1. Introduction
Ebola virus (EBOV), a member of the Filoviridae family, is an enveloped and negative-stranded RNA virus that causes severe hemorrhagic fever with high mortality rates in humans and other animals [
1,
2]. The genome of EBOV consists of seven genes encoding nucleoprotein (NP), RNA-dependent RNA polymerase (L), transmembrane glycoprotein (GP), matrix protein VP40, VP35, VP30, and VP24 [
3,
4]. Apart from the structural transmembrane GP, the transcription of the GP gene generates two nonstructural glycoproteins, the small soluble glycoprotein (ssGP) and the soluble glycoprotein (sGP), through transcriptional editing. The sGP, in turn, generates Δ-peptide as a result of its cleavage [
5,
6,
7]. The cleavage of GP by the cellular metalloprotease TNFα-converting enzyme (TACE) generally occurs during EBOV infection, giving rise to shed GP, which structurally resembles the full-length GP and therefore can misdirect host-neutralizing antibodies directed against the full-length GP by presenting alternative epitopes [
6,
8,
9].
GP can bind to multiple cell surface molecules, conferring on EBOV the ability to infect a wide range of cell types, including immune cells such as macrophages and dendritic cells, and other cells such as hepatocytes, epithelial cells, and endothelial cells (ECs) [
10,
11]. Vascular dysregulation leading to an increase in blood vessel permeability, loss of endothelial barrier integrity, and hemorrhage plays an important role in the severity of EBOV infection [
1,
12,
13]. Despite their susceptibility to EBOV in vitro, ECs are considered late viral targets in vivo, and the molecular mechanisms underlying their impairment during EBOV infection remain elusive to date [
8,
9]. In addition to direct viral infection, the vascular endothelium can be targeted indirectly via mediators such as cytokines, which are released upon infection of primary target cells, including immune cells, as well as virus-encoded GPs, which in turn can target ECs either directly or through the activation of primary target cells [
14,
15,
16,
17]. Among the proposed working models, only GP and the soluble shed GP have been found to play a key role in the activation of ECs and a decrease in their barrier function. Neither viral infection and replication nor other virus-encoded GPs, such as sGP, Δ-peptide, and ssGP, were found to be necessarily involved in the activation of ECs [
1,
8,
14,
17,
18]. As activation markers, the upregulation of cell adhesion molecules (CAMs), including intercellular adhesion molecules-1 (ICAM-1), was detected at the transcriptional level [
14] and then at the protein level [
1]. To confirm that EC activation depends on EBOV GP but does not require viral replication, Wahl-Jensen et al. [
1] generated virus-like particles (VLPs) formed by VP40 and GP or only VP40 and demonstrated that only VLPs formed by VP40 and GP were able to activate human umbilical vein endothelial cells (HUVECs). However, whether the EBOV GP-mediated activation and disruption of ECs could be the result of its direct interaction with the cells via some receptors or an indirect effect by some cellular mediators induced by EBOV GP remains unclear.
Existing evidence suggests that overexpression of ICAM-1 on ECs can lead to increased vascular permeability and loss of the endothelial barrier, which is mediated by the activation of the Rho/ROCK pathway [
19,
20] and rearrangement of the actin cytoskeleton [
21,
22]. The activation of the Rho/ROCK pathway by ICAM-1 has been reported to initiate a positive feedback loop that could result in the expression of more ICAM-1 and the recruitment of more leukocytes [
23] and that subsequently lead to diverse vasculopathies [
24,
25]. Junaid et al. [
13] recently reported that the inhibition of the Rho/ROCK pathway by RevitaCell Supplement suppressed Ebola VLP-induced permeability increase in HUVECs, suggesting a possible involvement of this molecular pathway in EBOV disease (EVD) pathogenesis.
In the present study, we hypothesized that suppressing the Rho/ROCK pathway and some cytoskeletal signaling molecules would prevent the overexpression of ICAM-1 and associated ECs disruption observed during EBOV infection. First, we confirmed the upregulation of ICAM-1 expression in HUVECs after exposure to Ebola VLPs bearing GP as well as TNF-α, but not Ebola VLPs lacking GP. In contrast to TNF-α treatment, Ebola VLPs bearing full-length GP induced ICAM-1 overexpression at late time points. We also found that only Ebola VLPs treatment induced significant cytotoxicity, mainly apoptosis. Moreover, screening of the cytoskeletal signaling inhibitors library identified focal adhesion kinase (FAK) inhibitors as potent inhibitors of ICAM-1 mediated by both TNF-α and Ebola VLPs bearing GP. Our results suggest that EBOV GP stimulates ECs to induce endothelial activation and dysfunction with the involvement of host cytoskeletal signaling molecules, which represent potential therapeutic targets for EBOV infection.
2. Materials and Methods
2.1. Plasmids and Compound Library
Expression plasmids for glycoprotein (GP), nucleoprotein (NP), and viral matrix protein 40 (VP40) of Ebola virus (Mayinga strain, 1976 outbreak), used for the generation of Ebola virus-like particles (VLPs), were provided by Dr. Yoshihiro Kawaoka (The University of Tokyo, Japan) and Dr. Thomas Hoenen (Friedrich Loeffler Institute, Germany). A library of pharmacologically active cytoskeletal signaling-related compounds was purchased from Selleck Chemicals (Houston, TX, USA). This library contains United States Food and Drug Administration (FDA)-approved and registered drugs, as well as preclinical compounds.
2.2. Cell Culture
Human umbilical vein endothelial cells (HUVECs) (cat# D10013, Takara, Shiga, Japan,) were cultured in collagen-coated dishes (Corning, AZ, USA) containing endothelial growth medium (EGM) (Promocell, Heidelberg, Germany) supplemented with 10% fetal bovine serum (FBS), 1% penicillin/streptomycin solution, and 0.1% amphotericin B (Gibco, Grand Island, NY, USA). The human embryonic kidney cell line (HEK293T) (cat# CRL11268, ATCC, Manassas, VA, USA), used for the preparation of Ebola VLPs was maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% FBS and 1% penicillin/streptomycin solution. All cells were incubated at 37 °C in a humidified 5% CO2 environment and were kept at a low passage number (below 20) for the experiments to maintain cell morphology and biological properties.
