
How RSV Proteins Join Forces to Overcome the Host Innate Immune Response

 ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
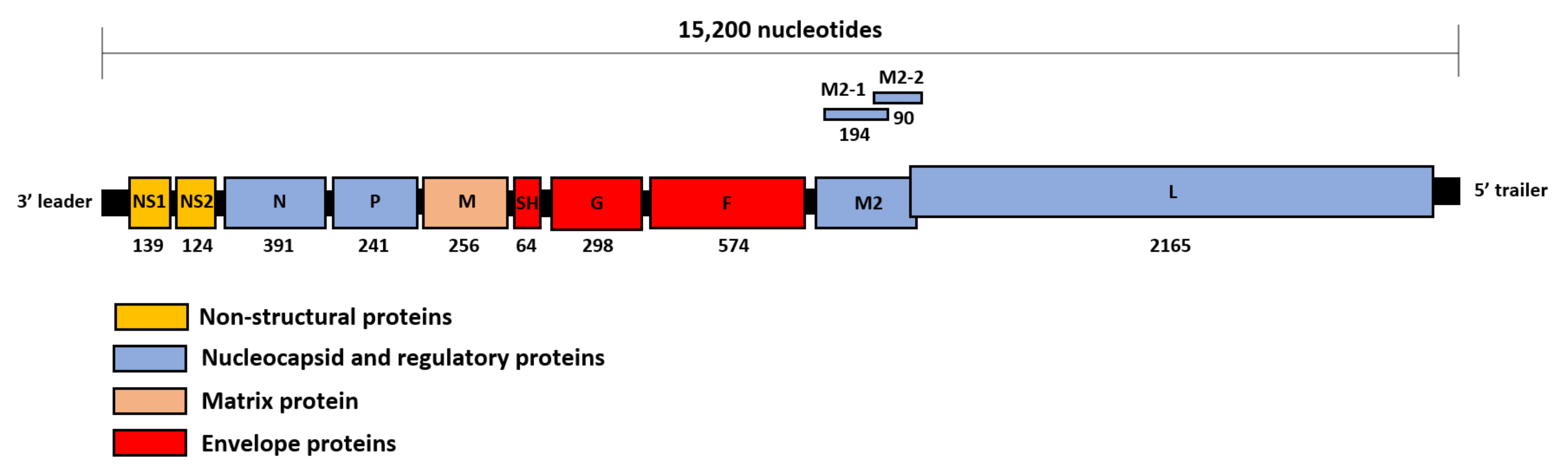
1. Introduction
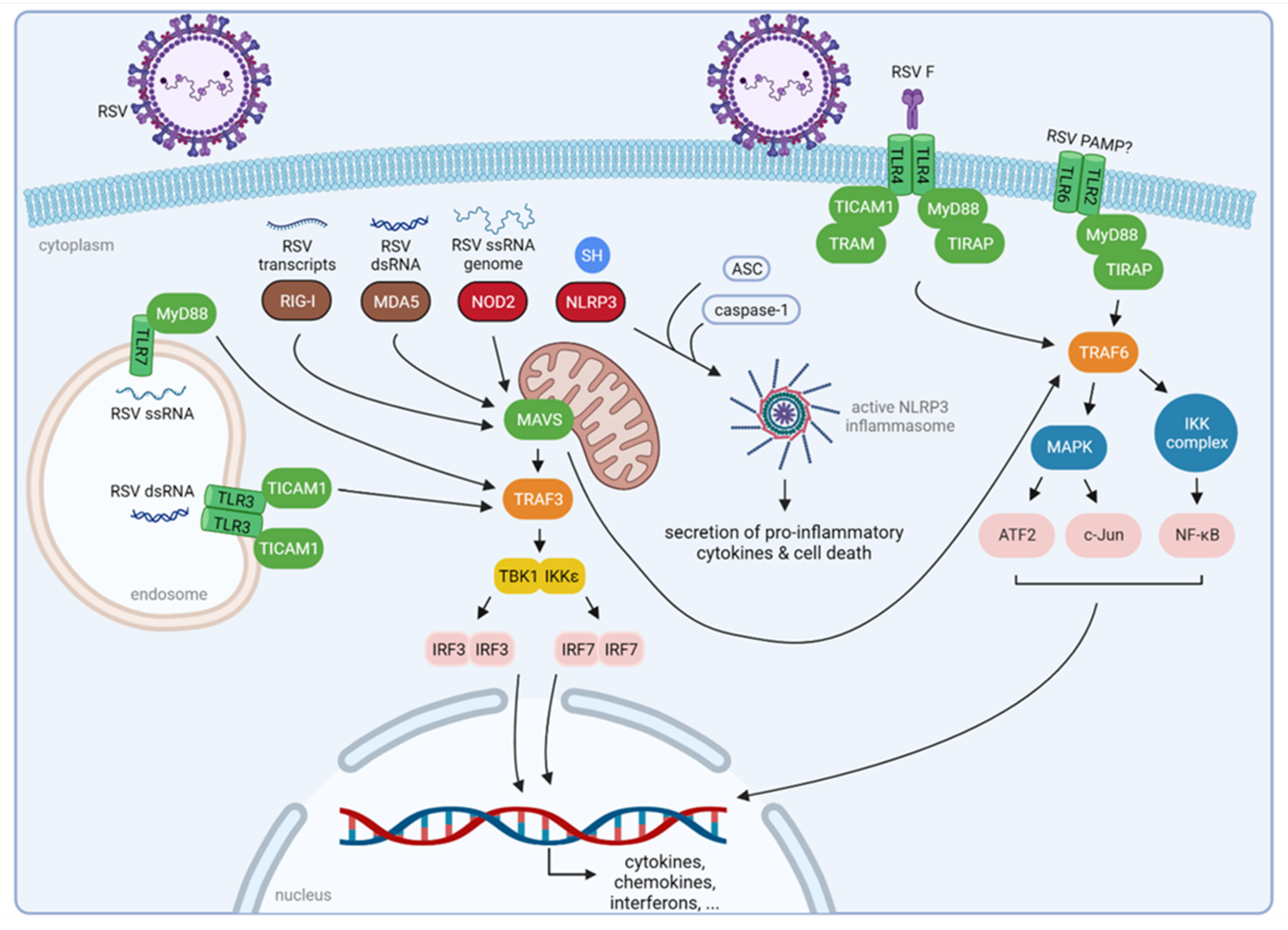
2. Innate Immune Recognition of RSV
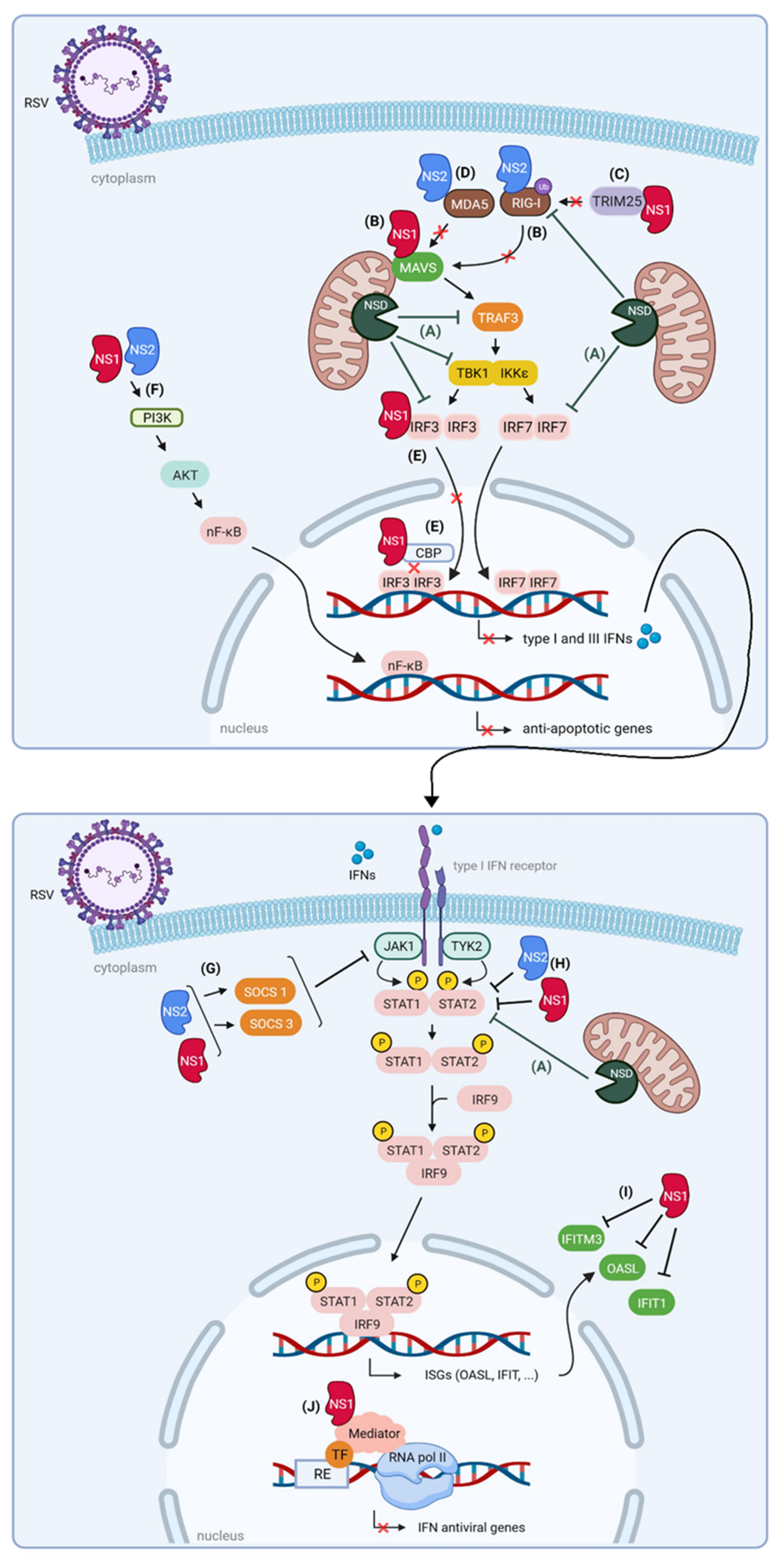
3. How RSV Proteins Join Forces to Overcome the Host Innate Immunity
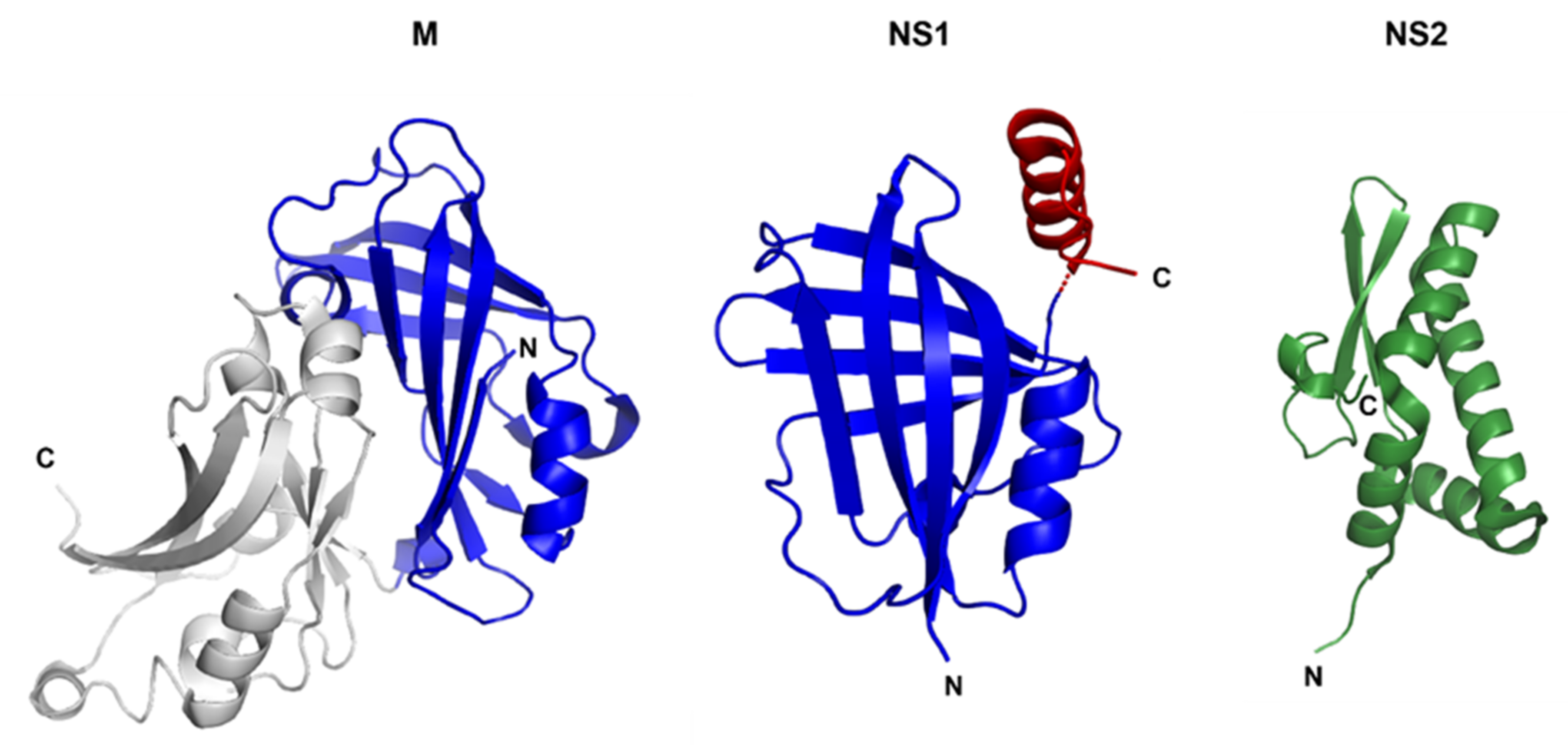
3.1. Non-Structural Protein 1 (NS1) and 2 (NS2)
3.1.1. NS1 and NS2 Impair the Interaction between RLRs and MAVS
3.1.2. NS1 and NS2 Suppress Signaling Downstream of MAVS
3.1.3. NS1 and NS2 Interfere with JAK/STAT Signaling
3.1.4. NS1 and NS2 Are Part of a Large Degradative Complex
3.1.5. Nuclear NS1 Modulates ISG Transcription

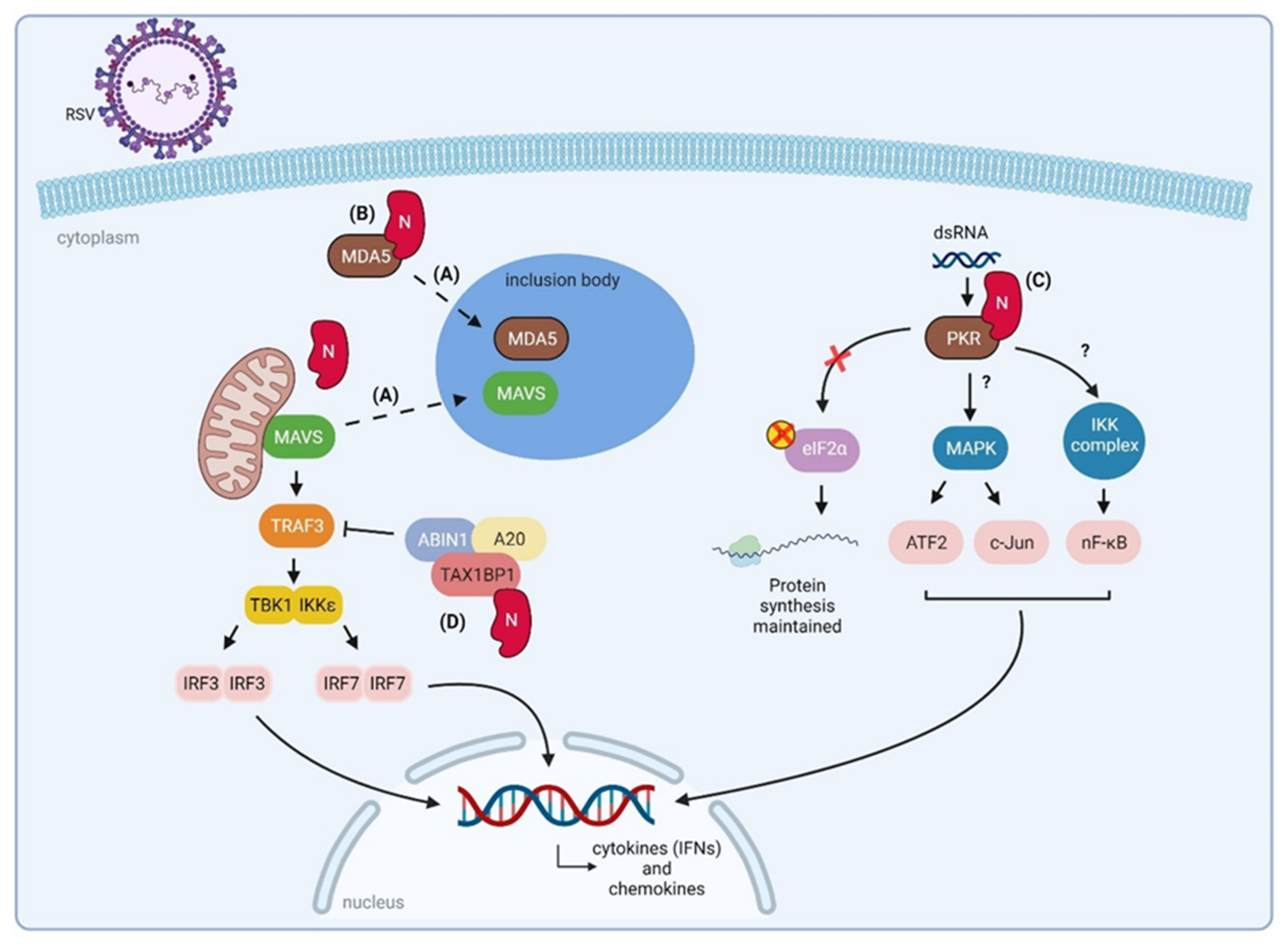
3.2. Nucleoprotein (N)
3.2.1. RSV N Sequesters Innate Immune Signaling Proteins to Inclusion Bodies
3.2.2. RSV N Interacts with PKR
3.2.3. RSV N Might Downregulate NF-κB and IRF3/7 Signaling
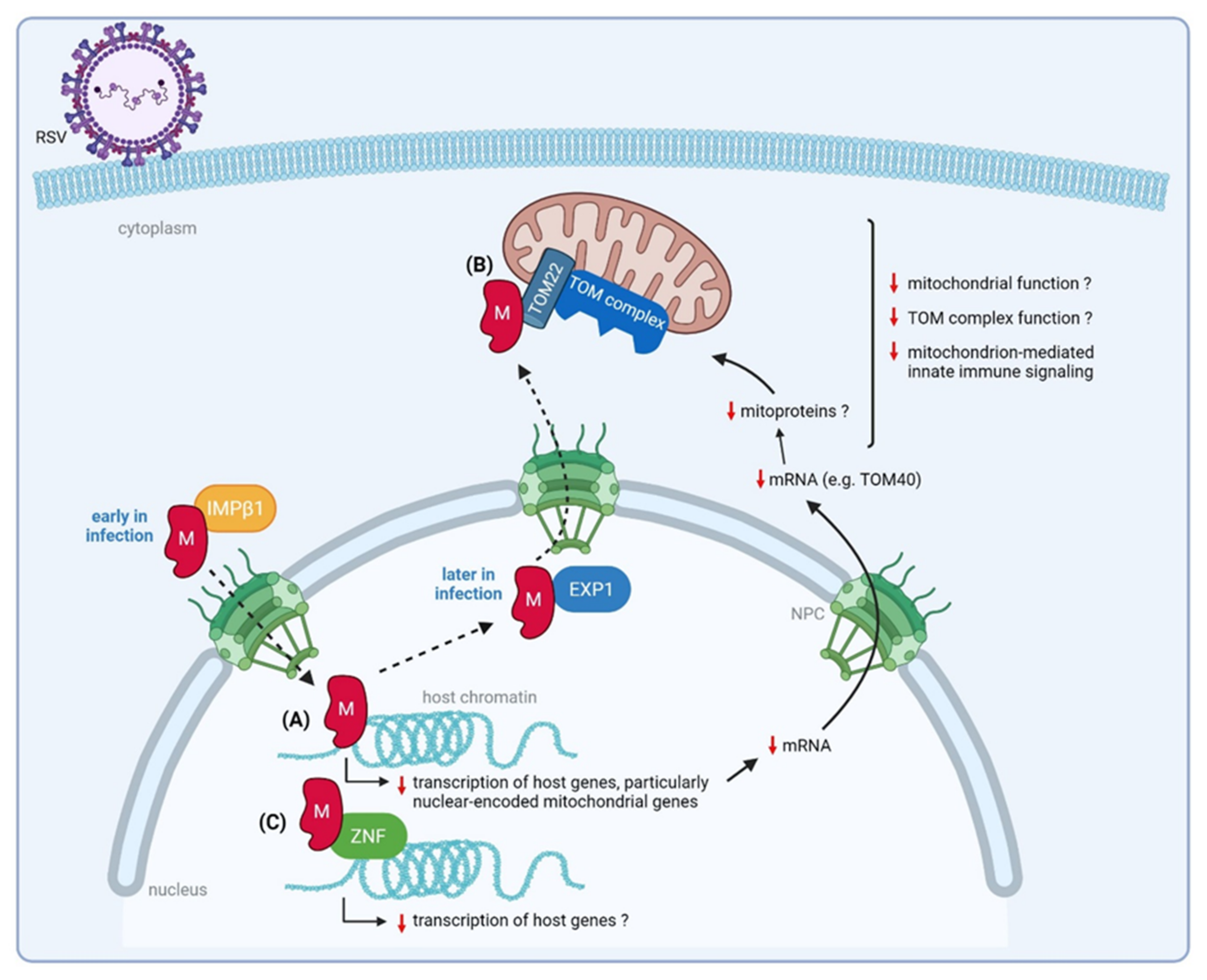
3.3. Matrix (M) Protein
3.3.1. Nuclear M Downregulates Transcription of Nuclear-Encoded Mitochondrial Genes
3.3.2. RSV M Interacts with a ZNF Protein
3.3.3. RSV M Interacts with Mitochondrial Proteins That Mediate Host Innate Immune Responses
3.4. Small Hydrophobic (SH) Protein
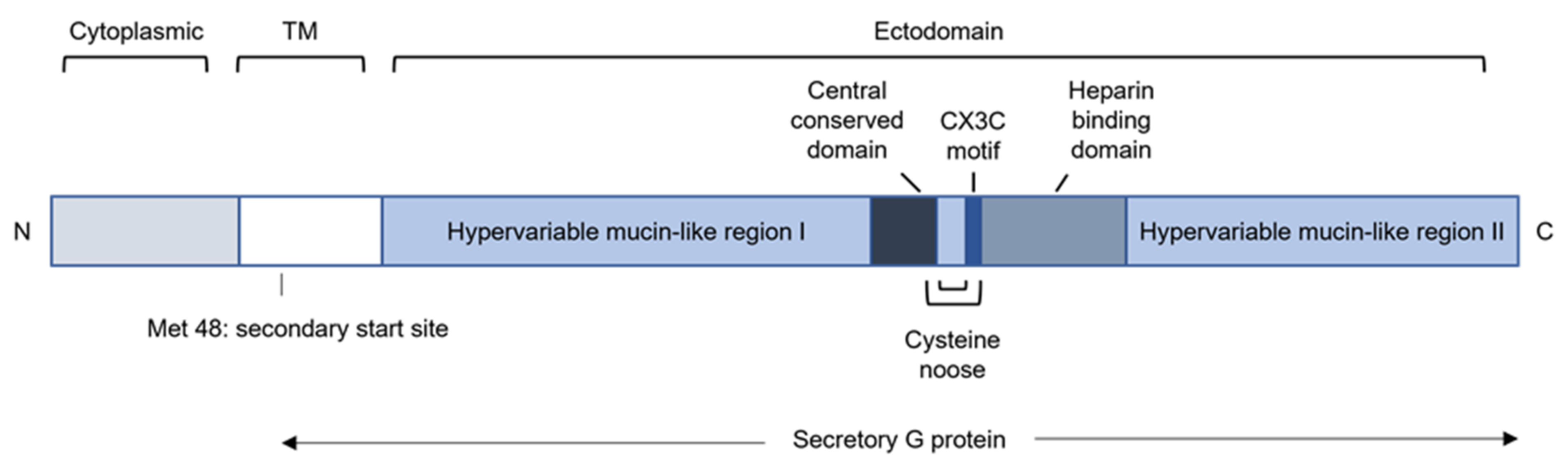
3.5. Glycoprotein (G)
3.5.1. RSV G Modulates Recruitment of Innate Immune Cells by Sequestering Chemokines
3.5.2. RSV G Modulates Cytokine Production
3.5.3. RSV G Modulates the Function of Innate Immune Cells
4. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Nair, H.; Nokes, D.J.; Gessner, B.D.; Dherani, M.; Madhi, S.A.; Singleton, R.J.; O’Brien, K.L.; Roca, A.; Wright, P.F.; Bruce, N.; et al. Global burden of acute lower respiratory infections due to respiratory syncytial virus in young children: A systematic review and meta-analysis. Lancet 2010, 375, 1545–1555. [Google Scholar] [CrossRef]
- Shi, T.; Mcallister, D.A.; Brien, K.L.O.; Simoes, E.A.F.; Madhi, S.A.; Gessner, B.D.; Polack, F.P.; Balsells, E.; Bont, L.; Breiman, R.F.; et al. Global, regional, and national disease burden estimates of acute lower respiratory infections due to respiratory syncytial virus in young children in 2015: A systematic review and modelling study. Lancet 2017, 390, 946–958. [Google Scholar] [CrossRef]
- Shi, T.; Denouel, A.; Tietjen, A.K.; Campbell, I.; Moran, E.; Li, X.; Campbell, H.; Demont, C.; Nyawanda, B.O.; Chu, H.Y.; et al. Global Disease Burden Estimates of Respiratory Syncytial Virus–Associated Acute Respiratory Infection in Older Adults in 2015: A Systematic Review and Meta-Analysis. J. Infect. Dis. 2020, 222, 577–583. [Google Scholar] [CrossRef] [PubMed]
- Collins, P.L.; Fearns, R.; Graham, B.S. Respiratory syncytial virus: Virology, reverse genetics, and pathogenesis of disease. Curr. Top. Microbiol. Immunol. 2013, 372, 3–38. [Google Scholar] [CrossRef]
- Seth, R.B.; Sun, L.; Chen, Z.J. Antiviral innate immunity pathways. Cell Res. 2006, 16, 141–147. [Google Scholar] [CrossRef]
- Weber, F. Antiviral Innate Immunity: Introduction. Encycl. Virol. 2021, 577–583. [Google Scholar] [CrossRef]
- Sokol, C.L.; Luster, A.D. The chemokine system in innate immunity. Cold Spring Harb. Perspect. Biol. 2015, 7, a016303. [Google Scholar] [CrossRef]
- Mesev, E.V.; Ledesma, R.A.; Ploss, A. Decoding type I and III interferon signalling during viral infection. Nat. Microbiol. 2019, 4, 914–924. [Google Scholar] [CrossRef]
- Sun, Y.; López, C.B. The innate immune response to RSV: Advances in our understanding of critical viral and host factors. Vaccine 2017, 35, 481–488. [Google Scholar] [CrossRef]
- Hall, C.B.; Walsh, E.E.; Long, C.E.; Schnabel, K.C. Immunity to and Frequency of Reinfection with Respiratory Syncytial Virus. J. Infect. Dis. 1991, 163, 693–698. [Google Scholar] [CrossRef]
- Henderson, F.W.; Collier, A.M.; Clyde, W.A.; Denny, F.W. Respiratory Syncytial Virus Infections, Reinfections and Immunity—A Prospective, Longitudinal Study in Young Children. N. Engl. J. Med. 1979, 300, 530–534. [Google Scholar] [CrossRef]
- Carty, M.; Guy, C.; Bowie, A.G. Detection of Viral Infections by Innate Immunity. Biochem. Pharmacol. 2021, 183, 114316. [Google Scholar] [CrossRef]
- Liu, P.; Jamaluddin, M.; Li, K.; Garofalo, R.P.; Casola, A.; Brasier, A.R. Retinoic Acid-Inducible Gene I Mediates Early Antiviral Response and Toll-Like Receptor 3 Expression in Respiratory Syncytial Virus-Infected Airway Epithelial Cells. J. Virol. 2007, 81, 1401–1411. [Google Scholar] [CrossRef]
- Loo, Y.-M.; Fornek, J.; Crochet, N.; Bajwa, G.; Perwitasari, O.; Martinez-Sobrido, L.; Akira, S.; Gill, M.A.; García-Sastre, A.; Katze, M.G.; et al. Distinct RIG-I and MDA5 Signaling by RNA Viruses in Innate Immunity. J. Virol. 2008, 82, 335–345. [Google Scholar] [CrossRef]
- Yoboua, F.; Martel, A.; Duval, A.; Mukawera, E.; Grandvaux, N. Respiratory Syncytial Virus-Mediated NF-κB p65 Phosphorylation at Serine 536 Is Dependent on RIG-I, TRAF6, and IKKβ. J. Virol. 2010, 84, 7267–7277. [Google Scholar] [CrossRef]
- Lifland, A.W.; Jung, J.; Alonas, E.; Zurla, C.; Crowe, J.E.; Santangelo, P.J. Human Respiratory Syncytial Virus Nucleoprotein and Inclusion Bodies Antagonize the Innate Immune Response Mediated by MDA5 and MAVS. J. Virol. 2012, 86, 8245–8258. [Google Scholar] [CrossRef]
- Demoor, T.; Petersen, B.C.; Morris, S.; Mukherjee, S.; Ptaschinski, C.; De Almeida Nagata, D.E.; Kawai, T.; Ito, T.; Akira, S.; Kunkel, S.L.; et al. IPS-1 Signaling Has a Nonredundant Role in Mediating Antiviral Responses and the Clearance of Respiratory Syncytial Virus. J. Immunol. 2012, 189, 5942–5953. [Google Scholar] [CrossRef]
