
Senecavirus A Enhances Its Adaptive Evolution via Synonymous Codon Bias Evolution


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Genome Sequence Acquisition of SVA
2.2. Experimental Tools and Parameters
2.2.1. Sequence Alignment and Phylogenetic Tree Construction
2.2.2. Construction of Median Joining Genetic Evolution Network Diagram
2.3. Codon Bias Index and Analysis Method
2.3.1. ENC and ENC Plot Analysis
2.3.2. Neutrality Plot Analysis
2.3.3. Parity Rule 2 (PR2) Analysis
2.3.4. SVA Encodes the Proportion of Synonymous Codons of the Same Protein
2.4. Statistical Analysis
3. Results
3.1. Determination of Interspecific Recombination Events
3.2. Phylogeny and MJ-Network Linear Genetic Evolution of SVA
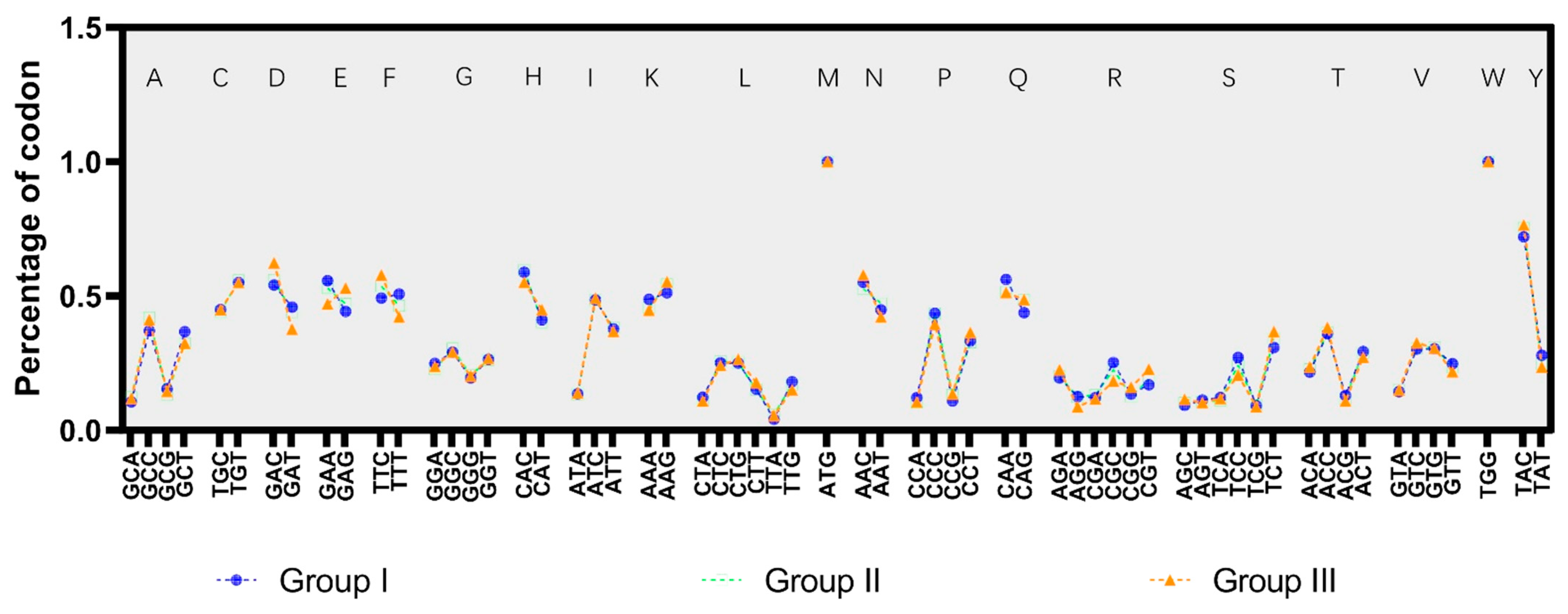
3.3. The Codon Usage Pattern of the SVA
3.4. Trend Change of GC Content in SVA Coding Region
3.5. Content of GC in the Third Base of the Synonymous Codon
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Hales, L.M.; Knowles, N.J.; Reddy, P.S.; Xu, L.; Hay, C.; Hallenbeck, P.L. Complete genome sequence analysis of Seneca Valley virus-001, a novel oncolytic picornavirus. J. Gen. Virol. 2008, 89 Pt 5, 1265–1275. [Google Scholar] [CrossRef] [PubMed]
- Montiel, N.; Buckley, A.; Guo, B.; Kulshreshtha, V.; VanGeelen, A.; Hoang, H.; Rademacher, C.; Yoon, K.; Lager, K.J.E. Vesicular Disease in 9-Week-Old Pigs Experimentally Infected with Senecavirus A. Emerg. Infect. Dis. 2016, 22, 1246–1248. [Google Scholar] [CrossRef] [PubMed]
- Hause, B.M.; Myers, O.; Duff, J.; Hesse, R.A. Senecavirus A in Pigs, United States, 2015. Emerg. Infect. Dis. 2016, 22, 1323–1325. [Google Scholar] [CrossRef] [PubMed]
- Hole, K.; Ambagala, T.; Nfon, C. Vesicular disease in pigs inoculated with a recent Canadian isolate of Senecavirus A. Can. J. Vet. Res. Rev. Can. Rech. Vet. 2019, 83, 242–247. [Google Scholar]
- Wu, Q.; Zhao, X.; Bai, Y.; Sun, B.; Xie, Q.; Ma, J. The First Identification and Complete Genome of Senecavirus A Affecting Pig with Idiopathic Vesicular Disease in China. Transbound. Emerg. Dis. 2017, 64, 1633–1640. [Google Scholar] [CrossRef]
- Saeng-Chuto, K.; Rodtian, P.; Temeeyasen, G.; Wegner, M.; Nilubol, D. The first detection of Senecavirus A in pigs in Thailand, 2016. Transbound. Emerg. Dis. 2018, 65, 285–288. [Google Scholar] [CrossRef]
- Laguardia-Nascimento, M.; Gasparini, M.R.; Sales, B.É.; Rivetti, A.V., Jr.; Sousa, N.M.; Oliveira, A.M.; Camargos, M.F.; Pinheiro de Oliveira, T.F.; Gonçalves, J.P.; Madureira, M.C.; et al. Molecular epidemiology of senecavirus A associated with vesicular disease in pigs in Brazil. Vet. J. 2016, 216, 207–209. [Google Scholar] [CrossRef]
- Canning, P.; Canon, A.; Bates, J.L.; Gerardy, K.; Linhares, D.C.; Piñeyro, P.E.; Schwartz, K.J.; Yoon, K.J.; Rademacher, C.J.; Holtkamp, D.; et al. Neonatal Mortality, Vesicular Lesions and Lameness Associated with Senecavirus A in a U.S. Sow Farm. Transbound. Emerg. Dis. 2016, 63, 373–378. [Google Scholar] [CrossRef]
