
Naïve Human Macrophages Are Refractory to SARS-CoV-2 Infection and Exhibit a Modest Inflammatory Response Early in Infection



{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells and Viruses
2.2. Treatments
2.3. Real-Time Quantitative Reverse Transcription PCR (qPCR)
2.4. Chemokine and Cytokine Detection
2.5. Statistical Analyses
3. Results and Discussions
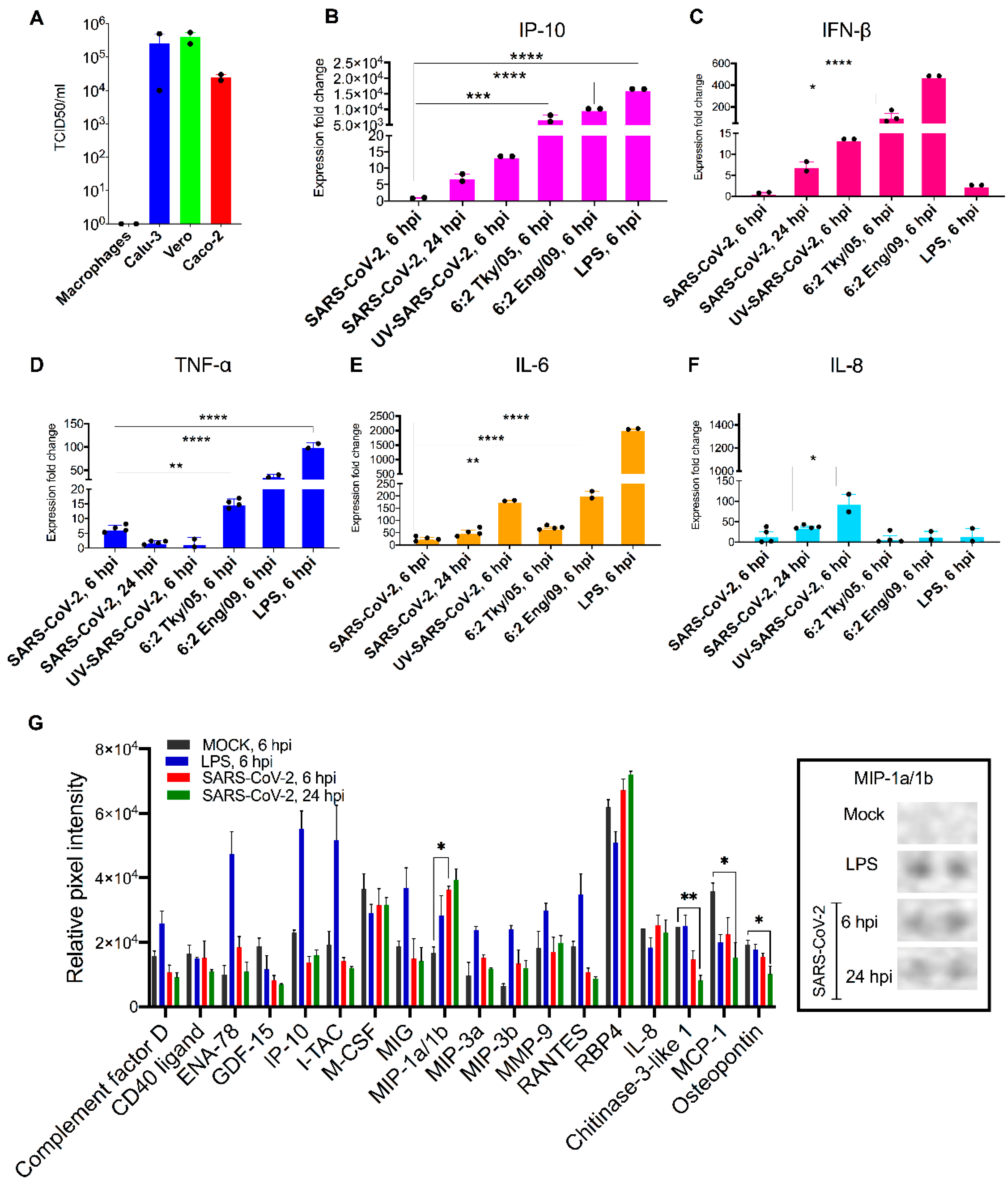
3.1. Macrophages Are Refractory to SARS-CoV-2 Infection
3.2. Macrophages Exhibit Modest Pro-Inflammatory Responses during SARS-CoV-2 Infection
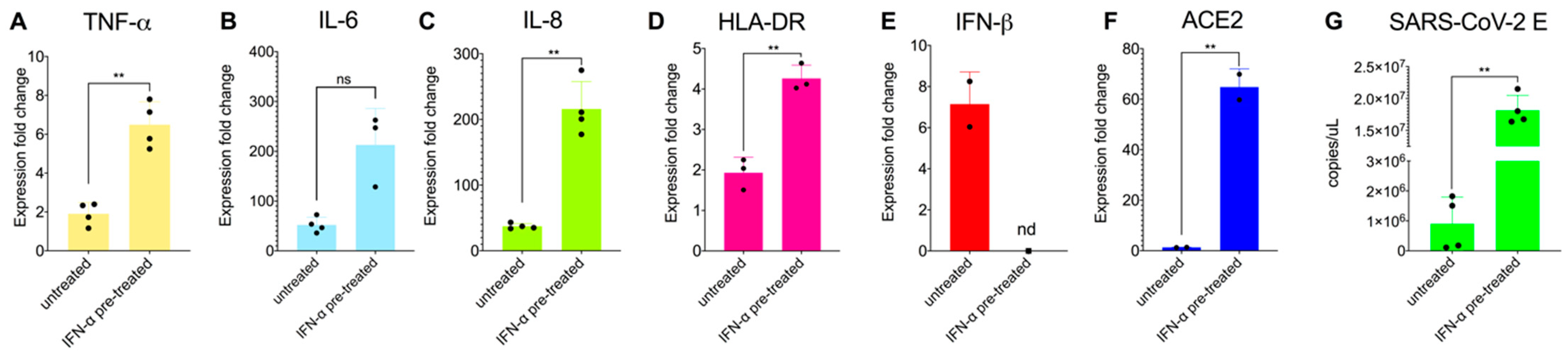
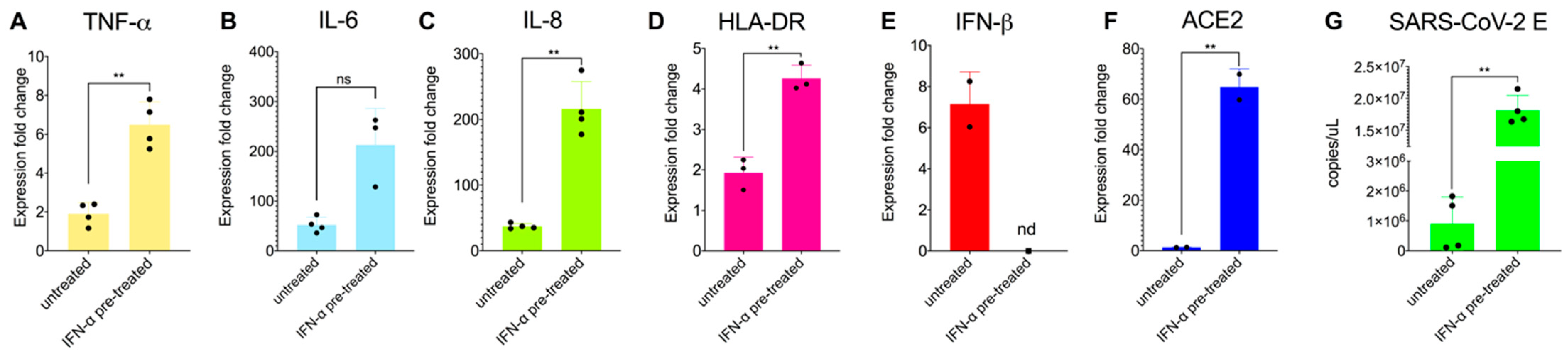
3.3. Priming Macrophages with IFNα Boosts Transcription of Proinflammatory Cytokines but Does Not Allow Productive Replication
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chen, Y.; Guo, Y.; Pan, Y.; Zhao, Z.J. Structure analysis of the receptor binding of 2019-nCoV. Biochem. Biophys Res. Commun. 2020, 525, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Kruger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Tay, M.Z.; Poh, C.M.; Renia, L.; MacAry, P.A.; Ng, L.F.P. The trinity of COVID-19: Immunity, inflammation and intervention. Nat. Rev. Immunol. 2020, 20, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Hikmet, F.; Mear, L.; Edvinsson, A.; Micke, P.; Uhlen, M.; Lindskog, C. The protein expression profile of ACE2 in human tissues. Mol. Syst. Biol. 2020, 16, e9610. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Jiang, Q.; Xia, X.; Liu, K.; Yu, Z.; Tao, W.; Gong, W.; Han, J.J. Individual variation of the SARS-CoV-2 receptor ACE2 gene expression and regulation. Aging Cell 2020, 19, e13168. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Zhao, Z.; Wang, Y.; Zhou, Y.; Ma, Y.; Zuo, W. Single-Cell RNA Expression Profiling of ACE2, the Receptor of SARS-CoV-2. Am. J. Respir. Crit. Care Med. 2020, 202, 756–759. [Google Scholar] [CrossRef]
- David, A.; Parkinson, N.; Peacock, T.P.; Pairo-Castineira, E.; Khanna, T.; Cobat, A.; Tenesa, A.; Sancho-Shimizu, V.; Casanova, J.L.; Abel, L.; et al. A common TMPRSS2 variant has a protective effect against severe COVID-19. Curr. Res. Transl. Med. 2022, 70, 103333. [Google Scholar] [CrossRef]
- Brodin, P. Immune determinants of COVID-19 disease presentation and severity. Nat. Med. 2021, 27, 28–33. [Google Scholar] [CrossRef]