- Bhoj, V.G.; Sun, Q.; Bhoj, E.J.; Somers, C.; Chen, X.; Torres, J.P.; Mejias, A.; Gomez, A.M.; Jafri, H.; Ramilo, O.; et al. MAVS and MyD88 are essential for innate immunity but not cytotoxic T lymphocyte response against respiratory syncytial virus. Proc. Natl. Acad. Sci. USA 2008, 105, 14046–14051. [Google Scholar] [CrossRef]
- Pichlmair, A.; Schulz, O.; Tan, C.-P.; Rehwinkel, J.; Kato, H.; Takeuchi, O.; Akira, S.; Way, M.; Schiavo, G.; Reis e Sousa, C. Activation of MDA5 Requires Higher-Order RNA Structures Generated during Virus Infection. J. Virol. 2009, 83, 10761–10769. [Google Scholar] [CrossRef]
- Yoneyama, M.; Kikuchi, M.; Natsukawa, T.; Shinobu, N.; Imaizumi, T.; Miyagishi, M.; Taira, K.; Akira, S.; Fujita, T. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat. Immunol. 2004, 5, 730–737. [Google Scholar] [CrossRef]
- Pichlmair, A.; Schulz, O.; Tan, C.P.; Näslund, T.I.; Liljeström, P.; Weber, F.; Reis e Sousa, C. RIG-I-Mediated Antiviral Responses to Single-Stranded RNA Bearing 5′-Phosphates. Science 2006, 314, 997–1001. [Google Scholar] [CrossRef] [PubMed]
- Sabbah, A.; Chang, T.H.; Harnack, R.; Frohlich, V.; Tominaga, K.; Dube, P.H.; Xiang, Y.; Bose, S. Activation of innate immune antiviral response by NOD2. Physiol. Behav. 2009, 10, 1073–1080. [Google Scholar] [CrossRef]
- Vissers, M.; Remijn, T.; Oosting, M.; de Jong, D.J.; Diavatopoulos, D.A.; Hermans, P.W.M.; Ferwerda, G. Respiratory syncytial virus infection augments NOD2 signaling in an IFN-β-dependent manner in human primary cells. Eur. J. Immunol. 2012, 42, 2727–2735. [Google Scholar] [CrossRef] [PubMed]
- Segovia, J.; Sabbah, A.; Mgbemena, V.; Tsai, S.Y.; Chang, T.H.; Berton, M.T.; Morris, I.R.; Allen, I.C.; Ting, J.P.Y.; Bose, S. TLR2/MyD88/NF-κB pathway, reactive oxygen species, potassium efflux activates NLRP3/ASC inflammasome during respiratory syncytial virus infection. PLoS ONE 2012, 7, e29695. [Google Scholar] [CrossRef]
- Davis, B.K.; Wen, H.; Ting, J.P.-Y. The Inflammasome NLRs in Immunity, Inflammation, and Associated Diseases. Annu. Rev. Immunol. 2011, 29, 707–735. [Google Scholar] [CrossRef]
- Broz, P.; Dixit, V.M. Inflammasomes: Mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 2016, 16, 407–420. [Google Scholar] [CrossRef]
- Triantafilou, K.; Kar, S.; Vakakis, E.; Kotecha, S.; Triantafilou, M. Human respiratory syncytial virus viroporin SH: A viral recognition pathway used by the host to signal inflammasome activation. Thorax 2013, 68, 66–75. [Google Scholar] [CrossRef]
- Fuentes, S.; Tran, K.C.; Luthra, P.; Teng, M.N.; He, B. Function of the Respiratory Syncytial Virus Small Hydrophobic Protein. J. Virol. 2007, 81, 8361–8366. [Google Scholar] [CrossRef]
- Russell, R.F.; McDonald, J.U.; Ivanova, M.; Zhong, Z.; Bukreyev, A.; Tregoning, J.S. Partial Attenuation of Respiratory Syncytial Virus with a Deletion of a Small Hydrophobic Gene Is Associated with Elevated Interleukin-1β Responses. J. Virol. 2015, 89, 8974–8981. [Google Scholar] [CrossRef]
- Pollock, N.; Taylor, G.; Jobe, F.; Guzman, E. Modulation of the transcription factor NF-κB in antigen-presenting cells by bovine respiratory syncytial virus small hydrophobic protein. J. Gen. Virol. 2017, 98, 1587–1599. [Google Scholar] [CrossRef]
- Kawasaki, T.; Kawai, T. Toll-like receptor signaling pathways. Front. Immunol. 2014, 5, 461. [Google Scholar] [CrossRef]
- Arnold, R.; König, W. Peroxisome proliferator-activated receptor-γ agonists inhibit the replication of respiratory syncytial virus (RSV) in human lung epithelial cells. Virology 2006, 350, 335–346. [Google Scholar] [CrossRef][Green Version]
- Murawski, M.R.; Bowen, G.N.; Cerny, A.M.; Anderson, L.J.; Haynes, L.M.; Tripp, R.A.; Kurt-Jones, E.A.; Finberg, R.W. Respiratory Syncytial Virus Activates Innate Immunity through Toll-Like Receptor. J. Virol. 2009, 83, 1492–1500. [Google Scholar] [CrossRef]
- Timur, H.; Yarovinsky, O.; Look, D.C.; Dayna, G.W.; Groskreutz, J.; Monick, M.M.; Powers, L.S. Respiratory Syncytial Virus Induces TLR3 Protein and Protein Kinase R, Leading to Increased Double-Stranded RNA Responsiveness in Airway Epithelial Cells. J. Immunol. 2006, 176, 1733–1740. [Google Scholar] [CrossRef]
- Rudd, B.D.; Burstein, E.; Duckett, C.S.; Li, X.; Lukacs, N.W. Differential Role for TLR3 in Respiratory Syncytial Virus-Induced Chemokine Expression. J. Virol. 2005, 79, 3350–3357. [Google Scholar] [CrossRef]
- Lukacs, N.W.; Smit, J.J.; Mukherjee, S.; Morris, S.B.; Nunez, G.; Lindell, D.M. Respiratory Virus-Induced TLR7 Activation Controls IL-17–Associated Increased Mucus via IL-23 Regulation. J. Immunol. 2010, 185, 2231–2239. [Google Scholar] [CrossRef]
- Park, B.S.; Lee, J.O. Recognition of lipopolysaccharide pattern by TLR4 complexes. Exp. Mol. Med. 2013, 45, e66. [Google Scholar] [CrossRef]
- Kurt-Jones, E.A.; Popova, L.; Kwinn, L.; Haynes, L.M.; Jones, L.P.; Tripp, R.A.; Walsh, E.E.; Freeman, M.W.; Golenbock, D.T.; Anderson, L.J.; et al. Pattern recognition receptors TLR4 and CD14 mediate response to respiratory syncytial virus. Nat. Immunol. 2000, 1, 398–401. [Google Scholar] [CrossRef]
- Haynes, L.M.; Moore, D.D.; Kurt-Jones, E.A.; Finberg, R.W.; Anderson, L.J.; Tripp, R.A. Involvement of Toll-Like Receptor 4 in Innate Immunity to Respiratory Syncytial Virus. J. Virol. 2001, 75, 10730–10737. [Google Scholar] [CrossRef]
- Ehl, S.; Bischoff, R.; Ostler, T.; Vallbracht, S.; Schulte-Mönting, J.; Poltorak, A.; Freudenberg, M. The role of Toll-like receptor 4 versus interleukin-12 in immunity to respiratory syncytial virus. Eur. J. Immunol. 2004, 34, 1146–1153. [Google Scholar] [CrossRef]