- Guo, B.; Piñeyro, P.E.; Rademacher, C.J.; Zheng, Y.; Li, G.; Yuan, J.; Hoang, H.; Gauger, P.C.; Madson, D.M.; Schwartz, K.J.; et al. Novel Senecavirus A in Swine with Vesicular Disease, United States, July 2015. Emerg. Infect. Dis. 2016, 22, 1325–1327. [Google Scholar] [CrossRef]
- Houston, E.; Temeeyasen, G.; Piñeyro, P.E. Comprehensive review on immunopathogenesis, diagnostic and epidemiology of Senecavirus A. Virus Res. 2020, 286, 198038. [Google Scholar] [CrossRef]
- Francis, M.J. Recent Advances in Vaccine Technologies. Vet. Clin. N. Am. Small Anim. Pract. 2018, 48, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Webster, R.G.; Webby, R.J. Influenza Virus: Dealing with a Drifting and Shifting Pathogen. Viral Immunol. 2018, 31, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Greninger, A.L. A decade of RNA virus metagenomics is (not) enough. Virus Res. 2018, 244, 218–229. [Google Scholar] [CrossRef] [PubMed]
- Mandary, M.B.; Masomian, M.; Poh, C.L. Impact of RNA Virus Evolution on Quasispecies Formation and Virulence. Int. J. Mol. Sci. 2019, 20, 4657. [Google Scholar] [CrossRef]
- Gonçalves-Carneiro, D.; Bieniasz, P.D. Mechanisms of Attenuation by Genetic Recoding of Viruses. Mbio 2021, 12, e02238-20. [Google Scholar] [CrossRef]
- de Visser, J.A.; Hermisson, J.; Wagner, G.P.; Ancel Meyers, L.; Bagheri-Chaichian, H.; Blanchard, J.L.; Chao, L.; Cheverud, J.M.; Elena, S.F.; Fontana, W.; et al. Perspective: Evolution and detection of genetic robustness. Evol. Int. J. Org. Evol. 2003, 57, 1959–1972. [Google Scholar] [CrossRef]
- Heaton, S.M. Harnessing host-virus evolution in antiviral therapy and immunotherapy. Clin. Transl. Immunol. 2019, 8, e1067. [Google Scholar] [CrossRef]
- McBride, R.C.; Ogbunugafor, C.B.; Turner, P.E. Robustness promotes evolvability of thermotolerance in an RNA virus. BMC Evol. Biol. 2008, 8, 231. [Google Scholar] [CrossRef]
- Liu, Y. A code within the genetic code: Codon usage regulates co-translational protein folding. Cell Commun. Signal 2020, 18, 145. [Google Scholar] [CrossRef]
- Boël, G.; Letso, R.; Neely, H.; Price, W.N.; Wong, K.H.; Su, M.; Luff, J.; Valecha, M.; Everett, J.K.; Acton, T.B.; et al. Codon influence on protein expression in E. coli correlates with mRNA levels. Nature 2016, 529, 358–363. [Google Scholar] [CrossRef]
- Fu, J.; Murphy, K.A.; Zhou, M.; Li, Y.H.; Lam, V.H.; Tabuloc, C.A.; Chiu, J.C.; Liu, Y. Codon usage affects the structure and function of the Drosophila circadian clock protein PERIOD. Genes Dev. 2016, 30, 1761–1775. [Google Scholar] [CrossRef] [PubMed]
- Komar, A.A. The Yin and Yang of codon usage. Hum. Mol. Genet. 2016, 25, R77–R85. [Google Scholar] [CrossRef] [PubMed]