- Galani, I.E.; Rovina, N.; Lampropoulou, V.; Triantafyllia, V.; Manioudaki, M.; Pavlos, E.; Koukaki, E.; Fragkou, P.C.; Panou, V.; Rapti, V.; et al. Untuned antiviral immunity in COVID-19 revealed by temporal type I/III interferon patterns and flu comparison. Nat. Immunol. 2021, 22, 32–40. [Google Scholar] [CrossRef]
- Huang, Q.; Wu, X.; Zheng, X.; Luo, S.; Xu, S.; Weng, J. Targeting inflammation and cytokine storm in COVID-19. Pharmacol. Res. 2020, 159, 105051. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.Y.; Li, B.R.; Ning, B.T. The Comparative Immunological Characteristics of SARS-CoV, MERS-CoV, and SARS-CoV-2 Coronavirus Infections. Front. Immunol. 2020, 11, 2033. [Google Scholar] [CrossRef]
- Peiris, J.S.; Hui, K.P.; Yen, H.L. Host response to influenza virus: Protection versus immunopathology. Curr. Opin. Immunol. 2010, 22, 475–481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryabkova, V.A.; Churilov, L.P.; Shoenfeld, Y. Influenza infection, SARS, MERS and COVID-19: Cytokine storm—The common denominator and the lessons to be learned. Clin. Immunol. 2021, 223, 108652. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Zhou, Y.H.; Yang, Z.Q. The cytokine storm of severe influenza and development of immunomodulatory therapy. Cell. Mol. Immunol. 2016, 13, 3–10. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.J.; Thomas, P.G. New fronts emerge in the influenza cytokine storm. Semin. Immunopathol. 2017, 39, 541–550. [Google Scholar] [CrossRef]
- Jaffer, U.; Wade, R.G.; Gourlay, T. Cytokines in the systemic inflammatory response syndrome: A review. HSR Proc. Intensive Care Cardiovasc. Anesth. 2010, 2, 161–175. [Google Scholar] [PubMed]
- Morris, G.; Bortolasci, C.C.; Puri, B.K.; Marx, W.; O’Neil, A.; Athan, E.; Walder, K.; Berk, M.; Olive, L.; Carvalho, A.F.; et al. The cytokine storms of COVID-19, H1N1 influenza, CRS and MAS compared. Can one sized treatment fit all? Cytokine 2021, 144, 155593. [Google Scholar] [CrossRef]
- Del Valle, D.M.; Kim-Schulze, S.; Huang, H.H.; Beckmann, N.D.; Nirenberg, S.; Wang, B.; Lavin, Y.; Swartz, T.H.; Madduri, D.; Stock, A.; et al. An inflammatory cytokine signature predicts COVID-19 severity and survival. Nat. Med. 2020, 26, 1636–1643. [Google Scholar] [CrossRef]