2.3. Ebola VLP Production and Purification
Ebola VLPs bearing GPs (designated VLPs) were generated as previously described [
26], with some modifications. In brief, confluent HEK293T cells were cotransfected with pCAGGS-EBOV-GP, pCAGGS-EBOV-VP40, and pCAGGS-EBOV-NP using polyethyleneimine (PEI MAX) (Polysciences, Inc., Warrington, PA, USA). Following transfection, cells were incubated overnight, and the culture medium was replaced with fresh DMEM. Supernatants were collected after 48 h and clarified by centrifugation at 2500 rpm for 15 min at 4 °C. Ebola VLPs without GPs (designated VLP
VP40/NP) were also generated by cotransfecting HEK293T cells with pCAGGS-EBOV-VP40 and pCAGGS-EBOV-NP. Transfection with pCAGGS alone served as a mock control. The collected supernatants containing Ebola VLPs and VLP
VP40/NP were layered onto 20% sucrose cushions and spun at 30,000 rpm at 4 °C for 2 h. The resultant pellets were resuspended in phosphate-buffered saline (PBS) and layered again onto 20–60% sucrose gradients for ultracentrifugation at 75,000 rpm for 2 h. The obtained fractions were harvested and analyzed by Western blotting, as described below. Further, EBOV protein-rich fractions were pooled and sedimented through a 20% sucrose cushion at 75,000 rpm for 30 min. The resulting pellet was resuspended in Opti-MEM serum-free medium (Thermo Fisher Scientific, Waltham, MA, USA), giving rise to purified Ebola VLPs and VLP
VP40/NP preparations. Purified Ebola VLPs and VLP
VP40/NP were quantified for GP content using Western blotting with recombinant GP lacking the transmembrane domain (EBOV rGPΔTM) (IBT Bioservices, Rockville, MD, USA) as the reference standard and anti-GP as the detection antibody. Unless otherwise stated, 10× diluted VLPs, corresponding to 9.2 µg/mL of GP, were used in most of the experiments, including drug screening.
2.4. Characterization of Ebola VLPs and Focal Adhesion Kinase Detection Using Western Blotting
Ebola VLPs and VLP
VP40/NP or the mock control particles were mixed with sodium dodecyl sulfate-containing buffer (Nacalai Tesque Inc., Kyoto, Japan), boiled for 5 min at 95 °C, and subjected to SDS-PAGE before being transferred onto a nitrocellulose blotting membrane (Amersham, Freiburg, Germany) overnight via Western blotting. The membranes were blocked in the blocking solution, made with the wash buffer (0.2 M Tris-HCl (pH 7.5), 8.76 g/L NaCl, and 0.25% Tween 20) containing 5% skim milk, for 1 h at 25 °C. After blocking, the membranes were first incubated overnight with mouse monoclonal anti-EBOV GP antibody provided by Dr. Ayato Takada (Hokkaido University, Japan) ([
27]; 1:5000), and for 3 h with rabbit anti-VP40 serum ([
28]; 1:5000) and rabbit polyclonal anti-EBOV NP antibody (IBT Bioservices; Rockville, MD, USA, 1:5000) separately. They were then probed with peroxidase-linked secondary antibodies, anti-mouse IgG-HRP (Merck, Darmstadt, Germany; 1:5000), or anti-rabbit IgG-HRP (Promega, Madison, WI, USA; 1:1000) for 1 h at room temperature. All antibodies were diluted in the blocking solution. The membranes were subsequently washed with the wash buffer three times for 10 min between primary and secondary antibody incubation, as well as after the secondary antibody incubation. Signals of bound secondary antibodies were detected using ECL prime chemiluminescent reagent (GE Healthcare, Chicago, IL, USA) and visualized using the LAS-3000 imaging system (Fujifilm, Tokyo, Japan). FAK was also detected in the cell lysates prepared from ECs treated with VLPs (10× dilution) and TNF-alpha (10 ng/mL) for 48 h, following the same protocol described above, using the rabbit monoclonal anti-FAK antibodies ([(D507U) XP, Cell signaling, Beverly, MA, USA; 1:1000; overnight]) and the corresponding secondary antibodies.
2.5. Treatment of Endothelial Cells (ECs) with Ebola VLPs or TNF-Alpha
ECs (2 × 104) per well were plated in collagen-coated 96-well plates (Corning, NY, USA) for 24 h. The cells were then treated with 10× diluted Ebola VLPs and VLPVP40/NP or, to study the dose-response effect, with 5×, 10×, 20×, 40×, 80×, and 160× diluted Ebola VLPs and VLPVP40/NP. As negative controls, ECs were either left untreated (mock) or treated with the mock control, which is the supernatant from the cells transfected with only pCAGGS. Cells treated with TNF-α at 10 ng/mL (Fujifilm Wako, Osaka, Japan) served as positive controls. Incubations were performed for up to 6, 12, 24, 48, and 72 h post-treatment at 37 °C in a humidified 5% CO2 environment. After incubation, the cells were fixed for immunofluorescence, as described below. In another set of experiments, confluent ECs were pretreated with RevitaCell Supplement (A2644501, Gibco, Grand Island, NY, USA, 1×) or increasing dilutions of focal adhesion kinase inhibitors from Selleck Chemicals (Houston, TX, USA) for 1 h before the addition of VLPs or TNF-α for 48 and 12 h incubation, respectively. The ECs were subsequently fixed for the evaluation of ICAM-1 expression through immunofluorescence assay.
2.6. Immunofluorescence Assay
For the evaluation of the expression level of ICAM-1 and CD31 (PECAM-1) in ECs, mouse monoclonal anti-ICAM-1 and anti-CD31 antibodies (MEM-111 and JC/70A, Abcam, Cambridge, UK) were used with 1000× dilution. After the appropriate treatment, the EC monolayers were fixed with 10% formalin overnight at 4 °C and then permeabilized with 0.2% Triton X-100 for 10 min at room temperature. Following permeabilization, the cells were blocked with 10% goat serum for 2 h at room temperature and incubated with specific primary anti-ICAM-1 and anti-CD31 antibodies overnight at 4 °C. A Corresponding secondary antibody conjugated to Alexa 488 (Thermo Fisher Scientific, Rockford, USA; 1:1000) was applied for 1 h at room temperature. Cells were washed with PBS four times for 5 min, and the resulting fluorescence was quantified with a SpectraMax iD5 microplate reader (Molecular Devices, San Jose, USA). Hoechst 33342 dye was added to stain cell nuclei, and cell images were obtained using a Cytation 5 imaging plate reader (BioTek Instruments, Winooski, VT, USA) with specific objectives.