- Frumkin, I.; Lajoie, M.J.; Gregg, C.J.; Hornung, G.; Church, G.M.; Pilpel, Y. Codon usage of highly expressed genes affects proteome-wide translation efficiency. Proc. Natl. Acad. Sci. USA 2018, 115, E4940–E4949. [Google Scholar] [CrossRef] [PubMed]
- Hanson, G.; Coller, J. Codon optimality, bias and usage in translation and mRNA decay. Nat. Rev. Mol. Cell Biol. 2018, 19, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Dolan, P.T.; Whitfield, Z.J.; Andino, R. Mechanisms and Concepts in RNA Virus Population Dynamics and Evolution. Annu. Rev. Virol. 2018, 5, 69–92. [Google Scholar] [CrossRef] [PubMed]
- Dagan, T. Phylogenomic networks. Trends Microbiol. 2011, 19, 483–491. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.P.; Varsani, A.; Roumagnac, P.; Botha, G.; Maslamoney, S.; Schwab, T.; Kelz, Z.; Kumar, V.; Murrell, B. RDP5: A computer program for analyzing recombination in, and removing signals of recombination from, nucleotide sequence datasets. Virus Evol. 2021, 7, veaa087. [Google Scholar] [CrossRef]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef]
- Bandelt, H.J.; Forster, P.; Röhl, A. Median-joining networks for inferring intraspecific phylogenies. Mol. Biol. Evol. 1999, 16, 37–48. [Google Scholar] [CrossRef]
- Wright, F. The ‘effective number of codons’ used in a gene. Gene 1990, 87, 23–29. [Google Scholar] [CrossRef]
- Sueoka, N. Directional mutation pressure and neutral molecular evolution. Proc. Natl. Acad. Sci. USA 1988, 85, 2653–2657. [Google Scholar] [CrossRef] [PubMed]
- Sueoka, N. Intrastrand parity rules of DNA base composition and usage biases of synonymous codons. J. Mol. Evol. 1995, 40, 318–325. [Google Scholar] [CrossRef] [PubMed]
- Lam, T.T.; Hon, C.C.; Tang, J.W. Use of phylogenetics in the molecular epidemiology and evolutionary studies of viral infections. Crit. Rev. Clin. Lab. Sci. 2010, 47, 5–49. [Google Scholar] [CrossRef] [PubMed]
- Wasik, B.R.; Turner, P.E. On the biological success of viruses. Annu. Rev. Microbiol. 2013, 67, 519–541. [Google Scholar] [CrossRef]
- Morley, V.J.; Turner, P.E. Dynamics of molecular evolution in RNA virus populations depend on sudden versus gradual environmental change. Evol. Int. J. Org. Evol. 2017, 71, 872–883. [Google Scholar] [CrossRef] [PubMed]
- Mattenberger, F.; Vila-Nistal, M.; Geller, R. Increased RNA virus population diversity improves adaptability. Sci. Rep. 2021, 11, 6824. [Google Scholar] [CrossRef]
- Cabanillas, L.; Arribas, M.; Lazaro, E. Evolution at increased error rate leads to the coexistence of multiple adaptive pathways in an RNA virus. BMC Evol. Biol. 2013, 13, 11. [Google Scholar] [CrossRef]
- Volz, E.M.; Koelle, K.; Bedford, T. Viral phylodynamics. PLoS Comput. Biol. 2013, 9, e1002947. [Google Scholar] [CrossRef]