- Jamilloux, Y.; Henry, T.; Belot, A.; Viel, S.; Fauter, M.; El Jammal, T.; Walzer, T.; Francois, B.; Seve, P. Should we stimulate or suppress immune responses in COVID-19? Cytokine and anti-cytokine interventions. Autoimmun. Rev. 2020, 19, 102567. [Google Scholar] [CrossRef]
- Xiong, Y.; Liu, Y.; Cao, L.; Wang, D.; Guo, M.; Jiang, A.; Guo, D.; Hu, W.; Yang, J.; Tang, Z.; et al. Transcriptomic characteristics of bronchoalveolar lavage fluid and peripheral blood mononuclear cells in COVID-19 patients. Emerg. Microbes Infect. 2020, 9, 761–770. [Google Scholar] [CrossRef]
- Tang, Y.; Liu, J.; Zhang, D.; Xu, Z.; Ji, J.; Wen, C. Cytokine Storm in COVID-19: The Current Evidence and Treatment Strategies. Front. Immunol. 2020, 11, 1708. [Google Scholar] [CrossRef] [PubMed]
- Hamidzadeh, K.; Christensen, S.M.; Dalby, E.; Chandrasekaran, P.; Mosser, D.M. Macrophages and the Recovery from Acute and Chronic Inflammation. Annu. Rev. Physiol. 2017, 79, 567–592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bost, P.; Giladi, A.; Liu, Y.; Bendjelal, Y.; Xu, G.; David, E.; Blecher-Gonen, R.; Cohen, M.; Medaglia, C.; Li, H.; et al. Host-Viral Infection Maps Reveal Signatures of Severe COVID-19 Patients. Cell 2020, 181, 1475–1488.e12. [Google Scholar] [CrossRef]
- Feng, Z.; Diao, B.; Wang, R.; Wang, G.; Wang, C.; Tan, Y.; Liu, L.; Wang, C.; Liu, Y.; Liu, Y.; et al. The Novel Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) Directly Decimates Human Spleens and Lymph Nodes. medRxiv 2020. [Google Scholar] [CrossRef]
- Jafarzadeh, A.; Chauhan, P.; Saha, B.; Jafarzadeh, S.; Nemati, M. Contribution of monocytes and macrophages to the local tissue inflammation and cytokine storm in COVID-19: Lessons from SARS and MERS, and potential therapeutic interventions. Life Sci. 2020, 257, 118102. [Google Scholar] [CrossRef] [PubMed]
- Chu, H.; Chan, J.F.; Yuen, T.T.; Shuai, H.; Yuan, S.; Wang, Y.; Hu, B.; Yip, C.C.; Tsang, J.O.; Huang, X.; et al. Comparative tropism, replication kinetics, and cell damage profiling of SARS-CoV-2 and SARS-CoV with implications for clinical manifestations, transmissibility, and laboratory studies of COVID-19: An observational study. Lancet Microbe 2020, 1, e14–e23. [Google Scholar] [CrossRef]