2.7. Apoptosis Detection
To detect apoptosis in cultured ECs following appropriate treatments, the CF488 terminal dUTP nick end labeling (TUNEL) apoptosis detection kit (Biotium, Fremont, CA, USA) was used according to the manufacturer’s instructions. Actinomycin D (Merck, Darmstadt, Germany) and ECs incubated with TUNEL reaction buffer without TdT enzyme were used as positive and negative controls, respectively. The fluorescence-positive cells representing apoptotic cells were quantified using the SpectraMax iD5 microplate reader, and images were acquired using a Cytation 5 imaging plate reader.
2.8. Cell Viability Assay
Cytotoxic effects of VLPs on ECs were assessed using the Cell Counting Kit-8 (Gold Biotechnology, St. Louis, MO, USA). Cells were seeded at a density of 2 × 104 cells per well into collagen-coated 96-well plates. After 24 h, the medium was removed, and the cultures were refed with 100 µL of medium alone as a negative control and with a medium containing either 10× diluted VLPs or to study the dose-response effect, VLPs at various concentrations. Non-seeded wells were treated similarly for blank measurements. The cytotoxic effect of TNF-α (10 ng/mL) was assessed following the same protocol. Forty-eight hours (or at a specific time point for the kinetic experiment) after the treatment, 10 µL of the Cell Counting Kit-8 reagent was added to each well, and the cultures were incubated at 37 °C for another 4 h before measuring the absorbance at 450 nm with a multi-mode microplate reader. The tetrazolium compound (WST-x8) contained in the assay reagent was reduced by live cells into a colored formazan that was measured at 450 nm, and its quantity was directly proportional to the number of live cells in the culture. In another set of experiments that were designed to confirm the cytotoxic effect of the focal adhesion kinase inhibitors, as well as the cytoskeletal signaling library compounds, the confluent ECs were pre-treated with 5 µM of library compounds and with serial dilutions of focal adhesion kinase inhibitors for 1 h before the addition of 10× diluted VLPs or TNF-α (10 ng/mL) for 48 and 12 h of incubation, respectively. Each experiment was performed in triplicates. Cell viability was calculated according to the manufacturer’s instructions and expressed as percentages. For the experiments using RevitaCell Supplement (A2644501, Gibco, Grand Island, NY, USA, 1×) to suppress the upregulation of VLPs- or TNF-α-mediated ICAM-1 expression, the viability of ECs was assessed at 48 and 12 h, respectively, as described above, following 1 h pretreatment with RevitaCell Supplement.
2.9. Drug Treatment Assays
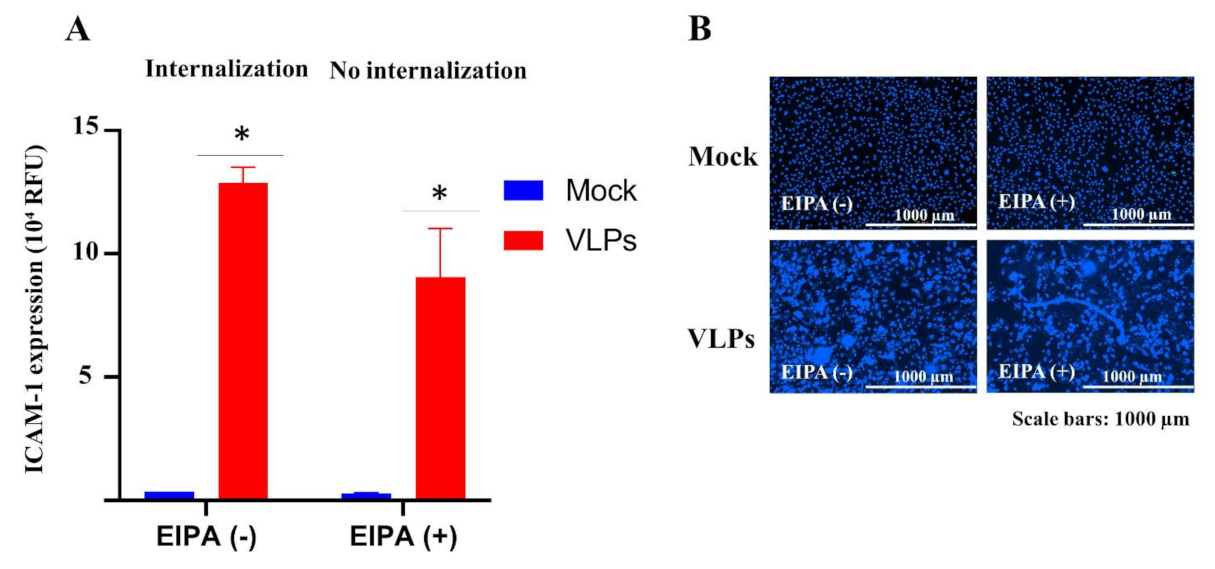
For screening in 96-well plates, ECs were cultured for 24 h, as described above. Cultured media were removed, and cells were pretreated with the vehicle (DMSO or water) or the library compound for 1 h at 37 °C before the addition of TNF-α (10 ng/mL) or 10× diluted VLPs for 12 or 48 h post-treatment incubation, respectively. Cells were then fixed with 10% formalin and subjected to an immunofluorescence assay as described above. All compounds were used at a final concentration of 5 µM during the screening, and their inhibition of VLPs- or TNF-α-mediated ICAM-1 expression was determined by fluorescence measurement. Percent inhibition of ICAM-1 expression for each compound was determined by normalization to either VLPs- or TNF-α-treated wells without compounds. Following the same protocol, the experiment for the inhibition of micropinocytosis was performed with 30 min of incubation at 37 °C with 10 µM EIPA [5-(N-ethyl-N-isopropyl) amiloride] before the addition of 10× diluted VLPs or the medium (control) for 48 h.
