
Inhibition of MAVS Aggregation-Mediated Type-I Interferon Signaling by Foot-and-Mouth Disease Virus VP3

 ,
,  ,
,  , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells and Viruses
2.2. Antibodies
2.3. Plasmids
2.4. Plasmid Transfection and Virus Infection
2.5. Virus Titer Determination
2.6. Enzyme-Linked Immunosorbent Assay (ELISA)
2.7. Quantitative Real-Time PCR
2.8. Luciferase Reporter Assay
2.9. Immunoprecipitation Assay
2.10. Immunoblot Analysis
2.11. Immunofluorescence and Confocal Microscopy
2.12. MAVS Aggregation Assay
2.13. Statistical Analysis
3. Results
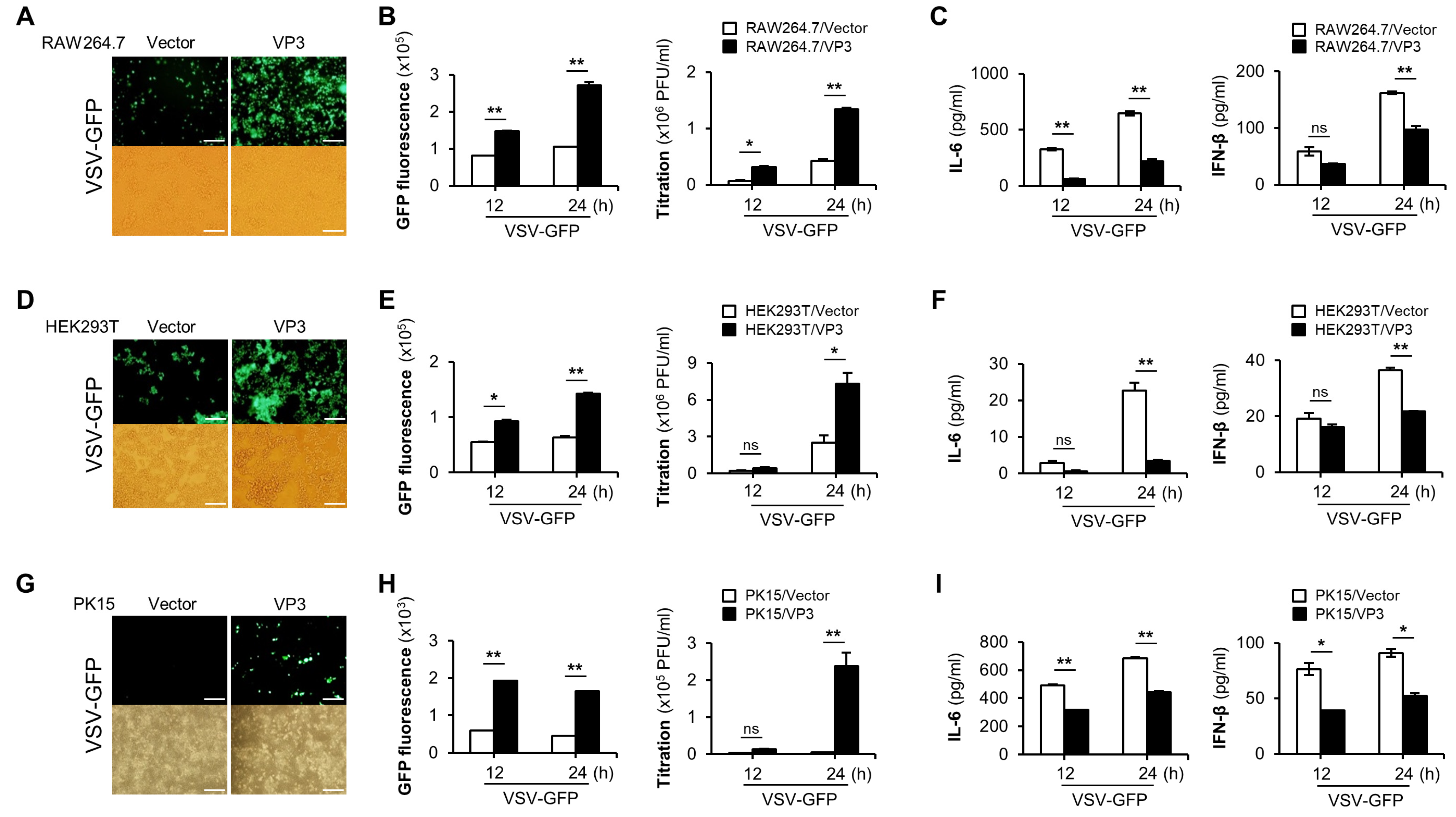
3.1. FMDV VP3 Negatively Regulate the Antiviral Responses
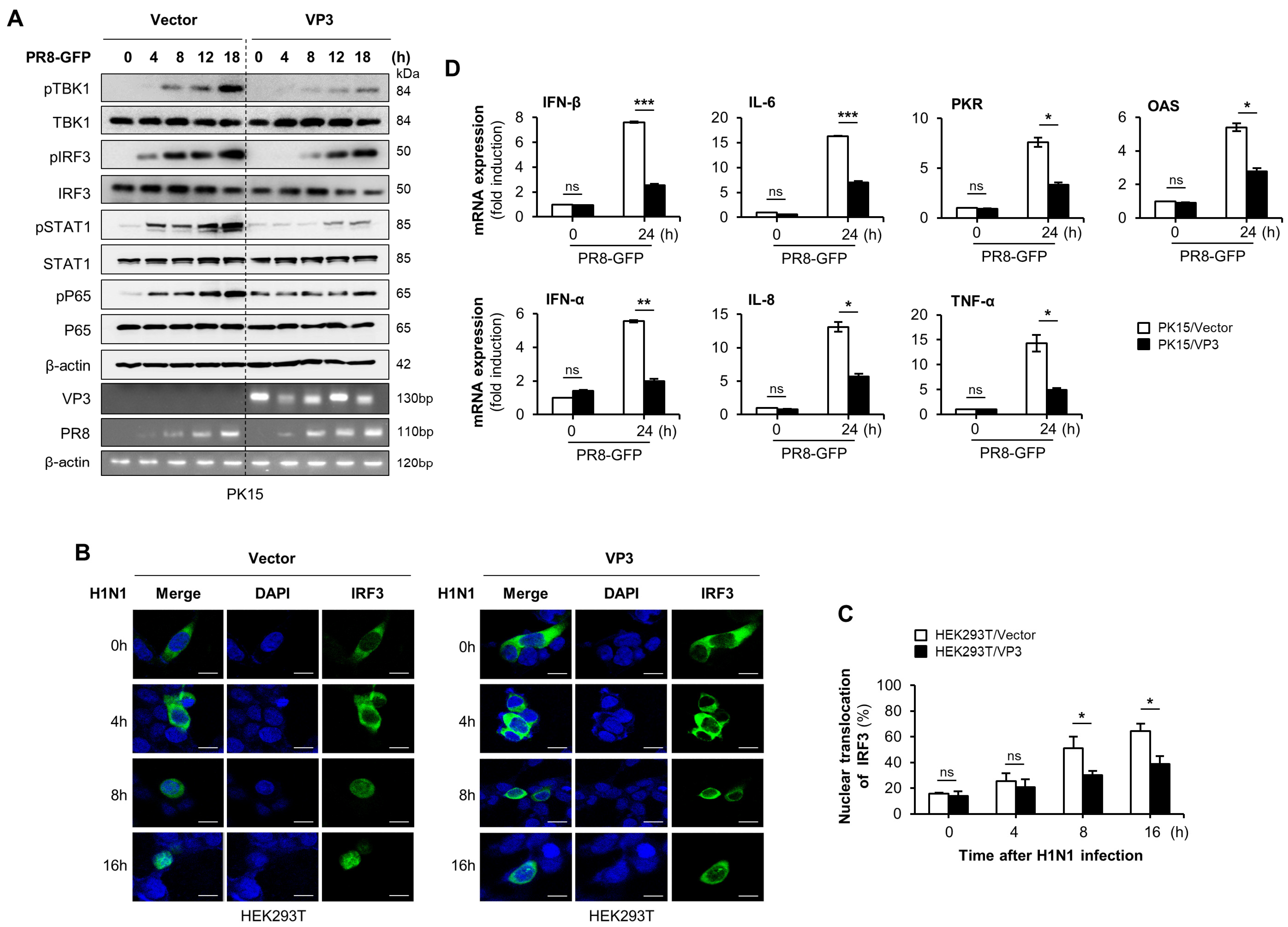
3.2. FMDV VP3 Suppress Virus-Mediated Type-I IFN Signaling and Antiviral Gene Transcription
3.3. FMDV VP3 Predominantly Targets the TM Domain of MAVS to Antagonize IFN-β Production
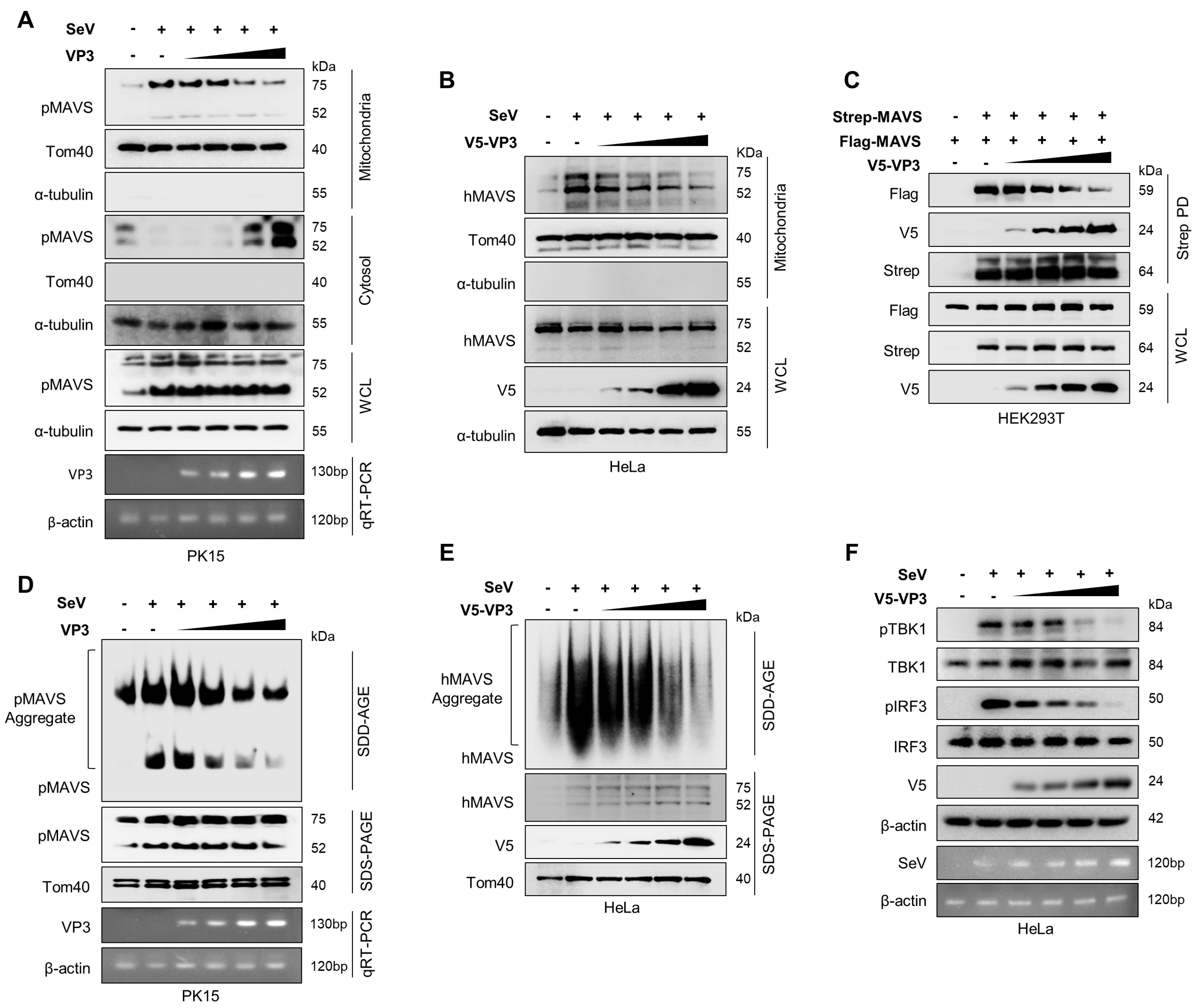
3.4. FMDV VP3 Inhibits Mitochondrial Localization, Self-Association, and Aggregation of MAVS
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jamal, S.M.; Belsham, G.J. Foot-and-mouth disease: Past, present and future. Vet. Res. 2013, 44, 116. [Google Scholar] [CrossRef] [Green Version]
- Grubman, M.J.; Baxt, B. Foot-and-mouth disease. Clin. Microbiol. Rev. 2004, 17, 465–493. [Google Scholar] [CrossRef] [Green Version]
- Sakamoto, K.; Kanno, T.; Yamakawa, M.; Yoshida, K.; Yamazoe, R.; Murakami, Y. Isolation of foot-and-mouth disease virus from Japanese black cattle in Miyazaki Prefecture, Japan, 2000. J. Vet. Med. Sci. 2002, 64, 91–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palmenberg, A.C. Proteolytic processing of picornaviral polyprotein. Annu. Rev. Microbiol. 1990, 44, 603–623. [Google Scholar] [CrossRef]
- Rowlands, D.J.; Mayo, M.; Mahy, B.W. The Molecular Biology of the Positive Strand RNA Viruses; Academic Press: London, UK, 1987. [Google Scholar]
- Belsham, G.J. Distinctive features of foot-and-mouth disease virus, a member of the picornavirus family; aspects of virus protein synthesis, protein processing and structure. Prog. Biophys. Mol. Biol. 1993, 60, 241–260. [Google Scholar] [CrossRef]
- Forss, S.; Strebel, K.; Beck, E.; Schaller, H. Nucleotide sequence and genome organization of foot-and-mouth disease virus. Nucleic Acids Res. 1984, 12, 6587–6601. [Google Scholar] [CrossRef] [Green Version]
