
IL-33 Induces an Antiviral Signature in Mast Cells but Enhances Their Permissiveness for Human Rhinovirus Infection

 , , and
, , and 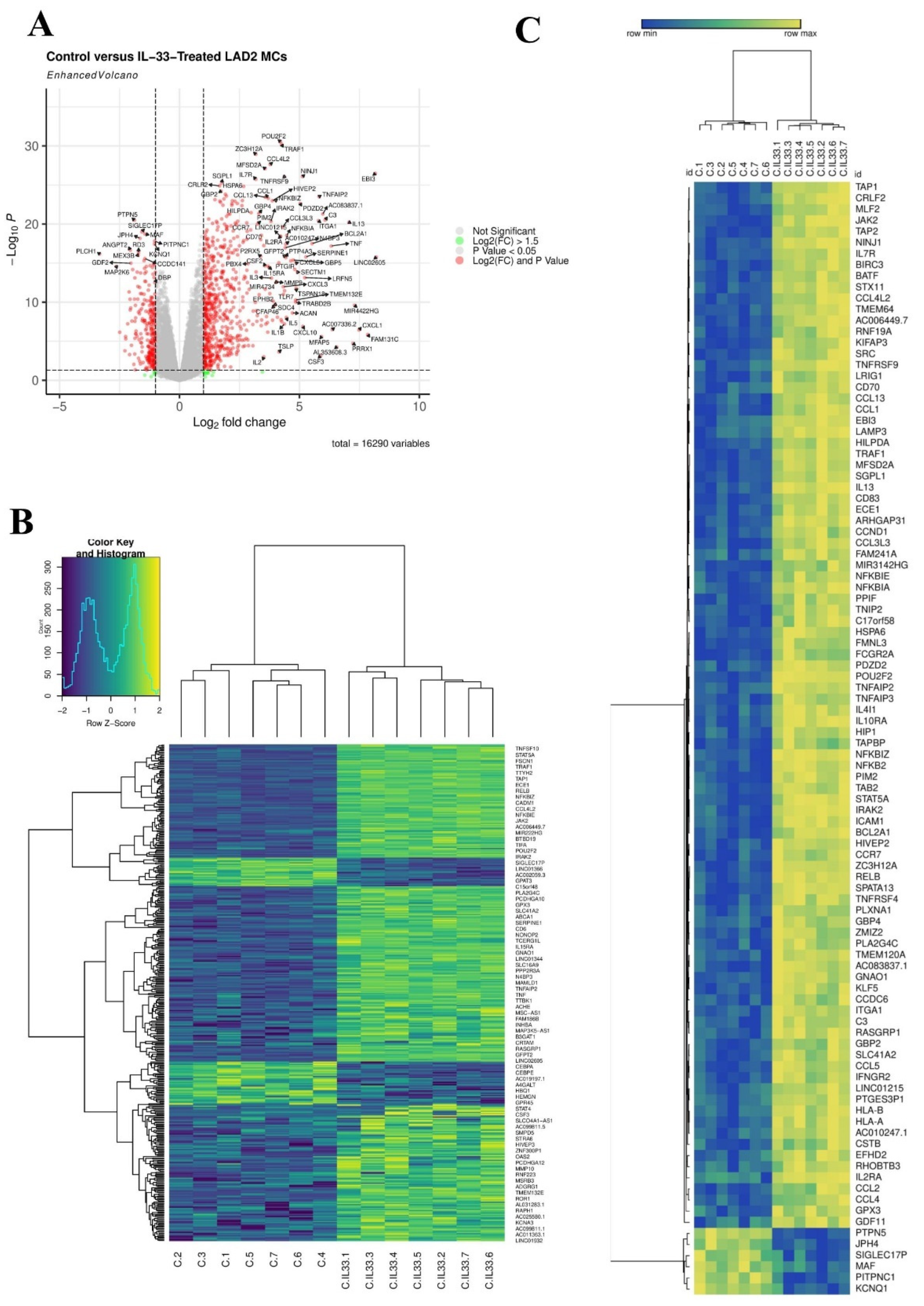{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Mast Cell Culture
2.2. Rhinovirus Stocks
2.3. Cell Treatments and Infections
2.4. mRNA Extraction, RNA Sequencing and Transcriptomic Analysis
2.5. RT-qPCR
2.6. Flow Cytometry
2.7. TCID50 Assay
2.8. Statistical Analysis
3. Results
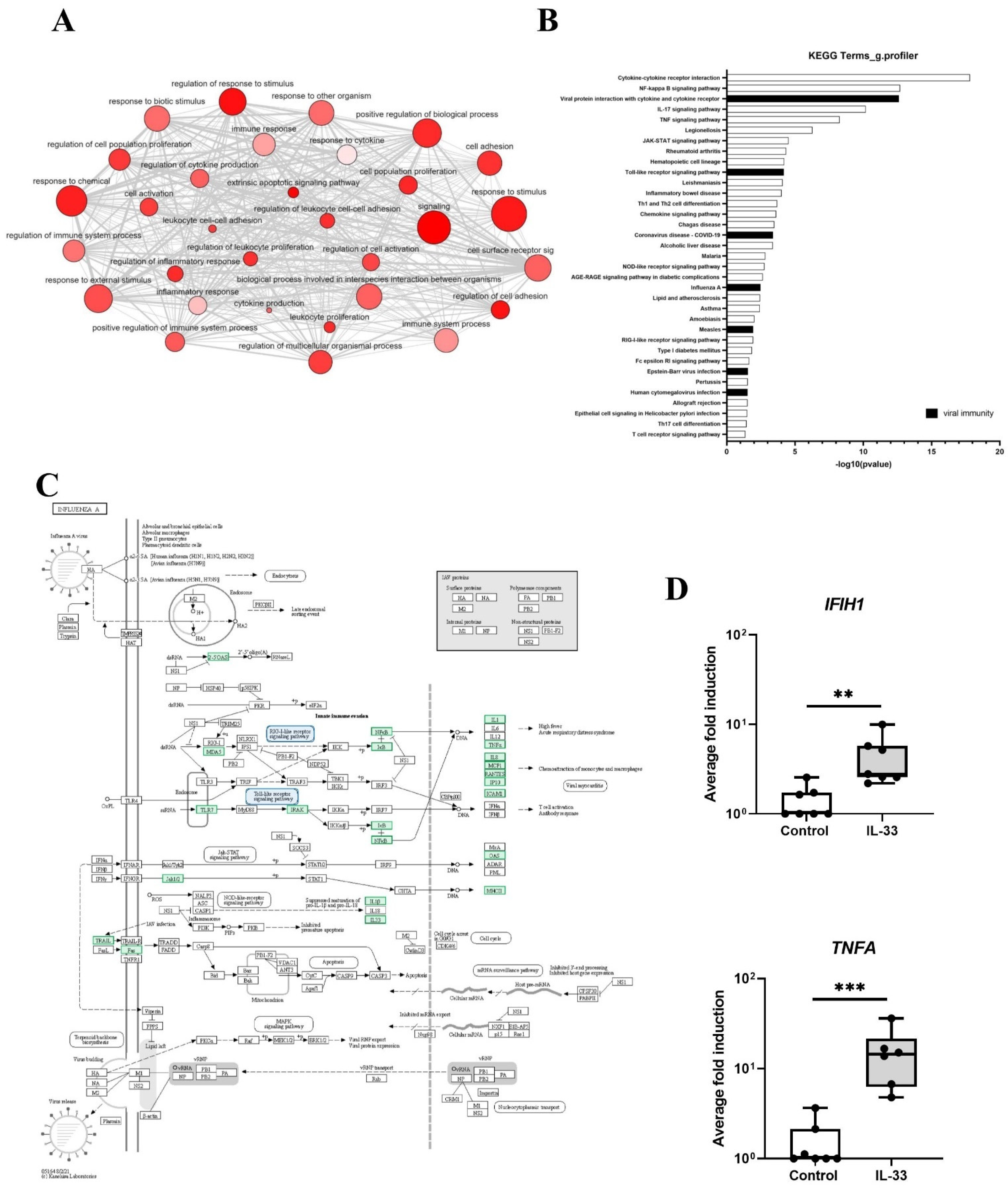
3.1. Transcriptomic Analysis of MCs Exposed to IL-33 Reveals an Antiviral Gene Signature
3.2. IL-33 Induces Interferons and IFN-Stimulated Genes in LAD2 MCs
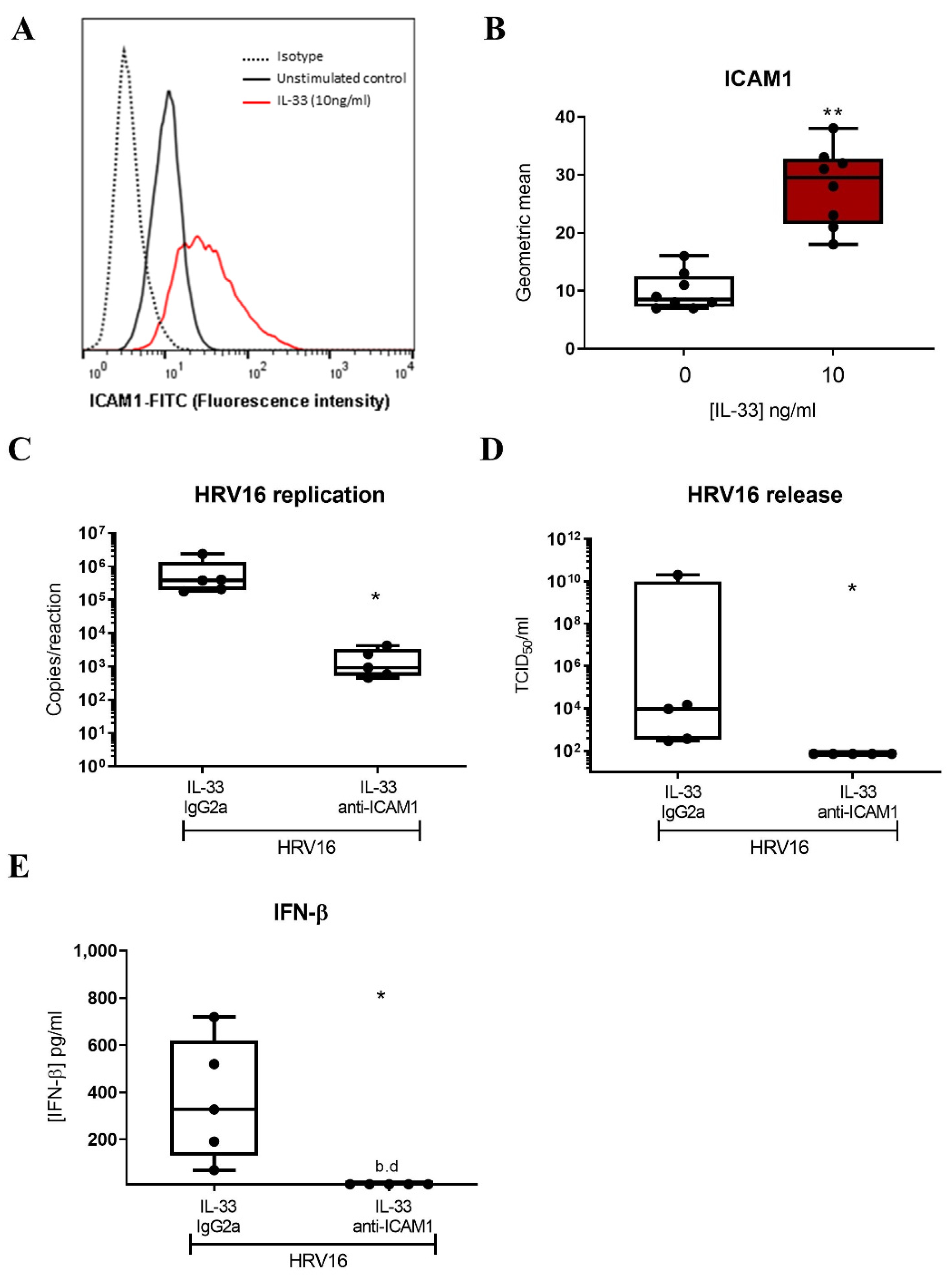
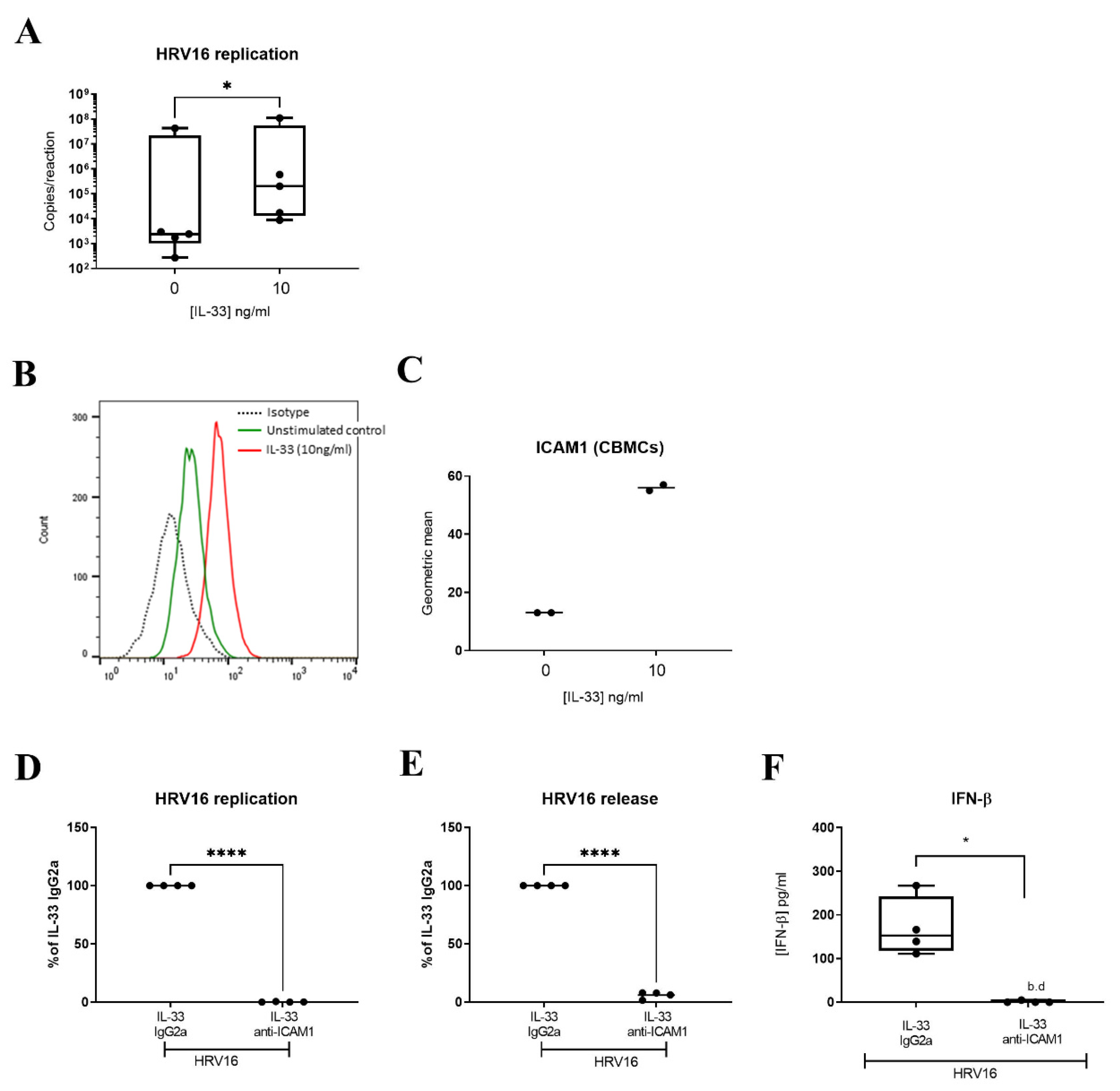
3.3. IL-33 Enhances HRV16 Replication and Release of Infectious Viral Particles through Increased ICAM-1 Expression
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Abraham, S.N.; St John, A.L. Mast cell-orchestrated immunity to pathogens. Nat. Rev. Immunol. 2010, 10, 440–452. [Google Scholar] [CrossRef] [PubMed]
- Metcalfe, D.D.; Baram, D.; Mekori, Y.A. Mast cells. Physiol. Rev. 1997, 77, 1033–1079. [Google Scholar] [CrossRef] [PubMed]
- Bradding, P.; Arthur, G. Mast cells in asthma—State of the art. Clin. Exp. Allergy 2016, 46, 194–263. [Google Scholar] [CrossRef]
- Bradding, P.; Walls, A.F.; Holgate, S.T. The role of the mast cell in the pathophysiology of asthma. J. Allergy Clin. Immunol. 2006, 117, 1277–1284. [Google Scholar] [CrossRef]
- Andersson, C.K.; Bergqvist, A.; Mori, M.; Mauad, T.; Bjermer, L.; Erjefalt, J.S. Mast cell-associated alveolar inflammation in patients with atopic uncontrolled asthma. J. Allergy Clin. Immunol. 2011, 127, 905–912.e1-7. [Google Scholar] [CrossRef] [PubMed]
- Dougherty, R.H.; Sidhu, S.S.; Raman, K.; Solon, M.; Solberg, O.D.; Caughey, G.H.; Woodruff, P.G.; Fahy, J.V. Accumulation of intraepithelial mast cells with a unique protease phenotype in T(H)2-high asthma. J. Allergy Clin. Immunol. 2010, 125, 1046–1053.e8. [Google Scholar] [CrossRef]
- Balzar, S.; Fajt, M.L.; Comhair, S.A.; Erzurum, S.C.; Bleecker, E.; Busse, W.W.; Castro, M.; Gaston, B.; Israel, E.; Schwartz, L.B.; et al. Mast cell phenotype, location, and activation in severe asthma. Data from the Severe Asthma Research Program. Am. J. Respir. Crit. Care Med. 2010, 183, 299–309. [Google Scholar] [CrossRef]