- Marr, N.; Turvey, S.E. Role of human TLR4 in respiratory syncytial virus-induced NF-κB activation, viral entry and replication. Innate Immun. 2012, 18, 856–865. [Google Scholar] [CrossRef]
- Scagnolari, C.; Midulla, F.; Pierangeli, A.; Moretti, C.; Bonci, E.; Berardi, R.; De Angelis, D.; Selvaggi, C.; Di Marco, P.; Girardi, E.; et al. Gene Expression of Nucleic Acid-Sensing Pattern Recognition Receptors in Children Hospitalized for Respiratory Syncytial Virus-Associated Acute Bronchiolitis. Clin. Vaccine Immunol. 2009, 16, 816–823. [Google Scholar] [CrossRef] [PubMed]
- Hall, C.B., Jr.; Douglas, G.; Simons, R.L.; Geiman, J.M. Interferon production in children with respiratory syncytial, influenza, and parainfluenza virus infections. J. Pediatr. 1978, 93, 28–32. [Google Scholar] [CrossRef]
- Midulla, F.; Tromba, V.; Russo, L.L.; Mileto, F.; Sabatino, G.; Sgarrella, M.; Panuska, J.R.; Manganozzi, L.; Korn, D.; Moretti, C. Cytokines in the nasal washes of children with respiratory syncytial virus bronchiolitis. Int. J. Immunopathol. Pharmacol. 2006, 19, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Isaacs, D. Production of interferon in respiratory syncytial virus bronchiolitis. Arch. Dis. Child. 1989, 64, 92–95. [Google Scholar] [CrossRef] [PubMed]
- Krilov, L.R.; Hendry, R.M.; Godfrey, E.; McIntosh, K. Respiratory Virus Infection of Peripheral Blood Monocytes: Correlation with Ageing of Cells and Interferon Production in vitro. J. Gen.Virol. 1987, 68, 1749–1753. [Google Scholar] [CrossRef]
- Okabayashi, T.; Kojima, T.; Masaki, T.; Yokota, S.-I.; Imaizumi, T.; Tsutsumi, H.; Himi, T.; Fujii, N.; Sawada, N. Type-III interferon, not type-I, is the predominant interferon induced by respiratory viruses in nasal epithelial cells. Virus Res. 2011, 160, 360–366. [Google Scholar] [CrossRef]
- Mordstein, M.; Neugebauer, E.; Ditt, V.; Jessen, B.; Rieger, T.; Falcone, V.; Sorgeloos, F.; Ehl, S.; Mayer, D.; Kochs, G.; et al. Lambda Interferon Renders Epithelial Cells of the Respiratory and Gastrointestinal Tracts Resistant to Viral Infections. J. Virol. 2010, 84, 5670–5677. [Google Scholar] [CrossRef]
- Spann, K.M.; Tran, K.-C.; Chi, B.; Rabin, R.L.; Collins, P.L. Suppression of the Induction of Alpha, Beta, and Gamma Interferons by the NS1 and NS2 Proteins of Human Respiratory Syncytial Virus in Human Epithelial Cells and Macrophages. J. Virol. 2004, 78, 4363–4369. [Google Scholar] [CrossRef]
- Monick, M.M.; Cameron, K.; Staber, J.; Powers, L.S.; Yarovinsky, T.O.; Koland, J.G.; Hunninghake, G.W. Activation of the Epidermal Growth Factor Receptor by Respiratory Syncytial Virus Results in Increased Inflammation and Delayed Apoptosis. J. Biol. Chem. 2005, 280, 2147–2158. [Google Scholar] [CrossRef]
- Kalinowski, A.; Galen, B.T.; Ueki, I.F.; Sun, Y.; Mulenos, A.; Osafo-Addo, A.; Clark, B.; Joerns, J.; Liu, W.; Nadel, J.A.; et al. Respiratory syncytial virus activates epidermal growth factor receptor to suppress interferon regulatory factor 1-dependent interferon-lambda and antiviral defense in airway epithelium. Mucosal Immunol. 2018, 11, 958–967. [Google Scholar] [CrossRef]
- Sedeyn Id, K.; Schepens, B.; Saelens, X. Respiratory syncytial virus nonstructural proteins 1 and 2: Exceptional disrupters of innate immune responses. PLoS Pathog. 2019, 15, e1007984. [Google Scholar] [CrossRef]
- Thornhill, E.M.; Verhoeven, D. Respiratory Syncytial Virus’s Non-structural Proteins: Masters of Interference. Front. Cell. Infect. Microbiol. 2020, 10, 225. [Google Scholar] [CrossRef]
- Levine, S.; Kaliaber-Franco, R.; Paradiso, P.R. Demonstration that glycoprotein G is the attachment protein of respiratory syncytial virus. J. Gen. Virol. 1987, 68, 2521–2524. [Google Scholar] [CrossRef]
- Arnold, R.; König, B.; Werchau, H.; König, W. Respiratory syncytial virus deficient in soluble G protein induced an increased proinflammatory response in human lung epithelial cells. Virology 2004, 330, 384–397. [Google Scholar] [CrossRef]
- König, H.-J.B.; Streckert, T.; Krusat, W.K. Respiratory syncytial virus G-protein modulates cytokine release from human peripheral blood mononuclear cells. J. Leukoc. Biol. 1996, 59, 403–406. [Google Scholar] [CrossRef]
- Polack, F.P.; Irusta, P.M.; Hoffman, S.J.; Schiatti, M.P.; Melendi, G.A.; Delgado, M.F.; Laham, F.R.; Thumar, B.; Hendry, R.M.; Melero, J.A.; et al. The cysteine-rich region of respiratory syncytial virus attachment protein inhibits innate immunity elicited by the virus and endotoxin. Proc. Natl. Acad. Sci. USA 2005, 102, 8996–9001. [Google Scholar] [CrossRef]
- Shingai, M.; Azuma, M.; Ebihara, T.; Sasai, M.; Funami, K.; Ayata, M.; Ogura, H.; Tsutsumi, H.; Matsumoto, M.; Seya, T. Soluble G protein of respiratory syncytial virus inhibits Toll-like receptor 3/4-mediated IFN-beta induction. Int. Immunol. 2008, 20, 1169–1180. [Google Scholar] [CrossRef]
- Garcia, J.; García-Barreno, B.; Martinez, I.; Melero, J.A. Cytoplasmic inclusions of respiratory syncytial virus-infected cells: Formation of inclusion bodies in transfected cells that coexpress the nucleoprotein, the phosphoprotein, and the 22K protein. Virology 1993, 195, 243–247. [Google Scholar] [CrossRef]
- Rincheval, V.; Lelek, M.; Gault, E.; Bouillier, C.; Sitterlin, D.; Blouquit-laye, S.; Galloux, M.; Zimmer, C.; Eleouet, J. Functional organization of cytoplasmic inclusion bodies in cells infected by respiratory syncytial virus. J. Gen. Virol. 2013, 94, 1734–1738. [Google Scholar] [CrossRef]