- Nei, M.; Kumar, S.; Takahashi, K. The optimization principle in phylogenetic analysis tends to give incorrect topologies when the number of nucleotides or amino acids used is small. Proc. Natl. Acad. Sci. USA 1998, 95, 12390–12397. [Google Scholar] [CrossRef]
- De Bruyn, A.; Martin, D.P.; Lefeuvre, P. Phylogenetic reconstruction methods: An overview. Methods Mol. Biol. 2014, 1115, 257–277. [Google Scholar]
- Elena, S.F.; Lenski, R.E. Evolution experiments with microorganisms: The dynamics and genetic bases of adaptation. Nat. Rev. Genet. 2003, 4, 457–469. [Google Scholar] [CrossRef] [PubMed]
- Nijhout, H.F.; Best, J.A.; Reed, M.C. Systems biology of robustness and homeostatic mechanisms. Wiley Interdiscip. Rev. Syst. Biol. Med. 2019, 11, e1440. [Google Scholar] [CrossRef] [PubMed]
- Masel, J.; Trotter, M.V. Robustness and evolvability. Trends Genet. 2010, 26, 406–414. [Google Scholar] [CrossRef] [PubMed]
- Elena, S.F. RNA virus genetic robustness: Possible causes and some consequences. Curr. Opin. Virol. 2012, 2, 525–530. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Xu, Q.; Tan, C.; Li, X.; Chi, X.; Cai, B.; Yu, Z.; Ma, Y.; Chen, J.L. Genomic analysis of codon usage shows influence of mutation pressure, natural selection, and host features on Senecavirus A evolution. Microb. Pathog. 2017, 112, 313–319. [Google Scholar] [CrossRef]
- Jenkins, G.M.; Holmes, E.C. The extent of codon usage bias in human RNA viruses and its evolutionary origin. Virus Res. 2003, 92, 1–7. [Google Scholar] [CrossRef]
- Liu, W.Q.; Zhang, J.; Zhang, Y.Q.; Zhou, J.H.; Chen, H.T.; Ma, L.N.; Ding, Y.Z.; Liu, Y. Compare the differences of synonymous codon usage between the two species within cardiovirus. Virol. J. 2011, 8, 325. [Google Scholar] [CrossRef][Green Version]
- Su, W.; Li, X.; Chen, M.; Dai, W.; Sun, S.; Wang, S.; Sheng, X.; Sun, S.; Gao, C.; Hou, A.; et al. Synonymous codon usage analysis of hand, foot and mouth disease viruses: A comparative study on coxsackievirus A6, A10, A16, and enterovirus 71 from 2008 to 2015. Infect. Genet. Evol. 2017, 53, 212–217. [Google Scholar] [CrossRef]
- Auewarakul, P. Composition bias and genome polarity of RNA viruses. Virus Res. 2005, 109, 33–37. [Google Scholar] [CrossRef]
- Chu, W.; Weerasekera, A.; Kim, C.H. On the conformational stability of the smallest RNA kissing complexes maintained through two G·C base pairs. Biochem. Biophys. Res. Commun. 2017, 483, 39–44. [Google Scholar] [CrossRef]
- Golden, M.; Murrell, B.; Martin, D.; Pybus, O.G.; Hein, J. Evolutionary Analyses of Base-Pairing Interactions in DNA and RNA Secondary Structures. Mol. Biol. Evol. 2020, 37, 576–592. [Google Scholar] [CrossRef] [PubMed]
- Dilucca, M.; Pavlopoulou, A.; Georgakilas, A.G.; Giansanti, A. Codon usage bias in radioresistant bacteria. Gene 2020, 742, 144554. [Google Scholar] [CrossRef] [PubMed]
- Hu, E.Z.; Lan, X.R.; Liu, Z.L.; Gao, J.; Niu, D.K. A positive correlation between GC content and growth temperature in prokaryotes. BMC Genom. 2022, 23, 110. [Google Scholar] [CrossRef] [PubMed]