- Hinks, T.S.C.; Zhou, X.; Staples, K.; Dimitrov, B.D.; Manta, A.; Petrossian, T.; Lum, P.; Smith, C.; Ward, J.; Howarth, P.; et al. Multidimensional endotypes of asthma: Topological data analysis of cross-sectional clinical, pathological, and immunological data. Lancet 2015, 385 (Suppl. 1), S42. [Google Scholar] [CrossRef]
- Hinks, T.S.; Zhou, X.; Staples, K.J.; Dimitrov, B.D.; Manta, A.; Petrossian, T.; Lum, P.Y.; Smith, C.G.; Ward, J.A.; Howarth, P.H.; et al. Innate and adaptive T cells in asthmatic patients: Relationship to severity and disease mechanisms. J. Allergy Clin. Immunol. 2015, 136, 323–333. [Google Scholar] [CrossRef]
- Tiotiu, A.; Badi, Y.; Kermani, N.Z.; Sanak, M.; Kolmert, J.; Wheelock, C.E.; Hansbro, P.M.; Dahlén, S.-E.; Sterk, P.J.; Djukanovic, R.; et al. Association of Differential Mast Cell Activation with Granulocytic Inflammation in Severe Asthma. Am. J. Respir. Crit. Care Med. 2022, 205, 397–411. [Google Scholar] [CrossRef] [PubMed]
- Kulka, M.; Alexopoulou, L.; Flavell, R.A.; Metcalfe, D.D. Activation of mast cells by double-stranded RNA: Evidence for activation through Toll-like receptor 3. J. Allergy Clin. Immunol. 2004, 114, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Burke, S.M.; Issekutz, T.B.; Mohan, K.; Lee, P.W.; Shmulevitz, M.; Marshall, J.S. Human mast cell activation with virus-associated stimuli leads to the selective chemotaxis of natural killer cells by a CXCL8-dependent mechanism. Blood 2008, 111, 5467–5476. [Google Scholar] [CrossRef] [PubMed]
- McAlpine, S.M.; Issekutz, T.B.; Marshall, J.S. Virus stimulation of human mast cells results in the recruitment of CD56+ T cells by a mechanism dependent on CCR5 ligands. FASEB J. 2011, 26, 1280–1289. [Google Scholar] [CrossRef]
- Al-Afif, A.; Alyazidi, R.; Oldford, S.A.; Huang, Y.Y.; King, C.A.; Marr, N.; Haidl, I.D.; Anderson, R.; Marshall, J.S. Respiratory syncytial virus infection of primary human mast cells induces the selective production of type I interferons, CXCL10, and CCL4. J. Allergy Clin. Immunol. 2015, 136, 1346–1354.e1. [Google Scholar] [CrossRef] [PubMed]
- Shirato, K.; Taguchi, F. Mast cell degranulation is induced by A549 airway epithelial cell infected with respiratory syncytial virus. Virology 2009, 386, 88–93. [Google Scholar] [CrossRef] [PubMed]