- Abassi, Z.; Knaney, Y.; Karram, T.; Heyman, S.N. The Lung Macrophage in SARS-CoV-2 Infection: A Friend or a Foe? Front. Immunol. 2020, 11, 1312. [Google Scholar] [CrossRef]
- Banerjee, A.; Nasir, J.A.; Budylowski, P.; Yip, L.; Aftanas, P.; Christie, N.; Ghalami, A.; Baid, K.; Raphenya, A.R.; Hirota, J.A.; et al. Isolation, Sequence, Infectivity, and Replication Kinetics of Severe Acute Respiratory Syndrome Coronavirus 2. Emerg. Infect. Dis. 2020, 26, 2054–2063. [Google Scholar] [CrossRef]
- Niles, M.A.; Gogesch, P.; Kronhart, S.; Ortega Iannazzo, S.; Kochs, G.; Waibler, Z.; Anzaghe, M. Macrophages and Dendritic Cells Are Not the Major Source of Pro-Inflammatory Cytokines Upon SARS-CoV-2 Infection. Front. Immunol. 2021, 12, 647824. [Google Scholar] [CrossRef]
- Boumaza, A.; Gay, L.; Mezouar, S.; Bestion, E.; Diallo, A.B.; Michel, M.; Desnues, B.; Raoult, D.; La Scola, B.; Halfon, P.; et al. Monocytes and macrophages, targets of SARS-CoV-2: The clue for Covid-19 immunoparalysis. J. Infect. Dis. 2021, 224, 395–406. [Google Scholar] [CrossRef]
- Thorne, L.G.; Reuschl, A.K.; Zuliani-Alvarez, L.; Whelan, M.V.X.; Turner, J.; Noursadeghi, M.; Jolly, C.; Towers, G.J. SARS-CoV-2 sensing by RIG-I and MDA5 links epithelial infection to macrophage inflammation. EMBO J. 2021, 40, e107826. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Chu, H.; Hou, Y.; Chai, Y.; Shuai, H.; Lee, A.C.; Zhang, X.; Wang, Y.; Hu, B.; Huang, X.; et al. Attenuated Interferon and Proinflammatory Response in SARS-CoV-2-Infected Human Dendritic Cells Is Associated With Viral Antagonism of STAT1 Phosphorylation. J. Infect. Dis. 2020, 222, 734–745. [Google Scholar] [CrossRef]
- Leach, D.A.; Mohr, A.; Giotis, E.S.; Cil, E.; Isac, A.M.; Yates, L.L.; Barclay, W.S.; Zwacka, R.M.; Bevan, C.L.; Brooke, G.N. The antiandrogen enzalutamide downregulates TMPRSS2 and reduces cellular entry of SARS-CoV-2 in human lung cells. Nat. Commun. 2021, 12, 4068. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Bradley, K.C.; Long, J.S.; Frise, R.; Ashcroft, J.W.; Hartgroves, L.C.; Shelton, H.; Makris, S.; Johansson, C.; Cao, B.; et al. Internal genes of a highly pathogenic H5N1 influenza virus determine high viral replication in myeloid cells and severe outcome of infection in mice. PLoS Pathog. 2018, 14, e1006821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kärber, G. Beitrag zur kollektiven Behandlung pharmakologischer Reihenversuche. Naunyn-Schmiedebergs Arch. Exp. Pathol. Pharmakol. 1931, 162, 480–483. [Google Scholar] [CrossRef]