2.10. Statistical Analysis
GraphPad Prism software, version 9.3.0. (463) (San Diego, CA, USA) was used for the determination of average values, standard errors, IC50, and CC50. Differences between groups were examined for statistical significance using either one-way or two-way ANOVA, with a p value < 0.05 considered as statistically significant. Relevant details of the analysis presented in individual figures are included in the accompanying figure legends.
4. Discussion
Dysfunction and injury of ECs constitute the major outcomes of EBOV infection and play key roles in disease development. Previous studies have indicated that EBOV GP is the main determinant of EC activation and cytotoxicity [
1,
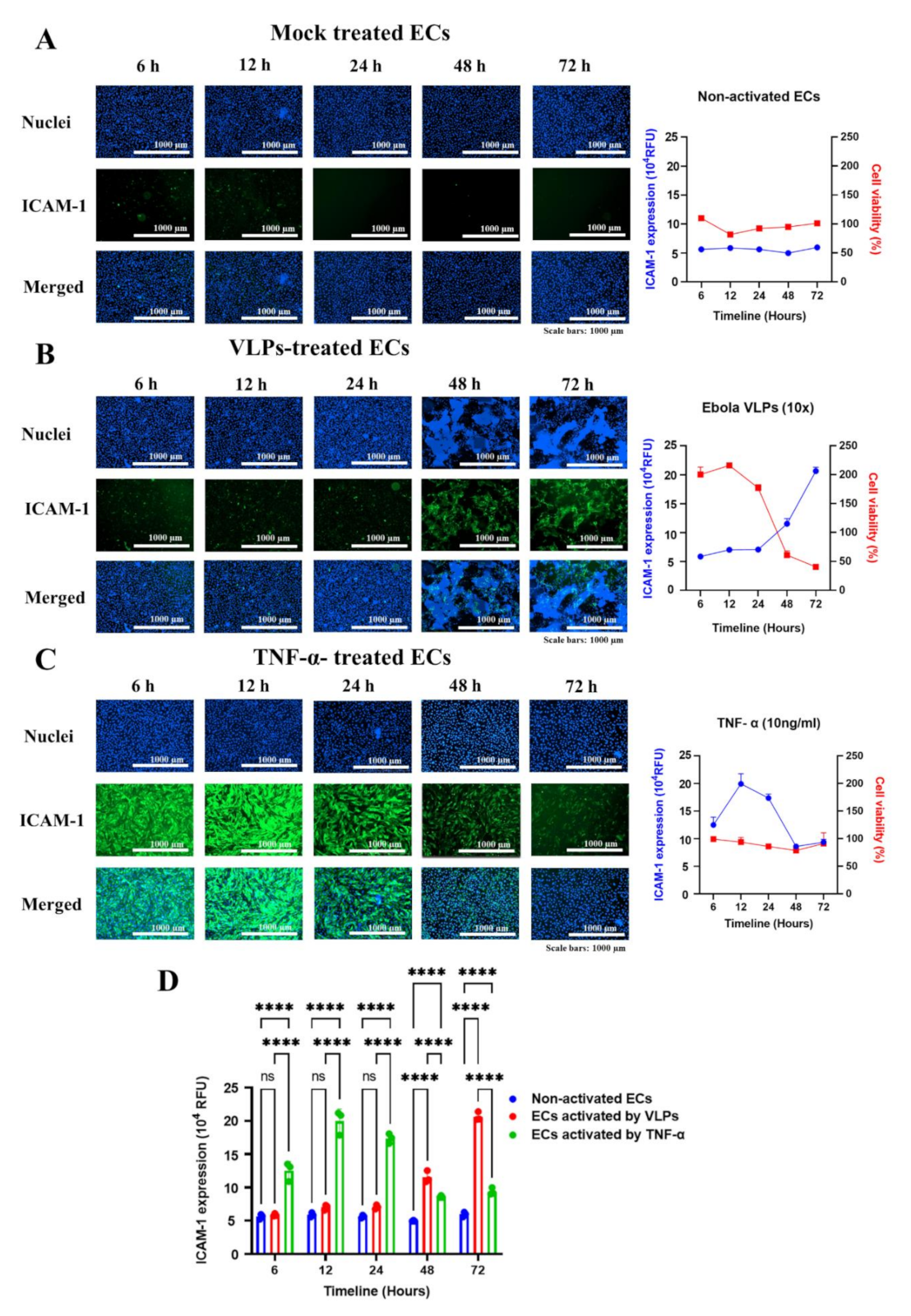
17]. However, the mechanisms underlying the interactions between EBOV GP and ECs are largely unknown. In this study, we found that, similar to TNF-α, Ebola VLPs bearing GP (referred to as VLPs) could activate ECs, as confirmed by the upregulation of ICAM-1 expression on the cell surface (
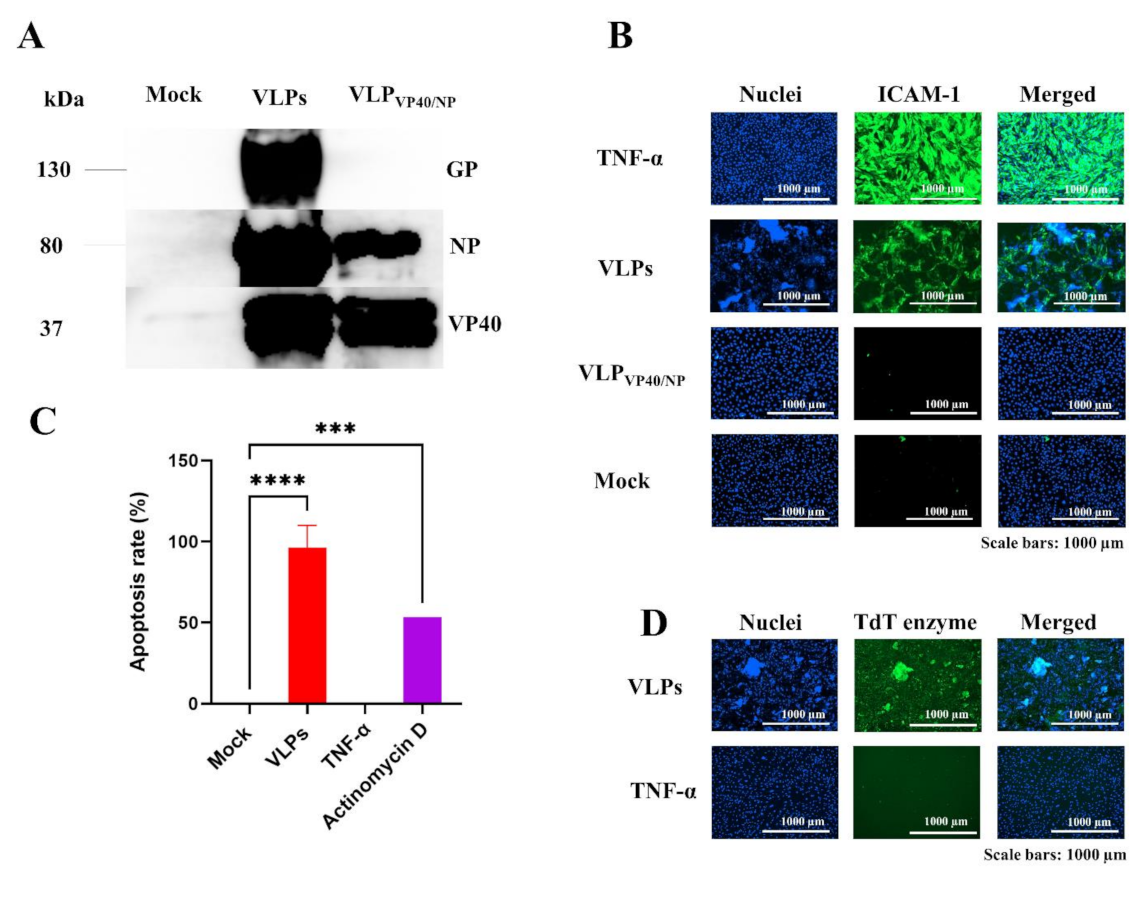
Figure 1B). In addition to the induction of ICAM-1 expression, the exposure of ECs to VLPs led to the appearance of cytopathic effects (
Figure 1B), which were due to apoptosis, while TNF-α activation of ECs was not associated with apoptosis and subsequent cytopathic effects (
Figure 1C–E). In contrast to VLPs, GP-deficient VLPs (referred to as VLP
VP40/NP) did not activate ECs, as shown by the absence of ICAM-1 expression (
