
Examining the Effects of an Anti-Salmonella Bacteriophage Preparation, BAFASAL®, on Ex-Vivo Human Gut Microbiome Composition and Function Using a Multi-Omics Approach

 , , , and
, , , and 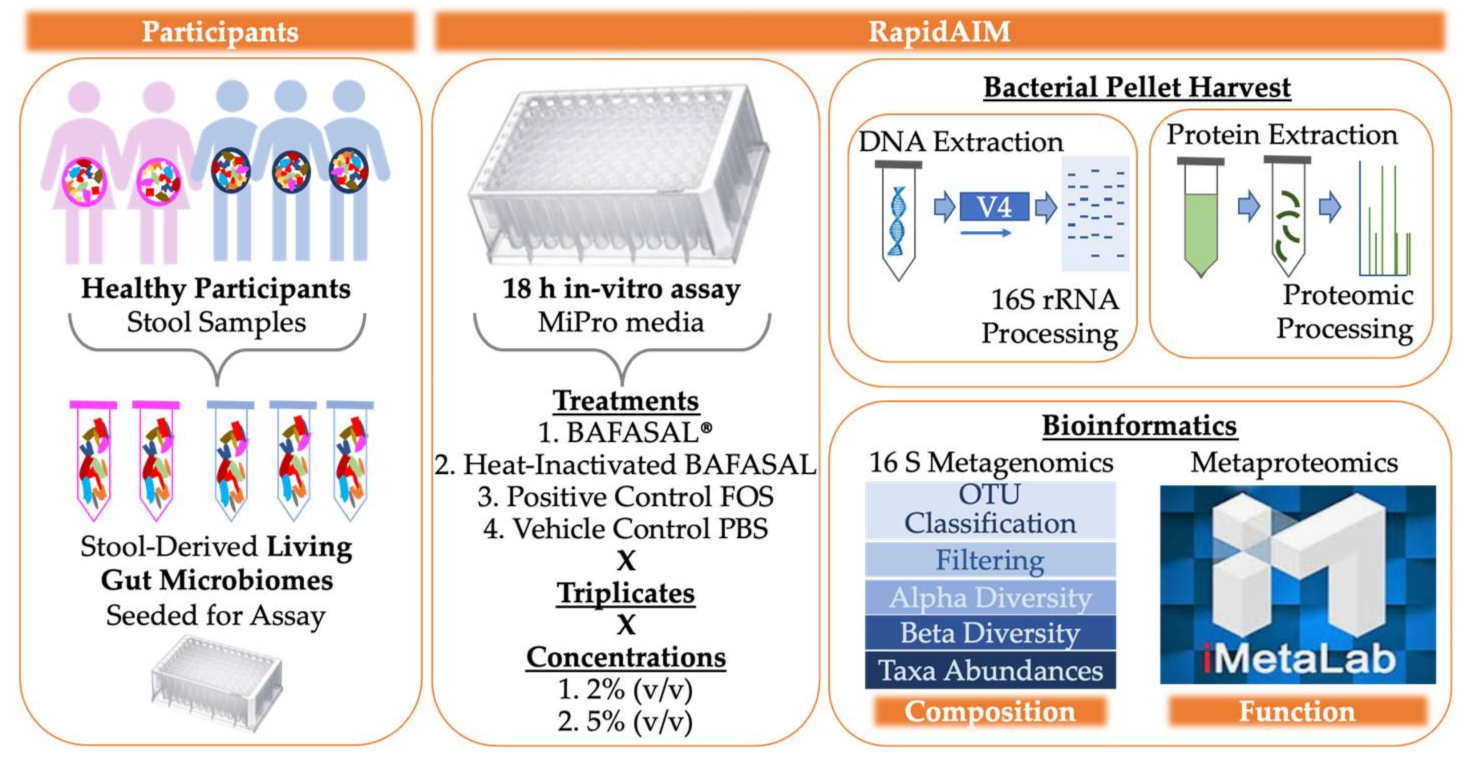{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. BAFASAL® Production
2.2. Stool Sample Preparation
2.3. Culturing of Microbiota and Treatments
2.4. Sample Processing
2.5. Metaproteomic Sample Processing and LS-MS/MS Analyses