- Ng, K.; Raheem, J.; Laurent, C.D.S.; Marcet, C.T.; Vliagoftis, H.; Befus, A.D.; Moon, T.C. Responses of human mast cells and epithelial cells following exposure to influenza A virus. Antivir. Res. 2019, 171, 104566. [Google Scholar] [CrossRef]
- Jackson, D.J.; Gangnon, R.E.; Evans, M.D.; Roberg, K.A.; Anderson, E.L.; Pappas, T.E.; Printz, M.C.; Lee, W.-M.; Shult, P.A.; Reisdorf, E.; et al. Wheezing rhinovirus illnesses in early life predict asthma development in high-risk children. Am. J. Respir. Crit. Care Med. 2008, 178, 667–672. [Google Scholar] [CrossRef]
- Johnston, S.L.; Pattemore, P.; Sanderson, G.; Smith, S.; Lampe, F.; Josephs, L.; Symington, P.; Toole, S.O.; Myint, S.H.; Tyrrell, D.A.J.; et al. Community study of role of viral infections in exacerbations of asthma in 9-11 year old children. BMJ 1995, 310, 1225–1229. [Google Scholar] [CrossRef]
- Zhu, J.; Message, S.D.; Qiu, Y.; Mallia, P.; Kebadze, T.; Contoli, M.; Ward, C.K.; Barnathan, E.S.; Mascelli, M.A.; Kon, O.M.; et al. Airway inflammation and illness severity in response to experimental rhinovirus infection in asthma. Chest 2014, 145, 1219–1229. [Google Scholar] [CrossRef]
- Djukanović, R.; Wilson, S.J.; Kraft, M.; Jarjour, N.N.; Steel, M.; Chung, K.F.; Bao, W.; Fowler-Taylor, A.; Matthews, J.; Busse, W.W.; et al. Effects of treatment with anti-immunoglobulin E antibody omalizumab on airway inflammation in allergic asthma. Am. J. Respir. Crit. Care Med. 2004, 170, 583–593. [Google Scholar] [CrossRef]
- Brusselle, G.; Michils, A.; Louis, R.; Dupont, L.; Van de Maele, B.; Delobbe, A.; Pilette, C.; Lee, C.; Gurdain, S.; Vancayzeele, S.; et al. “Real-life” effectiveness of omalizumab in patients with severe persistent allergic asthma: The PERSIST study. Respir. Med. 2009, 103, 1633–1642. [Google Scholar] [CrossRef] [PubMed]
- Solèr, M.; Matz, J.; Townley, R.; Buhlz, R.; O’Brien, J.; Fox, H.; Thirlwell, J.; Gupta, N.; Della Cioppa, G. The anti-IgE antibody omalizumab reduces exacerbations and steroid requirement in allergic asthmatics. Eur. Respir. J. 2001, 18, 254–261. [Google Scholar] [CrossRef] [PubMed]
- Akoto, C.; Davies, D.E.; Swindle, E.J. Mast cells are permissive for rhinovirus replication: Potential implications for asthma exacerbations. Clin. Exp. Allergy 2017, 47, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Schrauf, C.; Kirchberger, S.; Majdic, O.; Seyerl, M.; Zlabinger, G.; Stuhlmeier, K.M.; Sachet, M.; Seipelt, J.; Stöckl, J. The ssRNA genome of human rhinovirus induces a type I IFN response but fails to induce maturation in human monocyte-derived dendritic cells. J. Immunol. 2009, 183, 4440–4448. [Google Scholar] [CrossRef] [PubMed]