Figure 1B), suggesting that GP is required for vascular activation and disruption. These results are consistent with those of previous studies indicating that GP is the main determinant of vascular injury and cytotoxicity and that viral replication is not required for the vascular dysfunction observed during EBOV infection.
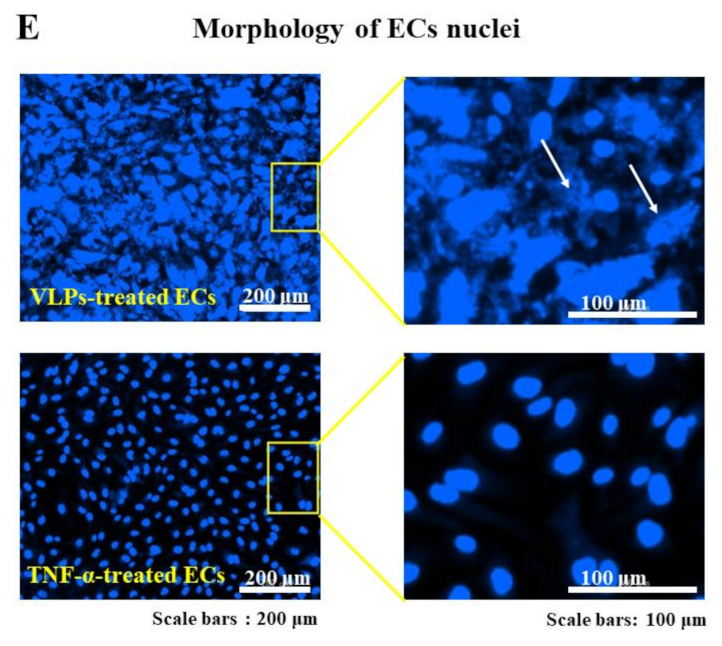
The induction of ICAM-1 expression on the surface of ECs and the associated apoptotic cell death occurred in a VLPs dose-dependent manner (
Figure 2A), and the level of the observed cytotoxicity was found to be directly dependent on the level of EBOV GP. Previously, it has been demonstrated that the cytotoxicity of EBOV is significantly increased by the overexpression of GP [
34], which has been further supported by Ray et al. [
35], who reported that the expression of EBOV GP in recombinant adenovirus (Ad-Ebola GP)-transduced primary human cardiac microvascular endothelial cells and HUVECs induced apoptotic cell death with related cytopathic effects in a dose-dependent manner, demonstrating the importance of GP expression level in the cytotoxic effect through apoptosis induction.
In agreement with these findings, we showed that ECs treated with 10× diluted VLPs, which contained approximately 9.2 µg/mL of GP, exhibited a strong cytopathic effect than that of ECs treated with 160× diluted VLPs, which contained a low amount of GP (
Figure 2).
The relevance of EBOV GP level on the induction of ECs apoptosis and disruption suggests a direct correlation between disease severity and the presence of a high level of shed GP and high viral load in the serum of EVD patients, as reported elsewhere [
36].