- Li, H.M.; Ghildyal, R.; Hu, M.; Tran, K.C.; Starrs, L.M.; Mills, J.; Teng, M.N.; Jans, D.A. Respiratory syncytial virus matrix protein-chromatin association is key to transcriptional inhibition in infected cells. Cells 2021, 10, 2786. [Google Scholar] [CrossRef]
- Kipper, S.; Hamad, S.; Caly, L.; Avrahami, D.; Bacharach, E.; Jans, D.A.; Gerber, D.; Bajorek, M. New Host Factors Important for Respiratory Syncytial Virus (RSV) Replication Revealed by a Novel Microfluidics Screen for Interactors of Matrix (M) Protein. Mol. Cell. Proteom. 2015, 14, 532. [Google Scholar] [CrossRef]
- Radhakrishnan, A.; Yeo, D.; Brown, G.; Myaing, M.Z.; Iyer, L.R.; Fleck, R.; Tan, B.H.; Aitken, J.; Sanmun, D.; Tang, K.; et al. Protein analysis of purified respiratory syncytial virus particles reveals an important role for heat shock protein 90 in virus particle assembly. Mol. Cell. Proteom. 2010, 9, 1829–1848. [Google Scholar] [CrossRef]
- Atreya, P.L.; Peeples, M.E.; Collins, P.L. The NS1 Protein of Human Respiratory Syncytial Virus Is a Potent Inhibitor of Minigenome Transcription and RNA Replication. J. Virol. 1998, 72, 1452–1461. [Google Scholar] [CrossRef]
- Uche, I.K.; Guerrero-Plata, A. Interferon-mediated response to human metapneumovirus infection. Viruses 2018, 10, 505. [Google Scholar] [CrossRef]
- Swedan, S.; Andrews, J.; Majumdar, T.; Musiyenko, A.; Barik, S. Multiple Functional Domains and Complexes of the Two Nonstructural Proteins of Human Respiratory Syncytial Virus Contribute to Interferon Suppression and Cellular Location. J. Virol. 2011, 85, 10090–10100. [Google Scholar] [CrossRef]
- Jin, H.; Cheng, X.; Traina-dorge, V.L.; Jung, H.; Zhou, H.; Soike, K.; Kemble, G. Evaluation of recombinant respiratory syncytial virus gene deletion mutants in African green monkeys for their potential as live attenuated vaccine candidates. Vaccine 2003, 21, 3647–3652. [Google Scholar] [CrossRef]
- Teng, M.N.; Collins, P.L. Altered Growth Characteristics of Recombinant Respiratory Syncytial Viruses Which Do Not Produce NS2 Protein. J. Virol. 1999, 73, 466–473. [Google Scholar] [CrossRef] [PubMed]
- Teng, M.N.; Whitehead, S.S.; Bermingham, A.; St. Claire, M.; Elkins, W.R.; Murphy, B.R.; Collins, P.L. Recombinant Respiratory Syncytial Virus That Does Not Express the NS1 or M2-2 Protein Is Highly Attenuated and Immunogenic in Chimpanzees. J. Virol. 2000, 74, 9317–9321. [Google Scholar] [CrossRef]
- Jin, H.; Zhou, H.; Cheng, X.; Tang, R.; Munoz, M.; Nguyen, N. Recombinant Respiratory Syncytial Viruses with Deletions in the NS1, NS2, SH, and M2-2 Genes Are Attenuated in Vitro and in Vivo. Virology 2000, 273, 210–218. [Google Scholar] [CrossRef] [PubMed]
- Schlender, J.; Bossert, B.; Buchholz, U.; Conzelmann, K.-K. Bovine Respiratory Syncytial Virus Nonstructural Proteins NS1 and NS2 Cooperatively Antagonize Alpha/Beta Interferon-Induced Antiviral Response. J. Virol. 2000, 74, 8234–8242. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Luthra, P.; Esaulova, E.; Agapov, E.; Yen, B.C.; Borek, D.M.; Edwards, M.R.; Mittal, A.; Jordan, D.S.; Ramanan, P.; et al. Structural basis for human respiratory syncytial virus NS1-mediated modulation of host responses. Nat. Microbiol. 2017, 2, 17101. [Google Scholar] [CrossRef] [PubMed]
- Pei, J.; Wagner, N.D.; Zou, A.J.; Chatterjee, S.; Borek, D.; Cole, A.R.; Kim, P.J.; Basler, C.F.; Otwinowski, Z.; Gross, M.L.; et al. Structural basis for IFN antagonism by human respiratory syncytial virus nonstructural protein. Proc. Natl. Acad. Sci. USA 2021, 118, e2020587118. [Google Scholar] [CrossRef]
- Boyapalle, S.; Wong, T.; Garay, J.; Teng, M.; San Juan-Vergara, H.; Mohapatra, S.; Mohapatra, S. Respiratory syncytial virus NS1 protein colocalizes with mitochondrial antiviral signaling protein MAVS following infection. PLoS ONE 2012, 7, e29386. [Google Scholar] [CrossRef]
- Ban, J.; Lee, N.R.; Lee, N.J.; Lee, J.K.; Quan, F.S.; Inn, K.S. Human Respiratory Syncytial Virus NS 1 Targets TRIM25 to Suppress RIG-I Ubiquitination and Subsequent RIG-I-Mediated Antiviral Signaling. Viruses 2018, 10, 716. [Google Scholar] [CrossRef]
- Ling, Z.; Tran, K.C.; Teng, M.N. Human Respiratory Syncytial Virus Nonstructural Protein NS2 Antagonizes the Activation of Beta Interferon Transcription by Interacting with RIG-I. J. Virol. 2009, 83, 3734–3742. [Google Scholar] [CrossRef]
- Goswami, R.; Majumdar, T.; Dhar, J.; Chattopadhyay, S.; Bandyopadhyay, S.K.; Verbovetskaya, V.; Sen, G.C.; Barik, S. Viral degradasome hijacks mitochondria to suppress innate immunity. Cell Res. 2013, 23, 1025–1042. [Google Scholar] [CrossRef]
- Swedan, S.; Musiyenko, A.; Barik, S. Respiratory Syncytial Virus Nonstructural Proteins Decrease Levels of Multiple Members of the Cellular Interferon Pathways. J. Virol. 2009, 83, 9682–9693. [Google Scholar] [CrossRef]
- Ren, J.; Liu, T.; Pang, L.; Li, K.; Garofalo, R.P.; Casola, A.; Bao, X. A novel mechanism for the inhibition of interferon regulatory factor-3-dependent gene expression by human respiratory syncytial virus NS1 protein. J. Gen. Virol. 2011, 92, 2153–2159. [Google Scholar] [CrossRef]
- Spann, K.M.; Tran, K.C.; Collins, P.L. Effects of Nonstructural Proteins NS1 and NS2 of Human Respiratory Syncytial Virus on Interferon Regulatory Factor 3, NF-B, and Proinflammatory Cytokines. J. Virol. 2005, 79, 5353–5362. [Google Scholar] [CrossRef]
- Bitko, V.; Shulyayeva, O.; Mazumder, B.; Musiyenko, A.; Ramaswamy, M.; Look, D.C.; Barik, S. Nonstructural Proteins of Respiratory Syncytial Virus Suppress Premature Apoptosis by an NF- B-Dependent, Interferon-Independent Mechanism and Facilitate Virus Growth. J. Virol. 2007, 81, 1786–1795. [Google Scholar] [CrossRef]