- Gern, J.E.; Vrtis, R.; Kelly, E.A.; Dick, E.C.; Busse, W.W. Rhinovirus produces nonspecific activation of lymphocytes through a monocyte-dependent mechanism. J. Immunol. 1996, 157, 1605–1612. [Google Scholar]
- Laza-Stanca, V.; Stanciu, L.A.; Message, S.D.; Edwards, M.R.; Gern, J.E.; Johnston, S.L. Rhinovirus replication in human macrophages induces NF-kappaB-dependent tumor necrosis factor alpha production. J. Virol. 2006, 80, 8248–8258. [Google Scholar] [CrossRef] [PubMed]
- Moffatt, M.F.; Gut, I.G.; Demenais, F.; Strachan, D.P.; Bouzigon, E.; Heath, S.; von Mutius, E.; Farrall, M.; Lathrop, M.; Cookson, W.O. A large-scale, consortium-based genomewide association study of asthma. N. Engl. J. Med. 2010, 363, 1211–1221. [Google Scholar] [CrossRef]
- Torgerson, D.G.; Ampleford, E.J.; Chiu, G.Y.; Gauderman, W.J.; Gignoux, C.R.; Graves, P.E.; Himes, B.E.; Levin, A.M.; Mathias, R.A.; Hancock, D.B. Meta-analysis of genome-wide association studies of asthma in ethnically diverse North American populations. Nat. Genet. 2011, 43, 887–892. [Google Scholar]
- Ramasamy, A.; Kuokkanen, M.; Vedantam, S.; Gajdos, Z.K.; Alves, A.C.; Lyon, H.N.; Ferreira, M.A.R.; Strachan, D.P.; Zhao, J.H.; Abramson, M.J.; et al. Genome-wide association studies of asthma in population-based cohorts confirm known and suggested loci and identify an additional association near HLA. PLoS ONE 2012, 7, e44008. [Google Scholar] [CrossRef]
- Portelli, M.A.; Dijk, F.N.; Ketelaar, M.E.; Shrine, N.; Hankinson, J.; Bhaker, S.; Grotenboer, N.S.; Obeidat, M.; Henry, A.P.; Billington, C.K.; et al. Phenotypic and functional translation of IL1RL1 locus polymorphisms in lung tissue and asthmatic airway epithelium. JCI Insight 2020, 5, e132446. [Google Scholar] [CrossRef]
- Préfontaine, D.; Lajoie-Kadoch, S.; Foley, S.; Audusseau, S.; Olivenstein, R.; Halayko, A.J.; Lemière, C.; Martin, J.G.; Hamid, Q. Increased expression of IL-33 in severe asthma: Evidence of expression by airway smooth muscle cells. J. Immunol. 2009, 183, 5094–5103. [Google Scholar] [CrossRef] [PubMed]