ICAM-1 expression is constitutively low at the surface of ECs and may be upregulated in response to stimuli, such as viral infection, oxidative stress, and pro-inflammatory cytokines, including interleukin-1β (IL1β), TNF-α, and interferon-γ (IFN-γ) [
37,
38]. Kinetic studies of ICAM-1 expression upon activation with TNF-α or VLPs revealed a difference in kinetics patterns. TNF-α-mediated upregulation of ICAM-1 occurred early around 6 h post-treatment and reached a peak at 12 h before decreasing around 48 h, with no cytopathic effect observed in ECs over time (
Figure 3C). Previous studies have shown that TNF-α can activate the endothelium and modulate its barrier function in tissue-culture models and in vivo while maintaining a relatively intact morphology [
2]. This observation was confirmed in our study, where the activation of the endothelium by TNF-α was not associated with a direct induction of ECs disruption and the appearance of cytopathic effects. In contrast, VLP-mediated upregulation of ICAM-1 appeared at a later time point (48 h post-treatment), with cells presenting injury and typical apoptotic morphology. A correlation between the levels of ECs disruption and the expression of ICAM-1 was observed (
Figure 3B). While the treatment of ECs with TNF-α did not affect cell viability and growth, ECs exposure to VLPs increased the survival rate of the cells by approximately 200% from 6 h, with a peak at 12 h before decreasing from 48 h (
Figure 3B, graph at the right). To the best of our knowledge, our study is the first to show that Ebola VLPs upregulate host cell proliferation within several hours following treatment before inducing apoptosis. We speculate that the EBOV VLP-induced abnormal cell proliferation might be the result of the cell stress response, which in turn has triggered the subsequent apoptosis after failing to cope with the insult caused by VLPs [
39]. More studies to understand the interplay between the GP on the VLPs, the ECs proliferation, and the apoptosis induction are needed.