- Moore, E.C.; Barber, J.; Tripp, R.A. Respiratory syncytial virus (RSV) attachment and nonstructural proteins modify the type I interferon response associated with suppressor of cytokine signaling (SOCS) proteins and IFN-stimulated gene-15 (ISG15). Virol. J. 2008, 5, 116. [Google Scholar] [CrossRef]
- Hashimoto, K.; Ishibashi, K.; Ishioka, K.; Zhao, D.; Sato, M.; Ohara, S.; Abe, Y.; Kawasaki, Y.; Sato, Y.; Yokota, S.; et al. RSV replication is attenuated by counteracting expression of the suppressor of cytokine signaling (SOCS) molecules. Virology 2009, 391, 162–170. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Zheng, J.; Zheng, K.; Hou, Y.; Zhao, F.; Zhao, D. Respiratory Syncytial Virus NS1 Protein Degrades STAT2 by Inducing SOCS1 Expression. Intervirology 2014, 57, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Elliott, J.; Lynch, O.T.; Suessmuth, Y.; Qian, P.; Boyd, C.R.; Burrows, J.F.; Buick, R.; Stevenson, N.J.; Touzelet, O.; Gadina, M.; et al. Respiratory Syncytial Virus NS1 Protein Degrades STAT2 by Using the Elongin-Cullin E3 Ligase. J. Virol. 2007, 81, 3428–3436. [Google Scholar] [CrossRef]
- Lo, M.S.; Brazas, R.M.; Holtzman, M.J. Respiratory Syncytial Virus Nonstructural Proteins NS1 and NS2 Mediate Inhibition of Stat2 Expression and Alpha/Beta Interferon Responsiveness. J. Virol. 2005, 79, 9315–9319. [Google Scholar] [CrossRef]
- Whelan, J.N.; Tran, K.C.; van Rossum, D.B.; Teng, M.N. Identification of Respiratory Syncytial Virus Nonstructural Protein 2 Residues Essential for Exploitation of the Host Ubiquitin System and Inhibition of Innate Immune Responses. J. Virol. 2016, 90, 6453–6463. [Google Scholar] [CrossRef]
- Wu, W.; Tran, K.C.; Teng, M.N.; Heesom, K.J.; Matthews, D.A.; Barr, J.N.; Hiscox, J.A. The Interactome of the Human Respiratory Syncytial Virus NS1 Protein Highlights Multiple Effects on Host Cell Biology. J. Virol. 2012, 86, 7777–7789. [Google Scholar] [CrossRef]
- Tan, Y.R.; Peng, D.; Chen, C.M.; Qin, X.Q. Nonstructural protein-1 of respiratory syncytial virus regulates HOX gene expression through interacting with histone. Mol. Biol. Rep. 2013, 40, 675–679. [Google Scholar] [CrossRef]
- Straub, C.P.; Lau, W.H.; Preston, F.M.; Headlam, M.J.; Gorman, J.J.; Collins, P.L.; Spann, K.M. Mutation of the elongin C binding domain of human respiratory syncytial virus non-structural protein 1 (NS1) results in degradation of NS1 and attenuation of the virus. Virol. J. 2011, 8, 252. [Google Scholar] [CrossRef]
- Munday, D.C.; Hiscox, J.A.; Barr, J.N. Quantitative proteomic analysis of A549 cells infected with human respiratory syncytial virus subgroup B using SILAC coupled to LC-MS/MS. Proteomics 2010, 10, 4320–4334. [Google Scholar] [CrossRef]
- Pei, J.; Beri, N.R.; Zou, A.J.; Hubel, P.; Dorando, H.K.; Bergant, V.; Andrews, R.D.; Pan, J.; Andrews, J.M.; Sheehan, K.C.F.; et al. Nuclear-localized human respiratory syncytial virus NS1 protein modulates host gene transcription. Cell Rep. 2021, 37, 109803. [Google Scholar] [CrossRef] [PubMed]
- Poss, Z.C.; Ebmeier, C.C.; Taatjes, D.J. The Mediator complex and transcription regulation. Crit. Rev. Biochem. Mol. Biol. 2013, 48, 575–608. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.W.; Wang, G. The Mediator complex: A master coordinator of transcription and cell lineage development. Development 2014, 141, 977–987. [Google Scholar] [CrossRef]
- Dhar, J.; Cuevas, R.A.; Goswami, R.; Zhu, J.; Sarkar, S.N.; Barik, S. 2′-5′-Oligoadenylate Synthetase-Like Protein Inhibits Respiratory Syncytial Virus Replication and Is Targeted by the Viral Nonstructural Protein 1. J. Virol. 2015, 89, 10115–10119. [Google Scholar] [CrossRef]
- Ribaudo, M.; Barik, S. The nonstructural proteins of Pneumoviruses are remarkably distinct in substrate diversity and specificity. Virol. J. 2017, 14, 215. [Google Scholar] [CrossRef]
- Allen, B.L.; Taatjes, D.J. The Mediator complex: A central integrator of transcription. Nat. Rev. Mol. Cell Biol. 2015, 16, 155–166. [Google Scholar] [CrossRef]
- Tawar, R.G.; Duquerroy, S.; Vonrhein, C.; Varela, P.F.; Damier-Piolle, L.; Castagne, N.; MacLellan, K.; Bedouelle, H.; Bricogne, G.; Bhella, D.; et al. Crystal structure of a nucleocapsid-like nucleoprotein-RNA complex of respiratory syncytial virus. Science 2009, 326, 1279–1283. [Google Scholar] [CrossRef]
- Bakker, S.E.; Duquerroy, S.; Galloux, M.; Loney, C.; Conner, E.; Eléouet, J.F.; Rey, F.A.; Bhella, D. The respiratory syncytial virus nucleoprotein-RNA complex forms a left-handed helical nucleocapsid. J. Gen. Virol. 2013, 94, 1734–1738. [Google Scholar] [CrossRef]
- Roux, K.J.; Kim, D.I.; Raida, M.; Burke, B. A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J. Cell Biol. 2012, 196, 801–810. [Google Scholar] [CrossRef]
- Eyckerman, S.; Titeca, K.; Van Quickelberghe, E.; Cloots, E.; Verhee, A.; Samyn, N.; De Ceuninck, L.; Timmerman, E.; De Sutter, D.; Lievens, S.; et al. Trapping mammalian protein complexes in viral particles. Nat. Commun. 2016, 7, 11416. [Google Scholar] [CrossRef] [PubMed]
- Groskreutz, D.J.; Babor, E.C.; Monick, M.M.; Varga, S.M.; Hunninghake, G.W. Respiratory syncytial virus limits α subunit of eukaryotic translation initiation factor 2 (eIF2α) phosphorylation to maintain translation and viral replication. J. Biol. Chem. 2010, 285, 24023–24031. [Google Scholar] [CrossRef] [PubMed]
- Gal-ben-ari, S.; Barrera, I.; Ehrlich, M.; Rosenblum, K. PKR: A Kinase to Remember. Front. Mol. Neurosci. 2019, 11, 480. [Google Scholar] [CrossRef] [PubMed]
- Kang, R.; Tang, D. PKR-Dependent Inflammatory Signals. Sci. Signal. 2012, 5, pe47. [Google Scholar] [CrossRef]