- Préfontaine, D.; Nadigel, J.; Chouiali, F.; Audusseau, S.; Semlali, A.; Chakir, J.; Martin, J.G.; Hamid, Q. Increased IL-33 expression by epithelial cells in bronchial asthma. J. Allergy Clin. Immunol. 2010, 125, 752–754. [Google Scholar] [CrossRef] [PubMed]
- Saglani, S.; Lui, S.; Ullmann, N.; Campbell, G.A.; Sherburn, R.T.; Mathie, S.A.; Denney, L.; Bossley, C.J.; Oates, T.; Walker, S.A.; et al. IL-33 promotes airway remodeling in pediatric patients with severe steroid-resistant asthma. J. Allergy Clin. Immunol. 2013, 132, 676–685.e13. [Google Scholar] [CrossRef] [PubMed]
- Traister, R.S.; Uvalle, C.E.; Hawkins, G.A.; Meyers, D.A.; Bleecker, E.R.; Wenzel, S.E. Phenotypic and genotypic association of epithelial IL1RL1 to human TH2-like asthma. J. Allergy Clin. Immunol. 2015, 135, 92–99. [Google Scholar] [CrossRef]
- Gordon, E.D.; Simpson, L.J.; Rios, C.L.; Ringel, L.; Lachowicz-Scroggins, M.E.; Peters, M.C.; Wesolowska-Andersen, A.; Gonzalez, J.R.; MacLeod, H.J.; Christian, L.S.; et al. Alternative splicing of interleukin-33 and type 2 inflammation in asthma. Proc. Natl. Acad. Sci. USA 2016, 113, 8765–8770. [Google Scholar] [CrossRef]
- Allakhverdi, Z.; Smith, D.E.; Comeau, M.R.; Delespesse, G. Cutting edge: The ST2 ligand IL-33 potently activates and drives maturation of human mast cells. J. Immunol. 2007, 179, 2051–2054. [Google Scholar]
- Jackson, D.J.; Makrinioti, H.; Rana, B.M.J.; Shamji, B.W.H.; Trujillo-Torralbo, M.-B.; Footitt, J.; Del-Rosario, J.; Telcian, A.G.; Nikonova, A.; Zhu, J.; et al. IL-33-dependent type 2 inflammation during rhinovirus-induced asthma exacerbations in vivo. Am. J. Respir. Crit. Care Med. 2014, 190, 1373–1382. [Google Scholar] [CrossRef]
- Wechsler, M.E.; Ruddy, M.K.; Pavord, I.D.; Israel, E.; Rabe, K.F.; Ford, L.B.; Maspero, J.F.; Abdulai, R.M.; Hu, C.-C.; Martincova, R.; et al. Efficacy and Safety of Itepekimab in Patients with Moderate-to-Severe Asthma. N. Engl. J. Med. 2021, 385, 1656–1668. [Google Scholar] [CrossRef]
- Kelsen, S.G.; Agache, I.O.; Soong, W.; Israel, E.; Chupp, G.L.; Cheung, D.S.; Theess, W.; Yang, X.; Staton, T.L.; Choy, D.F.; et al. Astegolimab (anti-ST2) efficacy and safety in adults with severe asthma: A randomized clinical trial. J. Allergy Clin. Immunol. 2021, 148, 790–798. [Google Scholar] [CrossRef]
