Complex Syst. 2006, 1695, 1–9. [Google Scholar]
- Tableau (version. 9.1). J. Med. Libr. Assoc. 2016, 104, 182. [CrossRef]
- BLAST: Basic Local Alignment Search Tool. Available online: https://blast.ncbi.nlm.nih.gov/Blast.cgi (accessed on 26 January 2022).
- Konishiid, T. Mutations in SARS-CoV-2 are on the increase against the acquired immunity. PLoS ONE 2022, 17, e0271305. [Google Scholar] [CrossRef]
- Pondé, R.A.A. Physicochemical effect of the N501Y, E484K/Q, K417N/T, L452R and T478K mutations on the SARS-CoV-2 spike protein RBD and its influence on agent fitness and on attributes developed by emerging variants of concern. Virology 2022, 572, 44. [Google Scholar] [CrossRef]
- Covid’s Delta Variant: What We Know. The New York Times. Available online: https://www.nytimes.com/2021/06/22/health/delta-variant-covid.html (accessed on 22 December 2021).
- Delta Surge Hits Southern States the Hardest | Best States |. US News. Available online: https://www.usnews.com/news/best-states/articles/2021-09-02/delta-surge-hits-southern-states-the-hardest (accessed on 1 February 2022).
- When Will the Delta Surge End? The New York Times. Available online: https://www.nytimes.com/2021/09/01/health/covid-delta-us-britain.html (accessed on 1 February 2022).



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Time Period | Total Number of Sequences | Number of Unique Sequences | Number of States Reported |
|---|---|---|---|
| T1: 1 Jan 2020 to 20 Mar 2020 | 4047 | 221 | 48 |
| T2: 21 Mar 2020 to 30 Apr 2020 | 5384 | 456 | 37 |
| T3: 1 May 2020 to 20 Sep 2020 | 5876 | 622 | 30 |
| T4: 21 Sep 2020 to 31 Dec 2020 | 5379 | 1035 | 49 |
| T5: 1 Jan 2021 to 31 Jan 2021 | 2932 | 787 | 46 |
| T6: 1 Feb 2021 to 28 Feb 2021 | 17,112 | 688 | 49 |
| T7: 1 Mar 2021 to 31 Mar 2021 | 26,375 | 736 | 50 |
| T8: 1 Apr 2021 to 30 Apr 2021 | 54,883 | 1478 | 50 |
| T9: 1 May 2021 to 31 May 2021 | 31,815 | 762 | 49 |
| T10: 1 Jun 2021 to 30 Jun 2021 | 8598 | 700 | 48 |
| T11: 1 Jul 2021 to 31 Jul 2021 | 5732 | 786 | 47 |
| Variant | Mutations |
|---|---|
| Alpha | ∆69, ∆70, ∆144, E484K, S494P, N501Y, A570D, D614G, P681H, T716I, S982A, D1118H, K1191N |
| Beta | D80A, D215G, ∆241-243, K417N, E484K, N501Y, D614G, A701V |
| Delta | T19R, V70F, T95I, G142D, ∆156-157, R158G, A222V, W258L, K417N, L452R, T478K, D614G, P681R, D950N |
| Gamma | L18F, T20N, P26S, D138Y, R190S, K417T, E484K, N501Y, D614G, H655Y, T1027I |
| Network Property | T6 | T8 | T11 | |||
|---|---|---|---|---|---|---|
| DiWANN | TBN | DiWANN | TBN | DiWANN | TBN | |
| Nodes | 689 | 689 | 1478 | 1478 | 786 | 786 |
| Edges | 2408 | 12,519 | 67,985 | 56,006 | 1995 | 21,215 |
| Avg. degree | 6.98 | 36.39 | 91.93 | 75.78 | 5.07 | 53.98 |
| Max degree | 688 | 265 | 1478 | 276 | 353 | 230 |
| Diameter | 9 | 10 | 18 | 11 | 8 | 7 |
| Clustering coeff. | 0.01 | 0.64 | 0.76 | 0.78 | 0.1 | 0.81 |
| No. of Comp. | 1 | 59 | 1 | 80 | 21 | 67 |
| Largest Comp. | 689 | 569 | 1478 | 1217 | 296 | 280 |
| Singleton nodes | 0 | 4 | 0 | 64 | 0 | 56 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Patil, S.S.; Catanese, H.N.; Brayton, K.A.; Lofgren, E.T.; Gebremedhin, A.H. Sequence Similarity Network Analysis Provides Insight into the Temporal and Geographical Distribution of Mutations in SARS-CoV-2 Spike Protein. Viruses 2022, 14, 1672. https://doi.org/10.3390/v14081672
Patil SS, Catanese HN, Brayton KA, Lofgren ET, Gebremedhin AH. Sequence Similarity Network Analysis Provides Insight into the Temporal and Geographical Distribution of Mutations in SARS-CoV-2 Spike Protein. Viruses. 2022; 14(8):1672. https://doi.org/10.3390/v14081672
Chicago/Turabian StylePatil, Shruti S., Helen N. Catanese, Kelly A. Brayton, Eric T. Lofgren, and Assefaw H. Gebremedhin. 2022. "Sequence Similarity Network Analysis Provides Insight into the Temporal and Geographical Distribution of Mutations in SARS-CoV-2 Spike Protein" Viruses 14, no. 8: 1672. https://doi.org/10.3390/v14081672
APA StylePatil, S. S., Catanese, H. N., Brayton, K. A., Lofgren, E. T., & Gebremedhin, A. H. (2022). Sequence Similarity Network Analysis Provides Insight into the Temporal and Geographical Distribution of Mutations in SARS-CoV-2 Spike Protein. Viruses, 14(8), 1672. https://doi.org/10.3390/v14081672