- Minor, R.A.C.; Limmon, G.V.; Miller-degraff, L.; Dixon, D.; Andrews, D.M.K.; Kaufman, R.J.; Imani, F. Double-Stranded RNA-Activated Protein Kinase Regulates Early Innate Immune Responses during Respiratory Syncytial Virus Infection. J. Interferon Cytokine Res. 2010, 30, 263–272. [Google Scholar] [CrossRef]
- Descamps, D.; De Oliveira, P.; Gonnin, L.; Madrières, S.; Fix, J.; Drajac, C. Depletion of TAX1BP1 Amplifies Innate Immune Responses during Respiratory Syncytial Virus Infection. J. Virol. 2021, 95, 912–921. [Google Scholar] [CrossRef]
- Martín-vicente, M.; González-sanz, R.; Cuesta, I.; Monzón, S.; Resino, S.; Martínez, I. Downregulation of A20 expression increases the immune response and apoptosis and reduces virus production in cells infected by the human respiratory syncytial virus. Vaccines 2020, 8, 100. [Google Scholar] [CrossRef]
- Harhaj, E.W.; Dixit, V.M. Deubiquitinases in the regulation of NF-κB signaling. Cell Res. 2011, 21, 22–39. [Google Scholar] [CrossRef]
- Davis, M.E.; Gack, M.U. Ubiquitination in the antiviral immune response. Virology 2015, 479–480, 52–65. [Google Scholar] [CrossRef]
- Verstrepen, L.; Verhelst, K.; Carpentier, I.; Beyaert, R. TAX1BP1, a ubiquitin-binding adaptor protein in innate immunity and beyond. Trends Biochem. Sci. 2011, 36, 347–354. [Google Scholar] [CrossRef]
- Gao, L.; Coope, H.; Grant, S.; Averil, M.; Ley, S.C.; Harhaj, E.W. ABIN1 Protein Cooperates with TAX1BP1 and A20 Proteins to Inhibit Antiviral Signaling. J. Biol. Chem. 2011, 286, 36592–36602. [Google Scholar] [CrossRef]
- Ghildyal, R.; Ho, A.; Jans, D.A. Central role of the respiratory syncytial virus matrix protein in infection. FEMS Microbiol. Rev. 2006, 30, 692–705. [Google Scholar] [CrossRef]
- Ghildyal, R.; Baulch-Brown, C.; Mills, J.; Meanger, J. The matrix protein of Human respiratory syncytial virus localises to the nucleus of infected cells and inhibits transcription. Arch. Virol. 2003, 148, 1419–1429. [Google Scholar] [CrossRef]
- Ghildyal, R.; Ho, A.; Dias, M.; Soegiyono, L.; Bardin, P.G.; Tran, K.C.; Teng, M.N.; Jans, D.A. The Respiratory Syncytial Virus Matrix Protein Possesses a Crm1-Mediated Nuclear Export Mechanism. J. Virol. 2009, 83, 5353–5362. [Google Scholar] [CrossRef]
- Shahriari, S.; Gordon, J.; Ghildyal, R. Host cytoskeleton in respiratory syncytial virus assembly and budding. Virol. J. 2016, 13, 161. [Google Scholar] [CrossRef]
- Mitra, R.; Baviskar, P.; Duncan-Decocq, R.R.; Patel, D.; Oomens, A.G.P. The Human Respiratory Syncytial Virus Matrix Protein Is Required for Maturation of Viral Filaments. J. Virol. 2012, 86, 4432–4443. [Google Scholar] [CrossRef]
- Förster, A.; Maertens, G.N.; Farrell, P.J.; Bajorek, M. Dimerization of Matrix Protein Is Required for Budding of Respiratory Syncytial Virus. J. Virol. 2015, 89, 4624–4635. [Google Scholar] [CrossRef]
- Battisti, A.J.; Meng, G.; Winkler, D.C.; McGinnes, L.W.; Plevka, P.; Steven, A.C.; Morrison, T.G.; Rossmann, M.G. Structure and assembly of a paramyxovirus matrix protein. Proc. Natl. Acad. Sci. USA 2012, 109, 13996–14000. [Google Scholar] [CrossRef]
- Hu, M.J.; Bogoyevitch, M.A.; Jans, D.A. Impact of respiratory syncytial virus infection on host functions: Implications for antiviral strategies. Physiol. Rev. 2020, 100, 1527–1594. [Google Scholar] [CrossRef]
- Barral-Arca, R.; Gómez-Carballa, A.; Cebey-López, M.; Bello, X.; Martinón-Torres, F.; Salas, A. A Meta-Analysis of Multiple Whole Blood Gene Expression Data Unveils a Diagnostic Host-Response Transcript Signature for Respiratory Syncytial Virus. Int. J. Mol. Sci. 2020, 21, 1831. [Google Scholar] [CrossRef]
- Munday, D.C.; Howell, G.; Barr, J.N.; Hiscox, J.A. Proteomic analysis of mitochondria in respiratory epithelial cells infected with human respiratory syncytial virus and functional implications for virus and cell biology. J. Pharm. Pharmacol. 2015, 67, 300–318. [Google Scholar] [CrossRef]
- Van Diepen, A.; Brand, H.K.; Sama, I.; Lambooy, L.H.J.; Van Den Heuvel, L.P.; Van Der Well, L.; Huynen, M.; Osterhaus, A.D.M.E.; Andeweg, A.C.; Hermans, P.W.M. Quantitative proteome profiling of respiratory virus-infected lung epithelial cells. J. Proteom. 2010, 73, 1680–1693. [Google Scholar] [CrossRef]
- Zhang, Y.; Jamaluddin, M.; Wang, S.; Tian, B.; Garofalo, R.P.; Casola, A.; Brasier, A.R. Ribavirin Treatment Up-Regulates Antiviral Gene Expression via the Interferon-Stimulated Response Element in Respiratory Syncytial Virus-Infected Epithelial Cells. J. Virol. 2003, 77, 5933–5947. [Google Scholar] [CrossRef]
- Zhang, Y.; Luxon, B.A.; Casola, A.; Garofalo, R.P.; Jamaluddin, M.; Brasier, A.R. Expression of Respiratory Syncytial Virus-Induced Chemokine Gene Networks in Lower Airway Epithelial Cells Revealed by cDNA Microarrays. J. Virol. 2001, 75, 9044–9058. [Google Scholar] [CrossRef]
- Tian, B.; Zhang, Y.; Luxon, B.A.; Garofalo, R.P.; Casola, A.; Sinha, M.; Brasier, A.R. Identification of NF-KB-Dependent Gene Networks in Respiratory Syncytial Virus-infected Cells. J. Virol. 2002, 76, 6800–6814. [Google Scholar] [CrossRef] [PubMed]
- Martı, I.; Lombardı, L.; Garcı, B.; Domı, O. Distinct gene subsets are induced at different time points after human respiratory syncytial virus infection of A549 cells. J. Gen. Virol. 2007, 88, 570–581. [Google Scholar] [CrossRef]
- Weinberg, S.E.; Sena, L.A.; Chandel, N.S. Mitochondria in the Regulation of Innate and Adaptive Immunity. Immunity 2015, 42, 406–417. [Google Scholar] [CrossRef] [PubMed]
- Jen, J.; Wang, Y. Zinc finger proteins in cancer progression. J. Biomed. Sci. 2016, 23, 1–9. [Google Scholar] [CrossRef]