The level of ICAM-1 expression in VLP-treated ECs was not upregulated before 48 h of incubation. Connolly-Andersen et al. observed the same kinetic pattern for ICAM-1 upregulation at the transcriptional level following the activation of HUVECs with various doses of Crimean-Congo hemorrhagic fever virus (CCHFV) [
38]. Wahl-Jensen et al. reported the upregulation of ICAM-1 at 12 and 24 h at the level of gene transcription after treatment of HUVECs with Ebola VLPs consisting of GP and VP40 and at the protein level after the infection of HUVECs with live EBOV [
1]. Whether the expression observed at the cell surface was a basal ICAM-1 expression, as no cytopathic effect was reported together with the ICAM-1 upregulation in HUVECs, needs to be clarified. One of the limitations of our assay was the evaluation of ECs activation only with the induction of ICAM-1 upregulation, which cannot provide more details such as ECs contraction and ultrastructural changes. Transmission electron microscopy of ECs undergoing apoptosis in the presence of the VLPs in comparison with the mock-treated ECs would provide a deep understanding of the VLPs’ effect on ECs.
The involvement of the Rho/ROCK pathway in the induction of hyperpermeability in HUVECs treated with Ebola VLPs [
13] led us to hypothesize that the inhibition of the Rho/ROCK pathway would suppress TNF-α- and VLP-induced ICAM-1 upregulation, as well as VLP-induced apoptotic disruption in ECs. Our hypothesis was verified for only the suppression of ICAM-1 expression (
Figure 5A,B). The kinetic patterns of TNF-α and VLP activities had an impact on the modulation of the Rho/ROCK pathway, as seen by RevitaCell suppression of TNF-α-induced ICAM-1 at 12 h (
Figure 5B) and VLP-induced ICAM-1 at 48 h (
Figure 5A). Several studies have reported that the inhibition of Rho/ROCK does not reverse the increase in EC permeability for several hours after TNF-α exposure and stimulation [
40,
41,