- Fry, E.; Stuart, D.; Rowlands, D. The structure of foot-and-mouth disease virus. In Foot-and-Mouth Disease Virus Current Topics in Microbiology and Immunology; Springer: Berlin/Heidelberg, Germany, 2005; pp. 71–101. [Google Scholar]
- Durbin, J.E.; Fernandez-Sesma, A.; Lee, C.-K.; Rao, T.D.; Frey, A.B.; Moran, T.M.; Vukmanovic, S.; García-Sastre, A.; Levy, D.E. Type I IFN modulates innate and specific antiviral immunity. J. Immunol. 2000, 164, 4220–4228. [Google Scholar] [CrossRef] [Green Version]
- Perry, A.K.; Gang, C.; Zheng, D.; Hong, T.; Cheng, G. The host type I interferon response to viral and bacterial infections. Cell Res. 2005, 15, 407–422. [Google Scholar] [CrossRef] [Green Version]
- Stetson, D.B.; Medzhitov, R. Type I interferons in host defense. Immunity 2006, 25, 373–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.-C.; Chathuranga, K.; Lee, J.-S. Intracellular sensing of viral genomes and viral evasion. Exp. Mol. Med. 2019, 51, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Kato, H.; Takeuchi, O.; Sato, S.; Yoneyama, M.; Yamamoto, M.; Matsui, K.; Uematsu, S.; Jung, A.; Kawai, T.; Ishii, K.J. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 2006, 441, 101–105. [Google Scholar] [CrossRef]
- Pichlmair, A.; e Sousa, C.R. Innate recognition of viruses. Immunity 2007, 27, 370–383. [Google Scholar] [CrossRef] [Green Version]
- Meylan, E.; Curran, J.; Hofmann, K.; Moradpour, D.; Binder, M.; Bartenschlager, R.; Tschopp, J. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature 2005, 437, 1167–1172. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Takahashi, K.; Sato, S.; Coban, C.; Kumar, H.; Kato, H.; Ishii, K.J.; Takeuchi, O.; Akira, S. IPS-1, an adaptor triggering RIG-I-and Mda5-mediated type I interferon induction. Nat. Immunol. 2005, 6, 981–988. [Google Scholar] [CrossRef] [PubMed]
- Loo, Y.-M.; Gale, M., Jr. Immune signaling by RIG-I-like receptors. Immunity 2011, 34, 680–692. [Google Scholar] [CrossRef] [Green Version]
- Dixit, E.; Boulant, S.; Zhang, Y.; Lee, A.S.; Odendall, C.; Shum, B.; Hacohen, N.; Chen, Z.J.; Whelan, S.P.; Fransen, M. Peroxisomes are signaling platforms for antiviral innate immunity. Cell 2010, 141, 668–681. [Google Scholar] [CrossRef] [Green Version]
- Horner, S.M.; Liu, H.M.; Park, H.S.; Briley, J.; Gale, M. Mitochondrial-associated endoplasmic reticulum membranes (MAM) form innate immune synapses and are targeted by hepatitis C virus. Proc. Natl. Acad. Sci. USA 2011, 108, 14590–14595. [Google Scholar] [CrossRef] [Green Version]