- Giotis, E.S.; Ross, C.S.; Robey, R.C.; Nohturfft, A.; Goodbourn, S.; Skinner, M.A. Constitutively elevated levels of SOCS1 suppress innate responses in DF-1 immortalised chicken fibroblast cells. Sci. Rep. 2017, 7, 17485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lau, S.K.P.; Lau, C.C.Y.; Chan, K.H.; Li, C.P.Y.; Chen, H.; Jin, D.Y.; Chan, J.F.W.; Woo, P.C.Y.; Yuen, K.Y. Delayed induction of proinflammatory cytokines and suppression of innate antiviral response by the novel Middle East respiratory syndrome coronavirus: Implications for pathogenesis and treatment. J. Gen. Virol. 2013, 94, 2679–2690. [Google Scholar] [CrossRef]
- Giotis, E.S.; Carnell, G.; Young, E.F.; Ghanny, S.; Soteropoulos, P.; Wang, L.F.; Barclay, W.S.; Skinner, M.A.; Temperton, N. Entry of the bat influenza H17N10 virus into mammalian cells is enabled by the MHC class II HLA-DR receptor. Nat. Microbiol. 2019, 4, 2035–2038. [Google Scholar] [CrossRef]
- Long, J.S.; Giotis, E.S.; Moncorge, O.; Frise, R.; Mistry, B.; James, J.; Morisson, M.; Iqbal, M.; Vignal, A.; Skinner, M.A.; et al. Species difference in ANP32A underlies influenza A virus polymerase host restriction. Nature 2016, 529, 101–104. [Google Scholar] [CrossRef] [Green Version]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhang, C.; Huang, F.; Yang, Y.; Wang, F.; Yuan, J.; Zhang, Z.; Qin, Y.; Li, X.; Zhao, D.; et al. Elevated plasma levels of selective cytokines in COVID-19 patients reflect viral load and lung injury. Natl. Sci. Rev. 2020, 7, 1003–1011. [Google Scholar] [CrossRef] [Green Version]
- Buckley, C.D.; Gilroy, D.W.; Serhan, C.N.; Stockinger, B.; Tak, P.P. The resolution of inflammation. Nat. Rev. Immunol. 2013, 13, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Guilliams, M.; Ginhoux, F.; Jakubzick, C.; Naik, S.H.; Onai, N.; Schraml, B.U.; Segura, E.; Tussiwand, R.; Yona, S. Dendritic cells, monocytes and macrophages: A unified nomenclature based on ontogeny. Nat. Rev. Immunol. 2014, 14, 571–578. [Google Scholar] [CrossRef]