- Bayrhuber, M.; Meins, T.; Habeck, M.; Becker, S.; Giller, K.; Villinger, S.; Vonrhein, C.; Griesinger, C.; Zweckstetter, M.; Zeth, K. Structure of the human voltage-dependent anion channel. Proc. Natl. Acad. Sci. USA 2008, 105, 15370–15375. [Google Scholar] [CrossRef]
- Pitt, A.S.; Buchanan, S.K. A biochemical and structural understanding of tom complex interactions and implications for human health and disease. Cells 2021, 10, 1164. [Google Scholar] [CrossRef]
- Yang, K.; Shi, H.; Qi, R.; Sun, S.; Tang, Y.; Zhang, B.; Wang, C. Hsp90 Regulates Activation of Interferon Regulatory Factor 3 and TBK-1 Stabilization in Sendai Virus-infected Cells. Mol. Biol. Cell 2006, 17, 1461–1471. [Google Scholar] [CrossRef]
- Liu, X.Y.; Wei, B.; Shi, H.X.; Shan, Y.F.; Wang, C. Tom70 mediates activation of interferon regulatory factor 3 on mitochondria. Cell Res. 2010, 20, 994–1011. [Google Scholar] [CrossRef]
- Den Brave, F.; Gupta, A.; Becker, T. Protein Quality Control at the Mitochondrial Surface. Front. Cell Dev. Biol. 2021, 9, 795685. [Google Scholar] [CrossRef]
- Bukreyev, A.; Whitehead, S.S.; Murphy, B.R.; Collins, P.L. Recombinant respiratory syncytial virus from which the entire SH gene has been deleted grows efficiently in cell culture and exhibits site-specific attenuation in the respiratory tract of the mouse. J. Virol. 1997, 71, 8973–8982. [Google Scholar] [CrossRef]
- Whitehead, S.S.; Bukreyev, A.; Teng, M.N.; Firestone, C.-Y.; St Claire, M.; Elkins, W.R.; Collins, P.L.; Murphy, B.R. Recombinant Respiratory Syncytial Virus Bearing a Deletion of either the NS2 or SH Gene Is Attenuated in Chimpanzees. J. Virol. 1999, 73, 3438–3442. [Google Scholar] [CrossRef]
- Techaarpornkul, S.; Barretto, N.; Peeples, M.E. Functional Analysis of Recombinant Respiratory Syncytial Virus Deletion Mutants Lacking the Small Hydrophobic and/or Attachment Glycoprotein Gene. J. Virol. 2001, 75, 6825–6834. [Google Scholar] [CrossRef]
- Gan, S.W.; Tan, E.; Lin, X.; Yu, D.; Wang, J.; Tan, G.M.Y.; Vararattanavech, A.; Yeo, C.Y.; Soon, C.H.; Soong, T.W.; et al. The small hydrophobic protein of the human respiratory syncytial virus forms pentameric ion channels. J. Biol. Chem. 2012, 287, 24671–24689. [Google Scholar] [CrossRef]
- Araujo, G.C.; Silva, R.H.T.; Scott, L.P.B.; Araujo, A.S.; Souza, F.P.; de Oliveira, R.J. Structure and functional dynamics characterization of the ion channel of the human respiratory syncytial virus (hRSV) small hydrophobic protein (SH) transmembrane domain by combining molecular dynamics with excited normal modes. J. Mol. Model. 2016, 22, 286. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Jain, N.; Limpanawat, S.; To, J.; Quistgaard, E.M.; Nordlund, P.; Thanabalu, T.; Torres, J. Interaction between human BAP31 and respiratory syncytial virus small hydrophobic (SH) protein. Virology 2015, 482, 105–110. [Google Scholar] [CrossRef]
- McLellan, J.S.; Ray, W.C.; Peeples, M.E. Structure and function of respiratory syncytial virus surface glycoproteins. Curr. Top. Microbiol. Immunol. 2013, 372, 83–104. [Google Scholar] [CrossRef]
- Hendricks, D.A.; Baradaran, K.; McIntosh, K.; Patterson, J.L. Appearance of a soluble form of the G protein of respiratory syncytial virus in fluids of infected cells. J. Gen. Virol. 1987, 68, 1705–1714. [Google Scholar] [CrossRef] [PubMed]
- Roberts, S.R.; Lichtenstein, D.; Ball, L.A.; Wertz, G.W. The membrane-associated and secreted forms of the respiratory syncytial virus attachment glycoprotein G are synthesized from alternative initiation codons. J. Virol. 1994, 68, 4538–4546. [Google Scholar] [CrossRef] [PubMed]
- Hendricks, D.A.; McIntosh, K.; Patterson, J.L. Further characterization of the soluble form of the G glycoprotein of respiratory syncytial virus. J. Virol. 1988, 62, 2228–2233. [Google Scholar] [CrossRef] [PubMed]
- Johnson, P.R.; Spriggs, M.K.; Olmsted, R.A.; Collins, P.L. The G glycoprotein of human respiratory syncytial viruses of subgroups A and B: Extensive sequence divergence between antigenically related proteins. Proc. Natl. Acad. Sci. USA 1987, 84, 5625–5629. [Google Scholar] [CrossRef]
- Feldman, S.A.; Hendry, R.M.; Beeler, J.A. Identification of a Linear Heparin Binding Domain for Human Respiratory Syncytial Virus Attachment Glycoprotein G. J. Virol. 1999, 73, 6610–6617. [Google Scholar] [CrossRef]
- Zhang, L.; Bukreyev, A.; Thompson, C.I.; Watson, B.; Peeples, M.E.; Collins, P.L.; Pickles, R.J. Infection of Ciliated Cells by Human Parainfluenza Virus Type 3 in an In Vitro Model of Human Airway Epithelium. J. Virol. 2005, 79, 1113–1124. [Google Scholar] [CrossRef]
- Johnson, S.M.; McNally, B.A.; Ioannidis, I.; Flano, E.; Teng, M.N.; Oomens, A.G.; Walsh, E.E.; Peeples, M.E. Respiratory Syncytial Virus Uses CX3CR1 as a Receptor on Primary Human Airway Epithelial Cultures. PLoS Pathog. 2015, 11, e1005318. [Google Scholar] [CrossRef]
- Jeong, K.; Piepenhagen, P.A.; Kishko, M.; DiNapoli, J.M.; Groppo, R.P.; Zhang, L.; Almond, J.; Kleanthous, H.; Delagrave, S.; Parrington, M. CX3CR1 is expressed in differentiated human ciliated airway cells and co-localizes with respiratory syncytial virus on cilia in a G protein-dependent manner. PLoS ONE 2015, 10, e0130517. [Google Scholar] [CrossRef]
- Nishimura, M.; Umehara, H.; Nakayama, T.; Yoneda, O.; Hieshima, K.; Kakizaki, M.; Dohmae, N.; Yoshie, O.; Imai, T. Dual Functions of Fractalkine/CX3C Ligand 1 in Trafficking of Perforin +/ Granzyme B + Cytotoxic Effector Lymphocytes That Are Defined by CX3CR1 Expression. J. Immunol. 2002, 168, 6173–6180. [Google Scholar] [CrossRef]