- Cayrol, C.; Girard, J.P. Interleukin-33 (IL-33): A critical review of its biology and the mechanisms involved in its release as a potent extracellular cytokine. Cytokine 2022, 156, 155891. [Google Scholar] [CrossRef]
- Liew, F.Y.; Girard, J.P.; Turnquist, H.R. Interleukin-33 in health and disease. Nat. Rev. Immunol. 2016, 16, 676–689. [Google Scholar] [CrossRef]
- Saluja, R.; Zoltowska, A.; Ketelaar, M.E.; Nilsson, G. IL-33 and Thymic Stromal Lymphopoietin in mast cell functions. Eur. J. Pharmacol. 2016, 778, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Kirshenbaum, A.S.; Akin, C.; Wu, Y.; Rottem, M.; Goff, J.P.; Beaven, M.A.; Rao, V.K.; Metcalfe, D.D. Characterization of novel stem cell factor responsive human mast cell lines LAD 1 and 2 established from a patient with mast cell sarcoma/leukemia; activation following aggregation of FcepsilonRI or FcgammaRI. Leuk. Res. 2003, 27, 677–682. [Google Scholar] [CrossRef]
- Gautier, V.; Cayrol, C.; Farache, D.; Roga, S.; Monsarrat, B.; Schiltz, O.; de Peredo, A.G.; Girard, J.-P. Extracellular IL-33 cytokine, but not endogenous nuclear IL-33, regulates protein expression in endothelial cells. Sci. Rep. 2016, 6, 34255. [Google Scholar] [CrossRef]
- Numata, T.; Ito, T.; Maeda, T.; Egusa, C.; Tsuboi, R. IL-33 promotes ICAM-1 expression via NF-kB in murine mast cells. Allergol. Int. 2016, 65, 158–165. [Google Scholar] [CrossRef] [PubMed]
- Farne, H.; Lin, L.; Jackson, D.J.; Rattray, M.; Simpson, A.; Custovic, A.; Joshi, S.; Wilson, P.A.; Williamson, R.; Edwards, M.R.; et al. In vivo bronchial epithelial interferon responses are augmented in asthma on day 4 following experimental rhinovirus infection. Thorax 2022, 77, 929–932. [Google Scholar] [CrossRef]
- Wark, P.A.; Johnston, S.; Bucchieri, F.; Powell, R.; Puddicombe, S.; Laza-Stanca, V.; Holgate, S.T.; Davies, D. Asthmatic bronchial epithelial cells have a deficient innate immune response to infection with rhinovirus. J. Exp. Med. 2005, 201, 937–947. [Google Scholar] [CrossRef]
- Andersson, C.K.; Iwasaki, J.; Cook, J.; Robinson, P.; Nagakumar, P.; Mogren, S.; Fleming, L.; Bush, A.; Saglani, S.; Lloyd, C.M. Impaired airway epithelial cell wound-healing capacity is associated with airway remodelling following RSV infection in severe preschool wheeze. Allergy 2020, 75, 3195–3207. [Google Scholar] [CrossRef]
- Battles, M.B.; McLellan, J.S. Respiratory syncytial virus entry and how to block it. Nat. Rev. Microbiol. 2019, 17, 233–245. [Google Scholar] [CrossRef]