- Kvedaraite, E.; Hertwig, L.; Sinha, I.; Ponzetta, A.; Hed Myrberg, I.; Lourda, M.; Dzidic, M.; Akber, M.; Klingstrom, J.; Folkesson, E.; et al. Major alterations in the mononuclear phagocyte landscape associated with COVID-19 severity. Proc. Natl. Acad. Sci. USA 2021, 118, e2018587118. [Google Scholar] [CrossRef] [PubMed]
- Marvin, S.A.; Russier, M.; Huerta, C.T.; Russell, C.J.; Schultz-Cherry, S. Influenza Virus Overcomes Cellular Blocks To Productively Replicate, Impacting Macrophage Function. J. Virol. 2017, 91, e01417-16. [Google Scholar] [CrossRef] [Green Version]
- Ntanasis-Stathopoulos, I.; Fotiou, D.; Terpos, E. CCL3 Signaling in the Tumor Microenvironment. Adv. Exp. Med. Biol. 2020, 1231, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Abers, M.S.; Delmonte, O.M.; Ricotta, E.E.; Fintzi, J.; Fink, D.L.; de Jesus, A.A.A.; Zarember, K.A.; Alehashemi, S.; Oikonomou, V.; Desai, J.V.; et al. An immune-based biomarker signature is associated with mortality in COVID-19 patients. JCI Insight 2021, 6, e144455. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, J.; Liu, C.; Su, L.; Zhang, D.; Fan, J.; Yang, Y.; Xiao, M.; Xie, J.; Xu, Y.; et al. IP-10 and MCP-1 as biomarkers associated with disease severity of COVID-19. Mol. Med. 2020, 26, 97. [Google Scholar] [CrossRef]
- Hayek, S.S.; Roderburg, C.; Blakely, P.; Launius, C.; Eugen-Olsen, J.; Tacke, F.; Ktena, S.; Keitel, V.; Luedde, M.; Giamarellos-Bourboulis, E.J.; et al. Circulating Osteopontin Levels and Outcomes in Patients Hospitalized for COVID-19. J. Clin. Med. 2021, 10, 3907. [Google Scholar] [CrossRef]
- Kamle, S.; Ma, B.; He, C.H.; Akosman, B.; Zhou, Y.; Lee, C.M.; El-Deiry, W.S.; Huntington, K.; Liang, O.; Machan, J.T.; et al. Chitinase 3-like-1 is a therapeutic target that mediates the effects of aging in COVID-19. JCI Insight 2021, 6, e148749. [Google Scholar] [CrossRef]
- Liu, S.Y.; Sanchez, D.J.; Aliyari, R.; Lu, S.; Cheng, G. Systematic identification of type I and type II interferon-induced antiviral factors. Proc. Natl. Acad. Sci. USA 2012, 109, 4239–4244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanco-Melo, D.; Nilsson-Payant, B.E.; Liu, W.C.; Uhl, S.; Hoagland, D.; Moller, R.; Jordan, T.X.; Oishi, K.; Panis, M.; Sachs, D.; et al. Imbalanced Host Response to SARS-CoV-2 Drives Development of COVID-19. Cell 2020, 181, 1036–1045. [Google Scholar] [CrossRef] [PubMed]