42]. Consistent with these reports, the inhibition of the Rho/ROCK pathway in our study did not influence TNF-α-induced ICAM-1 expression upregulation at 48 h after treatment. Furthermore, the inhibition of the Rho/ROCK pathway led to the abnormal cell proliferation of ECs exposed to VLPs at 12 h of incubation, suggesting a probable interaction with downstream effectors or pathways that might be specific to the VLPs and responsible for the observed apoptosis.
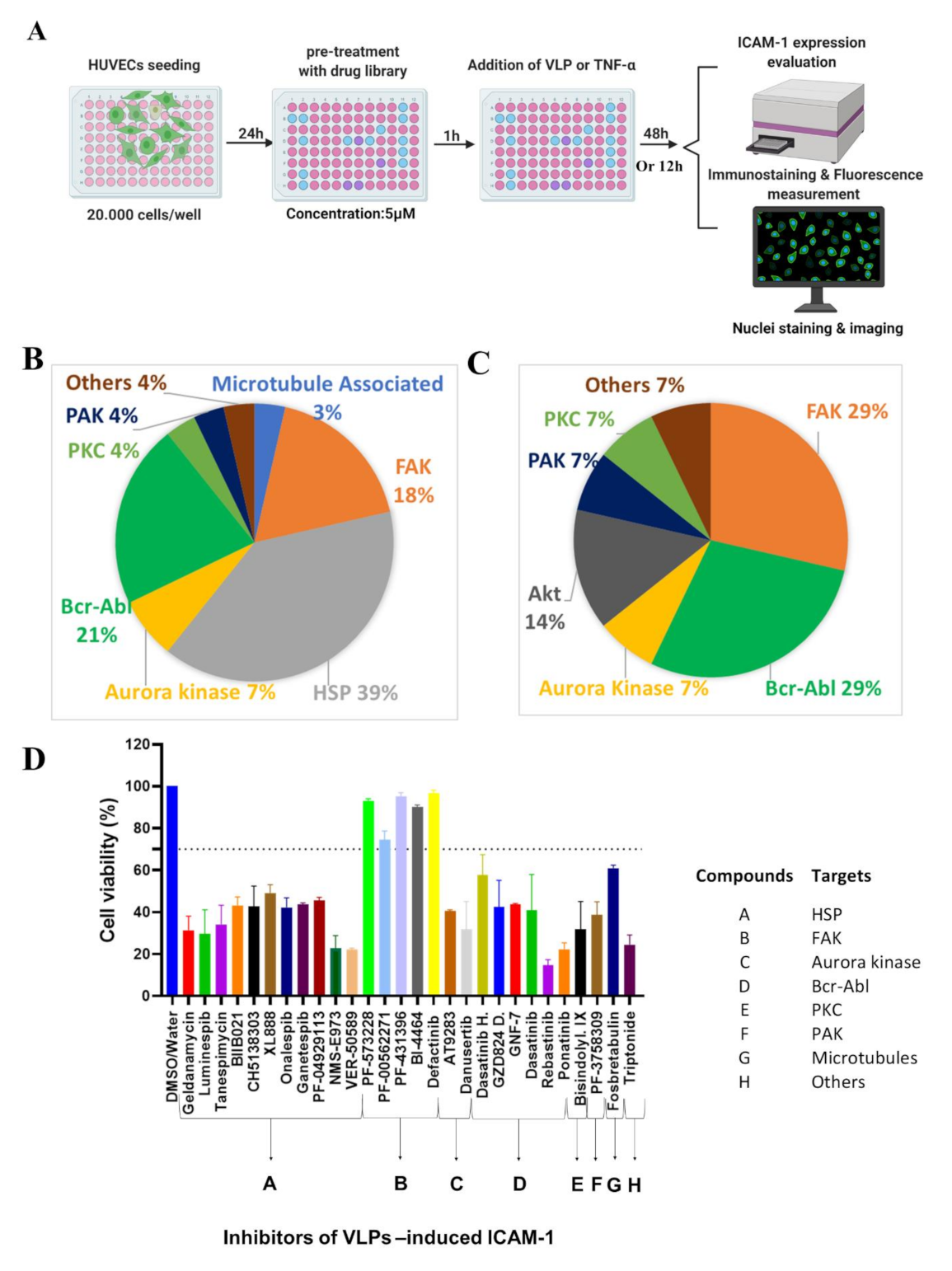
Since the Rho/ROCK pathway controls EC dysfunction through the regulation of cytoskeleton organization [
23], we screened the inhibitors of host cytoskeleton signaling pathways and identified FAK inhibitors as potent inhibitors of ICAM-1 upregulation mediated by both TNF-α and VLPs (
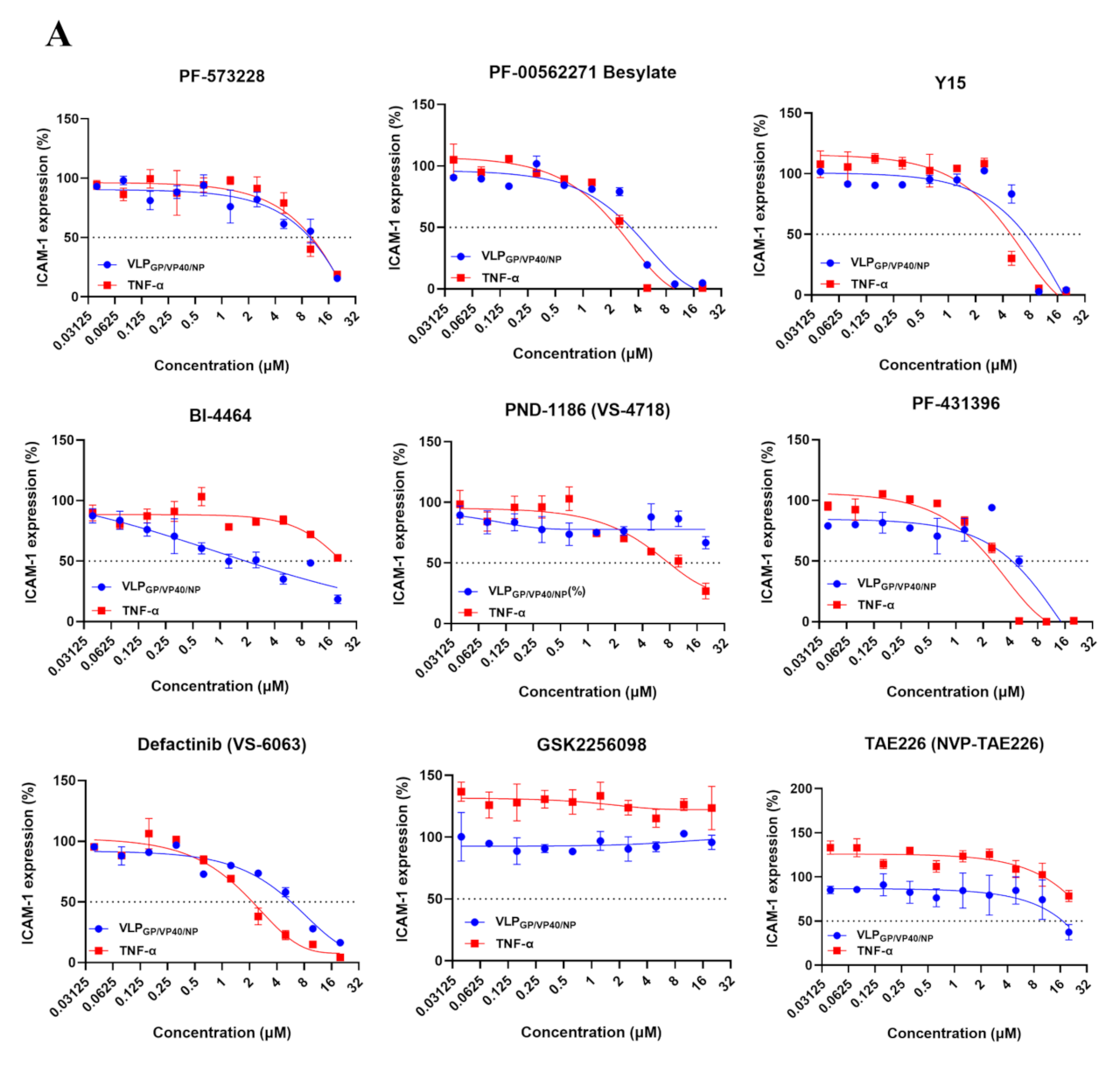
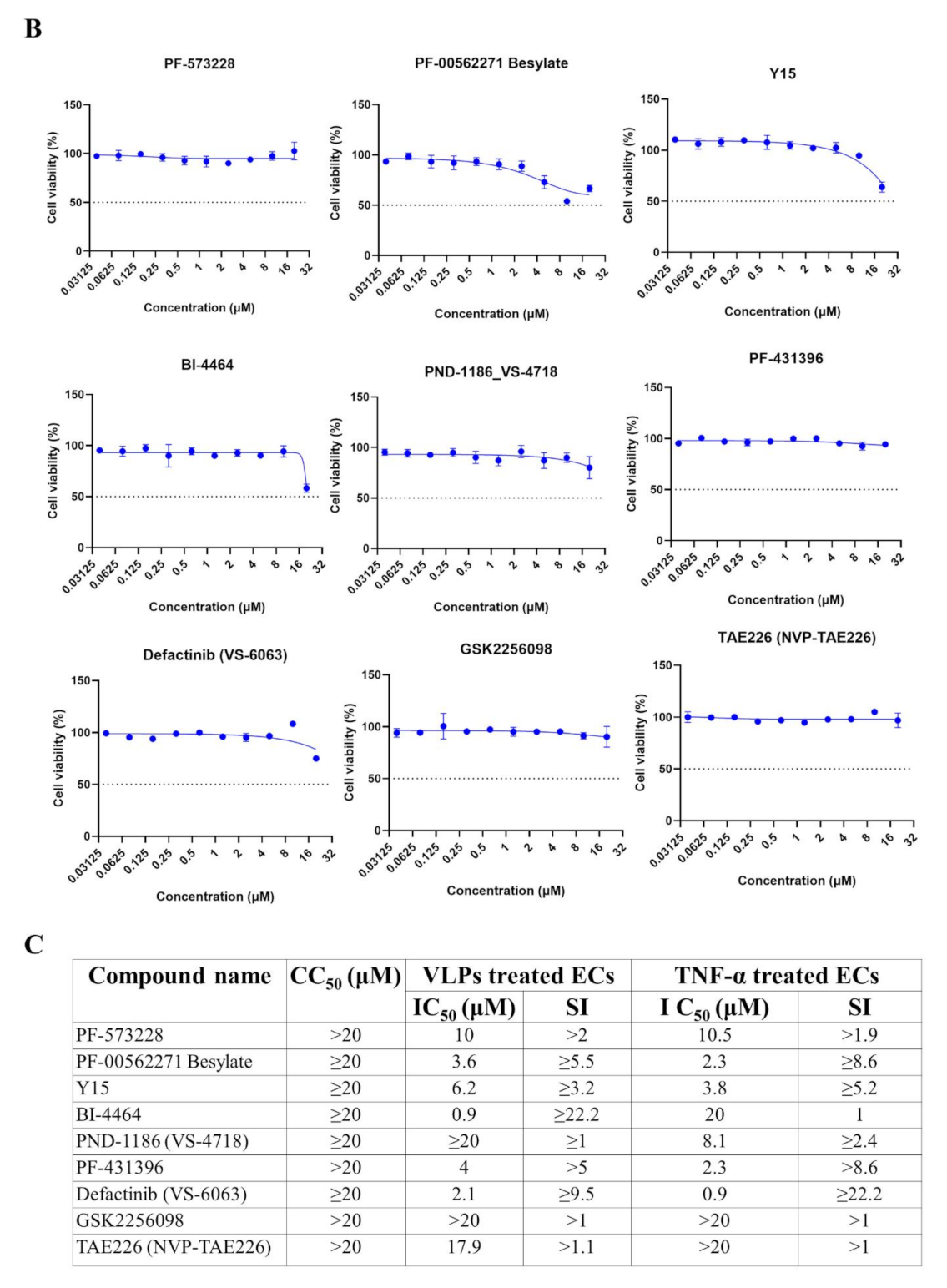
Figure 6B,C). Among the nine FAK inhibitors analyzed for their dose-response activity, BI-4464 was the most effective against VLPs-induced ICAM-1 expression, while Defactinib (VS-6063) showed potency against both VLPs- and TNF-α-induced ICAM-1 expression (
Figure 7).
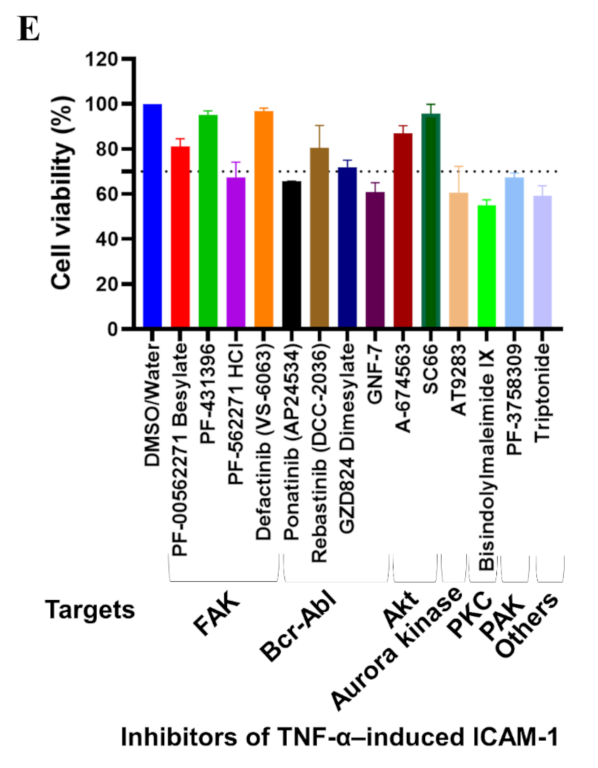
Unexpectedly, FAK inhibitors did not prevent Ebola VLPs-induced apoptosis in ECs, suggesting the possible existence of an additional host signaling pathway, acting either independently of FAK, or at triggering GP-mediated apoptosis at the upstream of FAK pathway associated with ICAM-1 induction. Further investigation of other identified compounds targets, including HSP and Bcr-Abl, may be key to unraveling the molecular mechanisms of GP-induced apoptosis. To the best of our knowledge, this is the first study identifying FAK as a therapeutic target for EVD pathogenesis. FAK is overexpressed in various cancers and has been found to play key roles in normal and cancer cellular functions, such as survival and proliferation [
43]. Notably, it has been reported as an anticancer target. As some of our identified compounds are in clinical test phases, the FAK inhibitors constitute potential therapeutic drugs or lead compounds for further development, which target disease pathogenesis related to endothelial barrier dysfunction in severe cases of EVD.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}