- Hou, F.; Sun, L.; Zheng, H.; Skaug, B.; Jiang, Q.-X.; Chen, Z.J. MAVS forms functional prion-like aggregates to activate and propagate antiviral innate immune response. Cell 2011, 146, 448–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seth, R.B.; Sun, L.; Ea, C.-K.; Chen, Z.J. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-κB and IRF3. Cell 2005, 122, 669–682. [Google Scholar] [CrossRef] [Green Version]
- Tang, E.D.; Wang, C.-Y. MAVS self-association mediates antiviral innate immune signaling. J. Virol. 2009, 83, 3420–3428. [Google Scholar] [CrossRef] [Green Version]
- Yang, B.; Zhang, X.; Zhang, D.; Hou, J.; Xu, G.; Sheng, C.; Choudhury, S.M.; Zhu, Z.; Li, D.; Zhang, K.; et al. Molecular mechanisms of immune escape for foot-and-mouth disease virus. Pathogens 2020, 9, 729. [Google Scholar] [CrossRef] [PubMed]
- Brewer, K.D.; Weir, G.M.; Dude, I.; Davis, C.; Parsons, C.; Penwell, A.; Rajagopalan, R.; Sammatur, L.; Bowen, C.V.; Stanford, M.M. Unique depot formed by an oil based vaccine facilitates active antigen uptake and provides effective tumour control. J. Biomed. Sci. 2018, 25, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Y.; Sun, S.-Q.; Guo, H.-C. Biological function of Foot-and-mouth disease virus non-structural proteins and non-coding elements. Virol. J. 2016, 13, 107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falk, M.; Grigera, P.; Bergmann, I.; Zibert, A.; Multhaup, G.; Beck, E. Foot-and-mouth disease virus protease 3C induces specific proteolytic cleavage of host cell histone H3. J. Virol. 1990, 64, 748–756. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Ross-Smith, N.; Proud, C.G.; Belsham, G.J. Cleavage of translation initiation factor 4AI (eIF4AI) but not eIF4AII by foot-and-mouth disease virus 3C protease: Identification of the eIF4AI cleavage site. FEBS Lett. 2001, 507, 1–5. [Google Scholar] [CrossRef]
- Wang, D.; Fang, L.; Li, P.; Sun, L.; Fan, J.; Zhang, Q.; Luo, R.; Liu, X.; Li, K.; Chen, H. The leader proteinase of foot-and-mouth disease virus negatively regulates the type I interferon pathway by acting as a viral deubiquitinase. J. Virol. 2011, 85, 3758–3766. [Google Scholar] [CrossRef] [Green Version]