- Hogg, J.R. Viral Evasion and Manipulation of Host RNA Quality Control Pathways. J. Virol. 2016, 90, 7010–7018. [Google Scholar] [CrossRef]
- Dickson, A.M.; Wilusz, J. Strategies for viral RNA stability: Live long and prosper. Trends Genet. 2011, 27, 286–293. [Google Scholar] [CrossRef]
- Hia, F.; Yang, S.F.; Shichino, Y.; Yoshinaga, M.; Murakawa, Y.; Vandenbon, A.; Fukao, A.; Fujiwara, T.; Landthaler, M.; Natsume, T.; et al. Codon bias confers stability to human mRNAs. EMBO Rep. 2019, 20, e48220. [Google Scholar] [CrossRef]
- Gelfman, S.; Ast, G. When epigenetics meets alternative splicing: The roles of DNA methylation and GC architecture. Epigenomics 2013, 5, 351–353. [Google Scholar] [CrossRef]
- Brahma, S.; Henikoff, S. Epigenome Regulation by Dynamic Nucleosome Unwrapping. Trends Biochem. Sci. 2020, 45, 13–26. [Google Scholar] [CrossRef]
- Li, Y.; Yang, X.; Wang, N.; Wang, H.; Yin, B.; Yang, X.; Jiang, W. GC usage of SARS-CoV-2 genes might adapt to the environment of human lung expressed genes. Mol. Genet. Genom. MGG 2020, 295, 1537–1546. [Google Scholar] [CrossRef]
- Huang, D.D. The potential of RNA interference-based therapies for viral infections. Curr. HIV/AIDS Rep. 2008, 5, 33–39. [Google Scholar] [CrossRef]
- Qureshi, A.; Tantray, V.G.; Kirmani, A.R.; Ahangar, A.G. A review on current status of antiviral siRNA. Rev. Med. Virol. 2018, 28, e1976. [Google Scholar] [CrossRef] [PubMed]
- Sidorenko, L.V.; Lee, T.F.; Woosley, A.; Moskal, W.A.; Bevan, S.A.; Merlo, P.A.O.; Walsh, T.A.; Wang, X.; Weaver, S.; Glancy, T.P.; et al. GC-rich coding sequences reduce transposon-like, small RNA-mediated transgene silencing. Nat. Plants 2017, 3, 875–884. [Google Scholar] [CrossRef] [PubMed]
- Pereira-Gomez, M.; Carrau, L.; Fajardo, A.; Moreno, P.; Moratorio, G. Altering Compositional Properties of Viral Genomes to Design Live-Attenuated Vaccines. Front. Microbiol. 2021, 12, 676582. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, S.; Cui, H.; Hu, Z.; Du, L.; Ran, X.; Wen, X. Senecavirus A Enhances Its Adaptive Evolution via Synonymous Codon Bias Evolution. Viruses 2022, 14, 1055. https://doi.org/10.3390/v14051055
Zhao S, Cui H, Hu Z, Du L, Ran X, Wen X. Senecavirus A Enhances Its Adaptive Evolution via Synonymous Codon Bias Evolution. Viruses. 2022; 14(5):1055. https://doi.org/10.3390/v14051055
Chicago/Turabian StyleZhao, Simiao, Huiqi Cui, Zhenru Hu, Li Du, Xuhua Ran, and Xiaobo Wen. 2022. "Senecavirus A Enhances Its Adaptive Evolution via Synonymous Codon Bias Evolution" Viruses 14, no. 5: 1055. https://doi.org/10.3390/v14051055
APA StyleZhao, S., Cui, H., Hu, Z., Du, L., Ran, X., & Wen, X. (2022). Senecavirus A Enhances Its Adaptive Evolution via Synonymous Codon Bias Evolution. Viruses, 14(5), 1055. https://doi.org/10.3390/v14051055