- Bar-On, L.; Birnberg, T.; Lewis, K.L.; Edelson, B.T.; Bruder, D.; Hildner, K.; Buer, J.; Murphy, K.M.; Reizis, B.; Jung, S. CX3CR1+ CD8α+ dendritic cells are a steady-state population related to plasmacytoid dendritic cells. Proc. Natl. Acad. Sci. USA 2010, 107, 14745–14750. [Google Scholar] [CrossRef]
- Imai, T.; Hieshima, K.; Haskell, C.; Baba, M.; Nagira, M.; Nishimura, M.; Kakizaki, M.; Takagi, S.; Nomiyama, H.; Schall, T.J.; et al. Identification and molecular characterization of fractalkine receptor CX3CR1, which mediates both leukocyte migration and adhesion. Cell 1997, 91, 521–530. [Google Scholar] [CrossRef]
- Tripp, R.A.; Jones, L.P.; Haynes, L.M.; Zheng, H.Q.; Murphy, P.M.; Anderson, L.J. CX3C chemokine mimicry by respiratory syncytial virus G glycoprotein. Nat. Immunol. 2001, 2, 732–738. [Google Scholar] [CrossRef]
- Tripp, R.A.; Jones, L.; Anderson, L.J. Respiratory Syncytial Virus G and/or SH Glycoproteins Modify CC and CXC Chemokine mRNA Expression in the BALB/c Mouse. J. Virol. 2000, 74, 6227–6229. [Google Scholar] [CrossRef]
- Johnson, T.R.; McLellan, J.S.; Graham, B.S. Respiratory Syncytial Virus Glycoprotein G Interacts with DC-SIGN and L-SIGN To Activate ERK1 and ERK2. J. Virol. 2012, 86, 1339–1347. [Google Scholar] [CrossRef]
- Tripp, R.A.; Moore, D.; Jones, L.; Sullender, W.; Winter, J.; Anderson, L.J. Respiratory Syncytial Virus G and/or SH Protein Alters Th1 Cytokines, Natural Killer Cells, and Neutrophils Responding to Pulmonary Infection in BALB/c Mice. J. Virol. 1999, 73, 7099–7107. [Google Scholar] [CrossRef]
- Rot, B.A.; Krieger, M.; Brunner, T.; Bischoff, S.C.; Schall, T.J.; Dahinden, C.A. RANTES and macrophage inflammatory protein-1 induce the migration and activation of normal human eosinophil granulocytes. J. Exp. Med. 1992, 176, 1489–1495. [Google Scholar] [CrossRef]
- Baggiolini, M.; Walz, A.; Kunkel, S.L. Neutrophil-activating peptide-1/interleukin 8, a novel cytokine that activates neutrophils. J. Clin. Investig. 1989, 84, 1045–1049. [Google Scholar] [CrossRef]
- Stark, J.M.; Godding, V.; Sedgwick, J.B.; Busse, W.W. Respiratory syncytial virus infection enhances neutrophil and eosinophil adhesion to cultured respiratory epithelial cells. Roles of CD18 and intercellular adhesion molecule-1. J. Immunol. 1996, 156, 4774–4782. [Google Scholar]
- Chini, B.A.; Fiedler, M.A.; Milligan, L.; Hopkins, T.; Stark, J.M. Essential Roles of NF-κB and C/EBP in the Regulation of Intercellular Adhesion Molecule-1 after Respiratory Syncytial Virus Infection of Human Respiratory Epithelial Cell Cultures. J. Virol. 1998, 72, 1623–1626. [Google Scholar] [CrossRef]
- Garofalo, R.; Sabry, M.; Jamaluddin, M.; Yu, R.K.; Casola, A.; Ogra, P.L.; Brasier, A.R. Transcriptional activation of the interleukin-8 gene by respiratory syncytial virus infection in alveolar epithelial cells: Nuclear translocation of the RelA transcription factor as a mechanism producing airway mucosal inflammation. J. Virol. 1996, 70, 8773–8781. [Google Scholar] [CrossRef]
- Thomas, L.H.; Friedland, J.S.; Sharland, M.; Becker, S. Respiratory Syncytial Virus-Induced RANTES Production from Human Bronchial Epithelial Cells Is Dependent on Nuclear Factor-κ B Nuclear Binding and Is Inhibited by Adenovirus-Mediated Expression of Inhibitor of κBα. J. Immunol. 1998, 161, 1007–1016. [Google Scholar] [PubMed]
- Schwarze, J.; Schauer, U. Enhanced virulence, airway inflammation and impaired lung function induced by respiratory syncytial virus deficient in secreted G protein. Thorax 2004, 59, 517–521. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Langedijk, J.P.M.; De Groot, B.L.; Berendsen, H.J.C.; Van Oirschot, J.T. Structural homology of the central conserved region of the attachment protein G of respiratory syncytial virus with the fourth subdomain of 55-kDa tumor necrosis factor receptor. Virology 1998, 243, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Chirkova, T.; Boyoglu-Barnum, S.; Gaston, K.A.; Malik, F.M.; Trau, S.P.; Oomens, A.G.P.; Anderson, L.J. Respiratory Syncytial Virus G Protein CX3C Motif Impairs Human Airway Epithelial and Immune Cell Responses. J. Virol. 2013, 87, 13466–13479. [Google Scholar] [CrossRef]
- Ross, R.J.; Zhou, M.; Shen, D.; Fariss, R.N.; Ding, X.; Bojanowski, C.M.; Tuo, J.; Chan, C. Immunological protein expression profile in Ccl2/Cx3cr1 deficient mice with lesions similar to age-related macular degeneration. Exp. Eye Res. 2008, 86, 675–683. [Google Scholar] [CrossRef]
- Ishida, Y.; Hayashi, T.; Goto, T.; Akimoto, S.; Mukaida, N.; Alerts, E. Essential Involvement of CX3CR1-Mediated Signals in the Bactericidal Host Defense during Septic Peritonitis. J. Immunol. 2008, 181, 4208–4218. [Google Scholar] [CrossRef]
- Openshaw, P.J.M.; Chiu, C.; Culley, F.J.; Johansson, C. Protective and Harmful Immunity to RSV Infection. Annu. Rev. Immunol. 2017, 35, 501–532. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Van Royen, T.; Rossey, I.; Sedeyn, K.; Schepens, B.; Saelens, X. How RSV Proteins Join Forces to Overcome the Host Innate Immune Response. Viruses 2022, 14, 419. https://doi.org/10.3390/v14020419
Van Royen T, Rossey I, Sedeyn K, Schepens B, Saelens X. How RSV Proteins Join Forces to Overcome the Host Innate Immune Response. Viruses. 2022; 14(2):419. https://doi.org/10.3390/v14020419
Chicago/Turabian StyleVan Royen, Tessa, Iebe Rossey, Koen Sedeyn, Bert Schepens, and Xavier Saelens. 2022. "How RSV Proteins Join Forces to Overcome the Host Innate Immune Response" Viruses 14, no. 2: 419. https://doi.org/10.3390/v14020419
APA StyleVan Royen, T., Rossey, I., Sedeyn, K., Schepens, B., & Saelens, X. (2022). How RSV Proteins Join Forces to Overcome the Host Innate Immune Response. Viruses, 14(2), 419. https://doi.org/10.3390/v14020419