- De Los Santos, T.; Diaz-San Segundo, F.; Grubman, M.J. Degradation of nuclear factor kappa B during foot-and-mouth disease virus infection. J. Virol. 2007, 81, 12803–12815. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Weiss, M.; Grubman, M.J.; de los Santos, T. Differential gene expression in bovine cells infected with wild type and leaderless foot-and-mouth disease virus. Virology 2010, 404, 32–40. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Fang, L.; Luo, R.; Ye, R.; Fang, Y.; Xie, L.; Chen, H.; Xiao, S. Foot-and-mouth disease virus leader proteinase inhibits dsRNA-induced type I interferon transcription by decreasing interferon regulatory factor 3/7 in protein levels. Biochem. Biophys. Res. Commun. 2010, 399, 72–78. [Google Scholar] [CrossRef]
- Wang, D.; Fang, L.; Bi, J.; Chen, Q.; Cao, L.; Luo, R.; Chen, H.; Xiao, S. Foot-and-mouth disease virus leader proteinase inhibits dsRNA-induced RANTES transcription in PK-15 cells. Virus Genes 2011, 42, 388–393. [Google Scholar] [CrossRef]
- Wang, D.; Fang, L.; Li, K.; Zhong, H.; Fan, J.; Ouyang, C.; Zhang, H.; Duan, E.; Luo, R.; Zhang, Z. Foot-and-mouth disease virus 3C protease cleaves NEMO to impair innate immune signaling. J. Virol. 2012, 86, 9311–9322. [Google Scholar] [CrossRef] [Green Version]
- Du, Y.; Bi, J.; Liu, J.; Liu, X.; Wu, X.; Jiang, P.; Yoo, D.; Zhang, Y.; Wu, J.; Wan, R. 3Cpro of foot-and-mouth disease virus antagonizes the interferon signaling pathway by blocking STAT1/STAT2 nuclear translocation. J. Virol. 2014, 88, 4908–4920. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Kim, A.-Y.; Choi, J.; Park, S.Y.; Park, S.H.; Kim, J.-S.; Lee, S.-I.; Park, J.-H.; Park, C.-K.; Ko, Y.-J. Foot-and-mouth disease virus evades innate immune response by 3C-targeting of MDA5. Cells 2021, 10, 271. [Google Scholar] [CrossRef]
- Zhu, Z.; Li, C.; Du, X.; Wang, G.; Cao, W.; Yang, F.; Feng, H.; Zhang, X.; Shi, Z.; Liu, H. Foot-and-mouth disease virus infection inhibits LGP2 protein expression to exaggerate inflammatory response and promote viral replication. Cell Death Dis. 2017, 8, e2747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, X.; Han, S.; Yan, D.; Gao, Y.; Wei, Y.; Liu, X.; Liao, Y.; Guo, H.; Sun, S. Foot-and-mouth disease virus infection suppresses autophagy and NF-к B antiviral responses via degradation of ATG5-ATG12 by 3C pro. Cell Death Dis. 2018, 8, e2561. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Zhu, Z.; Du, X.; Cao, W.; Yang, F.; Zhang, X.; Feng, H.; Li, D.; Zhang, K.; Liu, X. Foot-and-mouth disease virus induces lysosomal degradation of host protein kinase PKR by 3C proteinase to facilitate virus replication. Virology 2017, 509, 222–231. [Google Scholar] [CrossRef]