- Hadjadj, J.; Yatim, N.; Barnabei, L.; Corneau, A.; Boussier, J.; Smith, N.; Pere, H.; Charbit, B.; Bondet, V.; Chenevier-Gobeaux, C.; et al. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science 2020, 369, 718–724. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.M.; Shin, E.C. Type I and III interferon responses in SARS-CoV-2 infection. Exp. Mol. Med. 2021, 53, 750–760. [Google Scholar] [CrossRef]
- Channappanavar, R.; Fehr, A.R.; Vijay, R.; Mack, M.; Zhao, J.; Meyerholz, D.K.; Perlman, S. Dysregulated Type I Interferon and Inflammatory Monocyte-Macrophage Responses Cause Lethal Pneumonia in SARS-CoV-Infected Mice. Cell Host Microbe 2016, 19, 181–193. [Google Scholar] [CrossRef] [Green Version]
- Ziegler, C.G.K.; Allon, S.J.; Nyquist, S.K.; Mbano, I.M.; Miao, V.N.; Tzouanas, C.N.; Cao, Y.; Yousif, A.S.; Bals, J.; Hauser, B.M.; et al. SARS-CoV-2 Receptor ACE2 Is an Interferon-Stimulated Gene in Human Airway Epithelial Cells and Is Detected in Specific Cell Subsets across Tissues. Cell 2020, 181, 1016–1035.e9. [Google Scholar] [CrossRef] [PubMed]
- Ng, K.W.; Attig, J.; Bolland, W.; Young, G.R.; Major, J.; Wrobel, A.G.; Gamblin, S.; Wack, A.; Kassiotis, G. Tissue-specific and interferon-inducible expression of nonfunctional ACE2 through endogenous retroelement co-option. Nat. Genet. 2020, 52, 1294–1302. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Z.; Penn, R.; Barclay, W.S.; Giotis, E.S. Naïve Human Macrophages Are Refractory to SARS-CoV-2 Infection and Exhibit a Modest Inflammatory Response Early in Infection. Viruses 2022, 14, 441. https://doi.org/10.3390/v14020441
Zhang Z, Penn R, Barclay WS, Giotis ES. Naïve Human Macrophages Are Refractory to SARS-CoV-2 Infection and Exhibit a Modest Inflammatory Response Early in Infection. Viruses. 2022; 14(2):441. https://doi.org/10.3390/v14020441
Chicago/Turabian StyleZhang, Ziyun, Rebecca Penn, Wendy S. Barclay, and Efstathios S. Giotis. 2022. "Naïve Human Macrophages Are Refractory to SARS-CoV-2 Infection and Exhibit a Modest Inflammatory Response Early in Infection" Viruses 14, no. 2: 441. https://doi.org/10.3390/v14020441
APA StyleZhang, Z., Penn, R., Barclay, W. S., & Giotis, E. S. (2022). Naïve Human Macrophages Are Refractory to SARS-CoV-2 Infection and Exhibit a Modest Inflammatory Response Early in Infection. Viruses, 14(2), 441. https://doi.org/10.3390/v14020441