- Le Goffic, R.; Arshad, M.I.; Rauch, M.; L’Helgoualc’h, A.; Delmas, B.; Piquet-Pellorce, C.; Samson, M. Infection with influenza virus induces IL-33 in murine lungs. Am. J. Respir. Cell Mol. Biol. 2011, 45, 1125–1132. [Google Scholar] [CrossRef]
- Kim, C.W.; Yoo, H.J.; Park, J.H.; Oh, J.E.; Lee, H.K. Exogenous Interleukin-33 Contributes to Protective Immunity via Cytotoxic T-Cell Priming against Mucosal Influenza Viral Infection. Viruses 2019, 11, 840. [Google Scholar] [CrossRef]
- Martin, M.U. Special aspects of interleukin-33 and the IL-33 receptor complex. Semin. Immunol. 2013, 25, 449–457. [Google Scholar] [CrossRef]
- Gavala, M.L.; Bertics, P.J.; Gern, J.E. Rhinoviruses, allergic inflammation, and asthma. Immunol. Rev. 2011, 242, 69–90. [Google Scholar] [CrossRef]
- Nagarkar, D.R.; Ramirez-Carrozzi, V.; Choy, D.; Lee, K.; Soriano, R.; Jia, G.; Abbas, A.R.; Modrusan, Z.; Pappu, R.; Arron, J.R. IL-13 mediates IL-33-dependent mast cell and type 2 innate lymphoid cell effects on bronchial epithelial cells. J. Allergy Clin. Immunol. 2015, 136, 202–205. [Google Scholar] [CrossRef]
- Chhiba, K.D.; Hsu, C.L.; Berdnikovs, S.; Bryce, P.J. Transcriptional Heterogeneity of Mast Cells and Basophils upon Activation. J. Immunol. 2017, 198, 4868–4878. [Google Scholar] [CrossRef]
- Iikura, M.; Suto, H.; Kajiwara, N.; Oboki, K.; Ohno, T.; Okayama, Y.; Saito, H.; Galli, S.J.; Nakae, S. IL-33 can promote survival, adhesion and cytokine production in human mast cells. Lab. Investig. 2007, 87, 971–978. [Google Scholar] [CrossRef]
- Silver, M.R.; Margulis, A.; Wood, N.; Goldman, S.J.; Kasaian, M.; Chaudhary, D. IL-33 synergizes with IgE-dependent and IgE-independent agents to promote mast cell and basophil activation. Inflamm. Res. 2010, 59, 207–218. [Google Scholar] [CrossRef]
- Andrade, M.V.; Iwaki, S.; Ropert, C.; Gazzinelli, R.T.; Cunha-Melo, J.R.; Beaven, M.A. Amplification of cytokine production through synergistic activation of NFAT and AP-1 following stimulation of mast cells with antigen and IL-33. Eur. J. Immunol. 2011, 41, 760–772. [Google Scholar] [CrossRef]
- Jung, M.-Y.; Smrz, D.; Desai, A.; Bandara, G.; Ito, T.; Iwaki, S.; Kang, J.H.; Andrade, M.V.; Hilderbrand, S.C.; Brown, J.M.; et al. IL-33 induces a hyporesponsive phenotype in human and mouse mast cells. J. Immunol. 2013, 190, 531–538. [Google Scholar] [CrossRef]