- Zhu, Z.; Wang, G.; Yang, F.; Cao, W.; Mao, R.; Du, X.; Zhang, X.; Li, C.; Li, D.; Zhang, K. Foot-and-mouth disease virus viroporin 2B antagonizes RIG-I-mediated antiviral effects by inhibition of its protein expression. J. Virol. 2016, 90, 11106–11121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.-P.; Ru, Y.; Zhang, K.-S.; Wang, Y.; Liu, X.-T.; Li, D.; Zheng, H.-X. DDX56 cooperates with FMDV 3A to enhance FMDV replication by inhibiting the phosphorylation of IRF3. Cell. Signal. 2019, 64, 109393. [Google Scholar]
- Li, D.; Lei, C.; Xu, Z.; Yang, F.; Liu, H.; Zhu, Z.; Li, S.; Liu, X.; Shu, H.; Zheng, H. Foot-and-mouth disease virus non-structural protein 3A inhibits the interferon-β signaling pathway. Sci. Rep. 2016, 6, 21888. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.; Li, D.; Ru, Y.; Bai, J.; Ren, J.; Zhang, J.; Li, L.; Liu, X.; Zheng, H. Foot-and-mouth disease virus 3A protein causes upregulation of autophagy-related protein LRRC25 to inhibit the G3BP1-mediated RIG-like helicase-signaling pathway. J. Virol. 2020, 94, e02086-19. [Google Scholar] [CrossRef] [Green Version]
- Ekanayaka, P.; Lee, S.-Y.; Herath, T.U.; Kim, J.-H.; Kim, T.-H.; Lee, H.; Chathuranga, K.; Chathuranga, W.G.; Park, J.-H.; Lee, J.-S. Foot-and-mouth disease virus VP1 target the MAVS to inhibit type-I interferon signaling and VP1 E83K mutation results in virus attenuation. PLoS Pathog. 2020, 16, e1009057. [Google Scholar] [CrossRef]
- Li, X.; Wang, J.; Liu, J.; Li, Z.; Wang, Y.; Xue, Y.; Li, X.; Cao, H.; Zheng, S.J. Engagement of soluble resistance-related calcium binding protein (sorcin) with foot-and-mouth disease virus (FMDV) VP1 inhibits type I interferon response in cells. Vet. Microbiol. 2013, 166, 35–46. [Google Scholar] [CrossRef]
- Zhang, W.; Yang, F.; Zhu, Z.; Yang, Y.; Wang, Z.; Cao, W.; Dang, W.; Li, L.; Mao, R.; Liu, Y. Cellular DNAJA3, a novel VP1-interacting protein, inhibits foot-and-mouth disease virus replication by inducing lysosomal degradation of VP1 and attenuating its antagonistic role in the beta interferon signaling pathway. J. Virol. 2019, 93, e00588-19. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Yang, W.; Yang, F.; Liu, H.; Zhu, Z.; Lian, K.; Lei, C.; Li, S.; Liu, X.; Zheng, H. The VP3 structural protein of foot-and-mouth disease virus inhibits the IFN-β signaling pathway. FASEB J. 2016, 30, 1757–1766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, D.; Wei, J.; Yang, F.; Liu, H.-N.; Zhu, Z.-X.; Cao, W.-J.; Li, S.; Liu, X.-T.; Zheng, H.-X.; Shu, H.-B. Foot-and-mouth disease virus structural protein VP3 degrades Janus kinase 1 to inhibit IFN-γ signal transduction pathways. Cell Cycle 2016, 15, 850–860. [Google Scholar] [CrossRef] [Green Version]
- Baril, M.; Racine, M.-E.; Penin, F.; Lamarre, D. MAVS dimer is a crucial signaling component of innate immunity and the target of hepatitis C virus NS3/4A protease. J. Virol. 2009, 83, 1299–1311. [Google Scholar] [CrossRef] [Green Version]
- Lin, R.; Lacoste, J.; Nakhaei, P.; Sun, Q.; Yang, L.; Paz, S.; Wilkinson, P.; Julkunen, I.; Vitour, D.; Meurs, E. Dissociation of a MAVS/IPS-1/VISA/Cardif-IKKε molecular complex from the mitochondrial outer membrane by hepatitis C virus NS3-4A proteolytic cleavage. J. Virol. 2006, 80, 6072–6083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Wang, G.; Xu, Z.-G.; Tu, H.; Hu, F.; Dai, J.; Chang, Y.; Chen, Y.; Lu, Y.; Zeng, H. Lactate is a natural suppressor of RLR signaling by targeting MAVS. Cell 2019, 178, 176–189.e15. [Google Scholar] [CrossRef]
- Versteeg, G.A.; García-Sastre, A. Viral tricks to grid-lock the type I interferon system. Curr. Opin. Microbiol. 2010, 13, 508–516. [Google Scholar] [CrossRef]
- Schulz, K.S.; Mossman, K.L. Viral evasion strategies in type I IFN signaling–a summary of recent developments. Front. Immunol. 2016, 7, 498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Sastre, A. Ten strategies of interferon evasion by viruses. Cell Host Microbe 2017, 22, 176–184. [Google Scholar] [CrossRef]
- Rodriguez, J.J.; Parisien, J.-P.; Horvath, C.M. Nipah virus V protein evades alpha and gamma interferons by preventing STAT1 and STAT2 activation and nuclear accumulation. J. Virol. 2002, 76, 11476–11483. [Google Scholar] [CrossRef] [Green Version]
- Iborra, F.J.; Jackson, D.A.; Cook, P.R. Coupled transcription and translation within nuclei of mammalian cells. Science 2001, 293, 1139–1142. [Google Scholar] [CrossRef] [Green Version]
- Warke, R.V.; Martin, K.J.; Giaya, K.; Shaw, S.K.; Rothman, A.L.; Bosch, I. TRAIL is a novel antiviral protein against dengue virus. J. Virol. 2008, 82, 555–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pichlmair, A.; Lassnig, C.; Eberle, C.-A.; Górna, M.W.; Baumann, C.L.; Burkard, T.R.; Bürckstümmer, T.; Stefanovic, A.; Krieger, S.; Bennett, K.L. IFIT1 is an antiviral protein that recognizes 5′-triphosphate RNA. Nat. Immunol. 2011, 12, 624–630. [Google Scholar] [CrossRef]
- Komili, S.; Silver, P.A. Coupling and coordination in gene expression processes: A systems biology view. Nat. Rev. Genet. 2008, 9, 38–48. [Google Scholar] [CrossRef] [PubMed]
- Genuth, N.R.; Barna, M. Heterogeneity and specialized functions of translation machinery: From genes to organisms. Nat. Rev. Genet. 2018, 19, 431–452. [Google Scholar] [CrossRef] [PubMed]
- Baker, M.J.; Frazier, A.E.; Gulbis, J.M.; Ryan, M.T. Mitochondrial protein-import machinery: Correlating structure with function. Trends Cell Biol. 2007, 17, 456–464. [Google Scholar] [CrossRef] [PubMed]
- Vazquez, C.; Horner, S.M. MAVS coordination of antiviral innate immunity. J. Virol. 2015, 89, 6974–6977. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Gao, C. Regulation of MAVS activation through post-translational modifications. Curr. Opin. Immunol. 2018, 50, 75–81. [Google Scholar] [CrossRef]
- Moresco, E.M.Y.; La Vine, D.; Beutler, B. Prion-like behavior of MAVS in RIG-I signaling. Cell Res. 2011, 21, 1643–1645. [Google Scholar] [CrossRef] [Green Version]
- Sun, Q.; Sun, L.; Liu, H.-H.; Chen, X.; Seth, R.B.; Forman, J.; Chen, Z.J. The specific and essential role of MAVS in antiviral innate immune responses. Immunity 2006, 24, 633–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, Z.; Ding, T.; Zuo, Z.; Xu, Z.; Deng, J.; Wei, Z. Regulation of MAVS expression and signaling function in the antiviral innate immune response. Front. Immunol. 2020, 11, 1030. [Google Scholar] [CrossRef]
- Chen, Z.; Benureau, Y.; Rijnbrand, R.; Yi, J.; Wang, T.; Warter, L.; Lanford, R.E.; Weinman, S.A.; Lemon, S.M.; Martin, A. GB virus B disrupts RIG-I signaling by NS3/4A-mediated cleavage of the adaptor protein MAVS. J. Virol. 2007, 81, 964–976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoo, Y.-S.; Park, Y.-Y.; Kim, J.-H.; Cho, H.; Kim, S.-H.; Lee, H.-S.; Kim, T.-H.; Kim, Y.S.; Lee, Y.; Kim, C.-J.; et al. The mitochondrial ubiquitin ligase MARCH5 resolves MAVS aggregates during antiviral signalling. Nat. Commun. 2015, 6, 7910. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene a | Forward | Reverse |
|---|---|---|