- Sandig, H.; Jobbings, C.E.; Roldan, N.G.; Whittingham-Dowd, J.K.; Orinska, Z.; Takeuchi, O.; Akira, S.; Bulfone-Paus, S. IL-33 causes selective mast cell tolerance to bacterial cell wall products by inducing IRAK1 degradation. Eur. J. Immunol. 2013, 43, 979–988. [Google Scholar] [CrossRef]
- Turunen, R.; Koistinen, A.; Vuorinen, T.; Arku, B.; Söderlund-Venermo, M.; Ruuskanen, O.; Jartti, T. The first wheezing episode: Respiratory virus etiology, atopic characteristics, and illness severity. Pediatr. Allergy Immunol. 2014, 25, 796–803. [Google Scholar] [CrossRef]
- Grissell, T.V.; Powell, H.; Shafren, D.R.; Boyle, M.J.; Hensley, M.J.; Jones, P.D.; Whitehead, B.F.; Gibson, P.G. Interleukin-10 gene expression in acute virus-induced asthma. Am. J. Respir. Crit.Care Med. 2005, 172, 433–439. [Google Scholar] [CrossRef]
- Papadopoulos, N.G.; Christodoulou, I.; Rohde, G.; Agache, I.; Almqvist, C.; Bruno, A.; Bonini, S.; De Bont, L.G.M.; Bossios, A.; Bousquet, J.; et al. Viruses and bacteria in acute asthma exacerbations—A GA(2) LEN-DARE systematic review. Allergy 2011, 66, 458–468. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Akoto, C.; Willis, A.; Banas, C.F.; Bell, J.A.; Bryant, D.; Blume, C.; Davies, D.E.; Swindle, E.J. IL-33 Induces an Antiviral Signature in Mast Cells but Enhances Their Permissiveness for Human Rhinovirus Infection. Viruses 2022, 14, 2430. https://doi.org/10.3390/v14112430
Akoto C, Willis A, Banas CF, Bell JA, Bryant D, Blume C, Davies DE, Swindle EJ. IL-33 Induces an Antiviral Signature in Mast Cells but Enhances Their Permissiveness for Human Rhinovirus Infection. Viruses. 2022; 14(11):2430. https://doi.org/10.3390/v14112430
Chicago/Turabian StyleAkoto, Charlene, Anna Willis, Chiara F. Banas, Joseph A. Bell, Dean Bryant, Cornelia Blume, Donna E. Davies, and Emily J. Swindle. 2022. "IL-33 Induces an Antiviral Signature in Mast Cells but Enhances Their Permissiveness for Human Rhinovirus Infection" Viruses 14, no. 11: 2430. https://doi.org/10.3390/v14112430
APA StyleAkoto, C., Willis, A., Banas, C. F., Bell, J. A., Bryant, D., Blume, C., Davies, D. E., & Swindle, E. J. (2022). IL-33 Induces an Antiviral Signature in Mast Cells but Enhances Their Permissiveness for Human Rhinovirus Infection. Viruses, 14(11), 2430. https://doi.org/10.3390/v14112430