| pIFN-β | AAATCGCTCTCCTGATGTGT | TGCTCCTTTGTTGGTATCG |
| pIL-6 | CACCGGTCTTGTGGAGTTTC | GTGGTGGCTTTGTCTGGATT |
| pPKR | GAGAAGGTAGAGCGTGAAG | CCAGCAACCGTAGTAGAG |
| pOAS | CTGTCGTTGGACGATGTATGCT | CAGCCGGGTCCAGAATCA |
| pIFN-α | GCCTCCTGCACCAGTTCTACA | TGCATGACACAGGCTTCCA |
| pIL-8 | TTTCTGCAGCTCTCTGTGAGG | CTGCTGTTGTTGTTGCTTCTC |
| pTNF-α | CCACGTTGTAGCCAATGTC | CTGGGAGTAGATGAGGTACAG |
| pβ-actin | CTCGATCATGAAGTGCGACG | GTGATCTCCTTCTGCATCCTGT |
| mIFN-β | TCCAAGAAAGGACGAACATTCG | TGCGGACATCTCCCACGTCAA |
| mISG-15 | CAATGGCCTGGGACCTAAA | CTTCTTCAGTTCTGACACCGTCA |
| mISG-56 | AGAGAACAGCTACCACCTTT | TGGACCTGCTCTGAGATTCT |
| mPKR | GCCAGATGCACGGAGTAGCC | GAAAACTTGGCCAAATCCACC |
| mOAS | GAGGCGGTTGGCTGAAGAGG | GAGGAAGGCTGGCTGTGATTGG |
| mGAPDH | TGACCACAGTCCATGCCATC | GACGGACACATTGGGGGTAG |
| hIFN-β | CATCAACTATAAGCAGCTCCA | TTCAAGTGGAGAGCAGTTGAG |
| hIL-6 | CCACACAGACAGCCACTCACC | CTACATTTGCCGAAGAGCCCTC |
| hISG-15 | GAGAGGCAGCGAACTCATCT | CTTCAGCTCTGACACCGACA |
| hISG-56 | AAGGCAGGCTGTCCGCTTA | TCCTGTCCTTCATCCTGAAGCT |
| hTNF-α | ATGAGCACTGAAAGCAT | TCGACGGGGAGTCGAACT |
| hβ-actin | CCAACCGCGAGAAGATGACC | GATCTTCATGAGGTAGTCAGT |
| PR8-NS1 | CAAACGAGTTGCAGACCAAG | TCTTGATGTCCAGACCGAGA |
| SeV-C | GGAGGAAGAGAGTCGCTCTC | TCCTTGGGGAGTGTTGATGG |
| FMDV-VP3 | TAAGACCTCGGACCCCGTTTACG | ATACGGTACCCCGTTGAAATCGA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ekanayaka, P.; Lee, B.-H.; Weerawardhana, A.; Chathuranga, K.; Park, J.-H.; Lee, J.-S. Inhibition of MAVS Aggregation-Mediated Type-I Interferon Signaling by Foot-and-Mouth Disease Virus VP3. Viruses 2021, 13, 1776. https://doi.org/10.3390/v13091776
Ekanayaka P, Lee B-H, Weerawardhana A, Chathuranga K, Park J-H, Lee J-S. Inhibition of MAVS Aggregation-Mediated Type-I Interferon Signaling by Foot-and-Mouth Disease Virus VP3. Viruses. 2021; 13(9):1776. https://doi.org/10.3390/v13091776
Chicago/Turabian StyleEkanayaka, Pathum, Byeong-Hoon Lee, Asela Weerawardhana, Kiramage Chathuranga, Jong-Hyeon Park, and Jong-Soo Lee. 2021. "Inhibition of MAVS Aggregation-Mediated Type-I Interferon Signaling by Foot-and-Mouth Disease Virus VP3" Viruses 13, no. 9: 1776. https://doi.org/10.3390/v13091776
APA StyleEkanayaka, P., Lee, B.-H., Weerawardhana, A., Chathuranga, K., Park, J.-H., & Lee, J.-S. (2021). Inhibition of MAVS Aggregation-Mediated Type-I Interferon Signaling by Foot-and-Mouth Disease Virus VP3. Viruses, 13(9), 1776. https://doi.org/10.3390/v13091776