2.6. Metaproteomic Data Analysis
2.7. Metagenomic DNA Extraction and 16S rDNA-V4 Amplicon Sequencing
2.8. Processing for 16S rRNA Sequencing and Analyses
3. Results
3.1. Experimental Set-Up
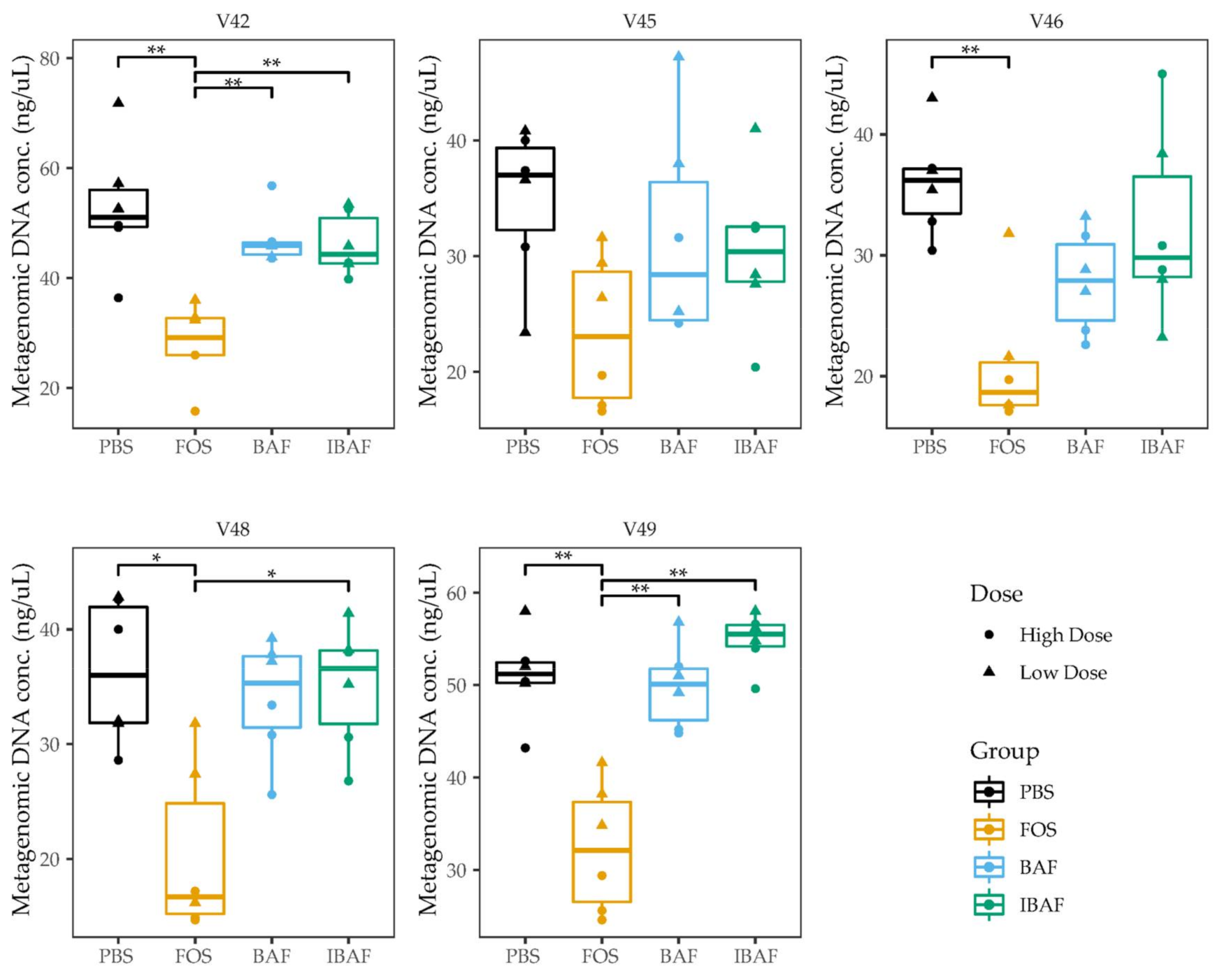
3.2. Microbial Biomass
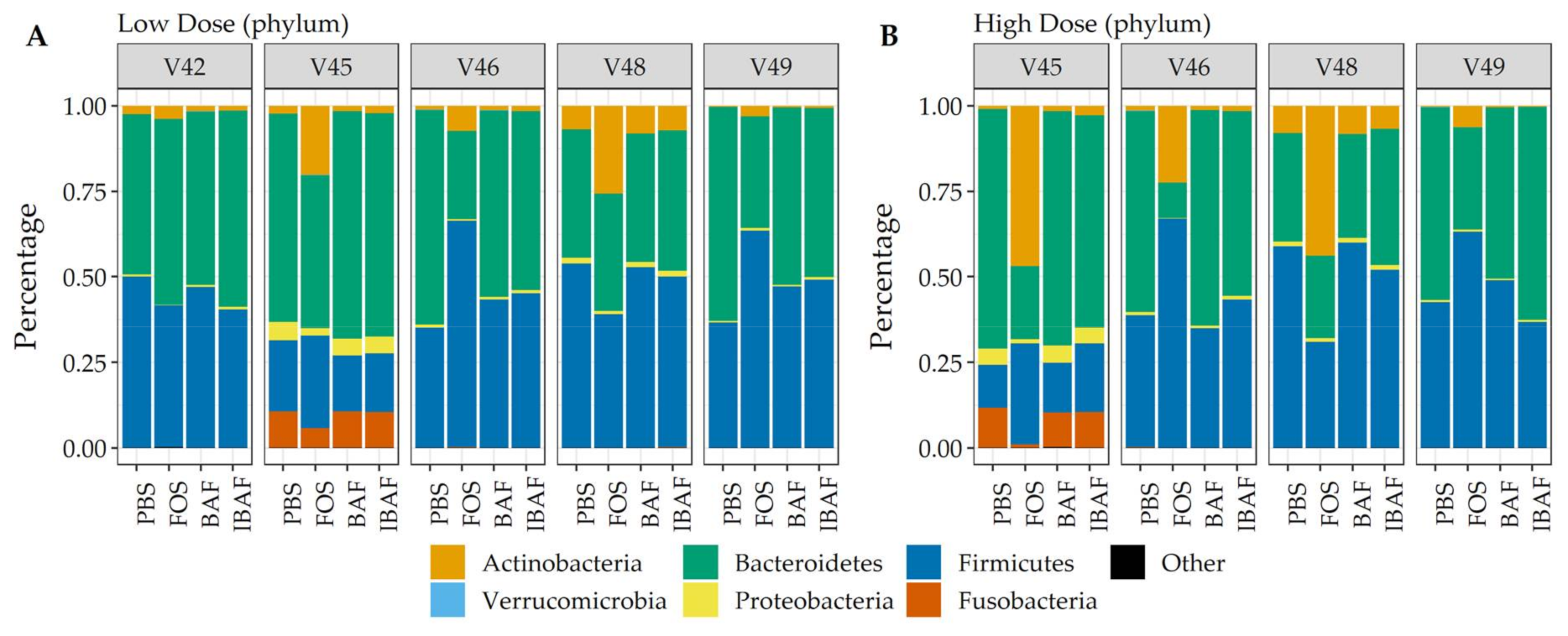
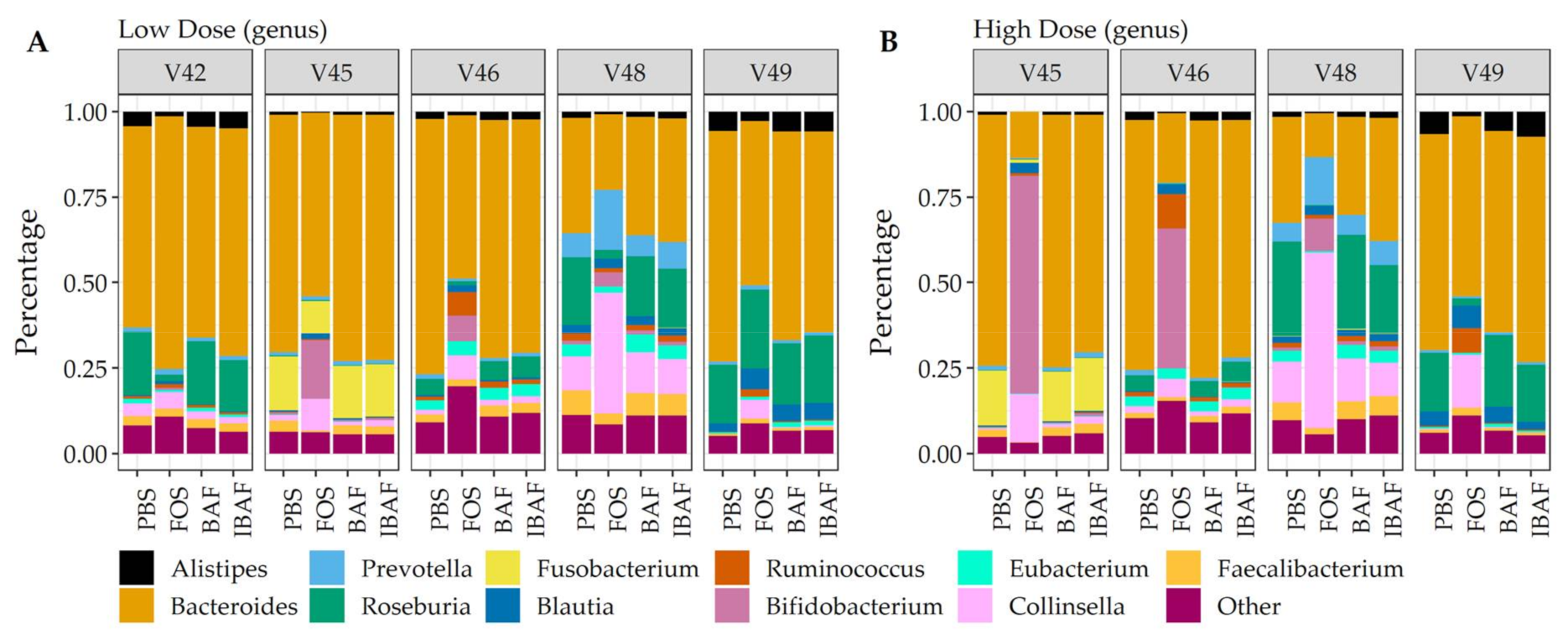
3.3. Compositional Analyses
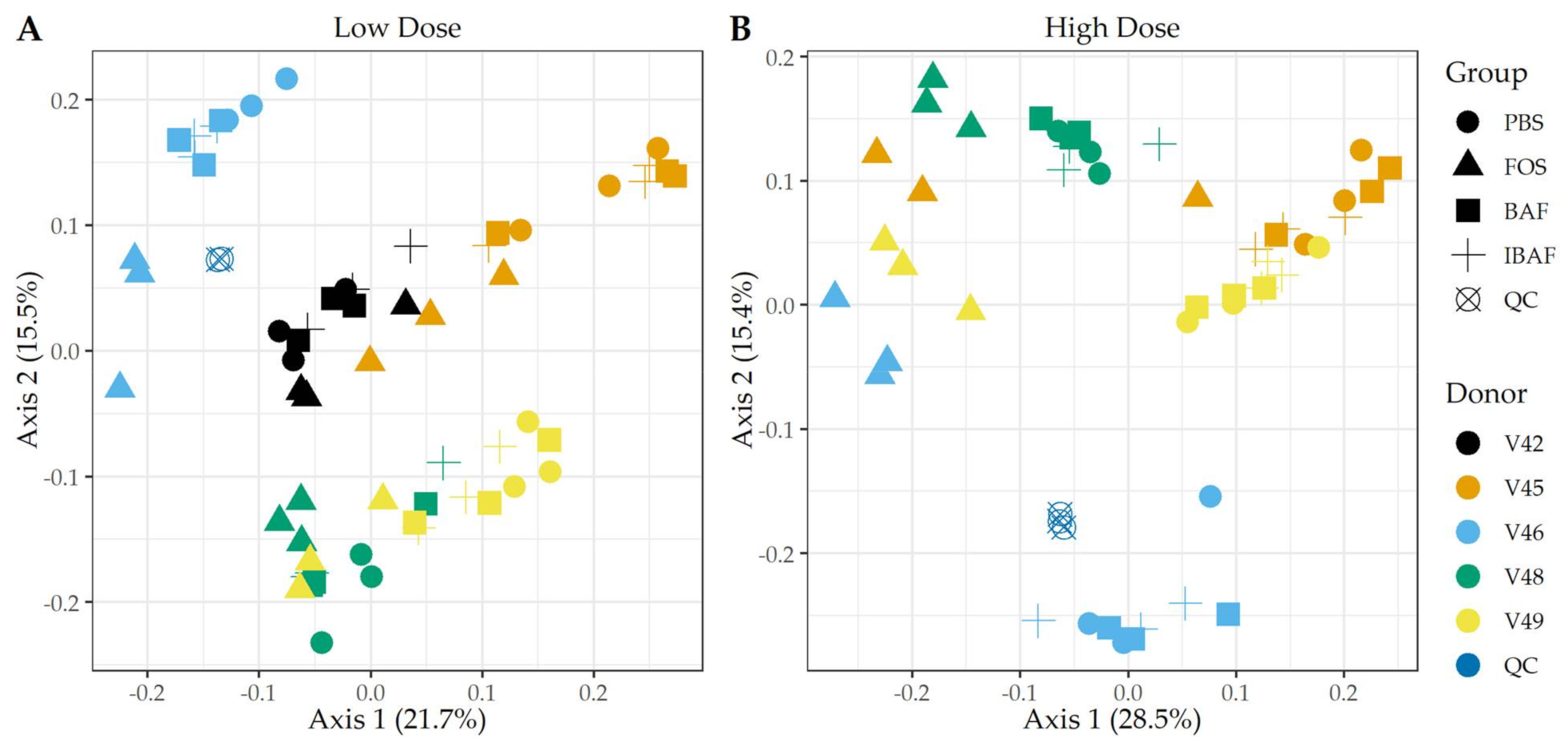
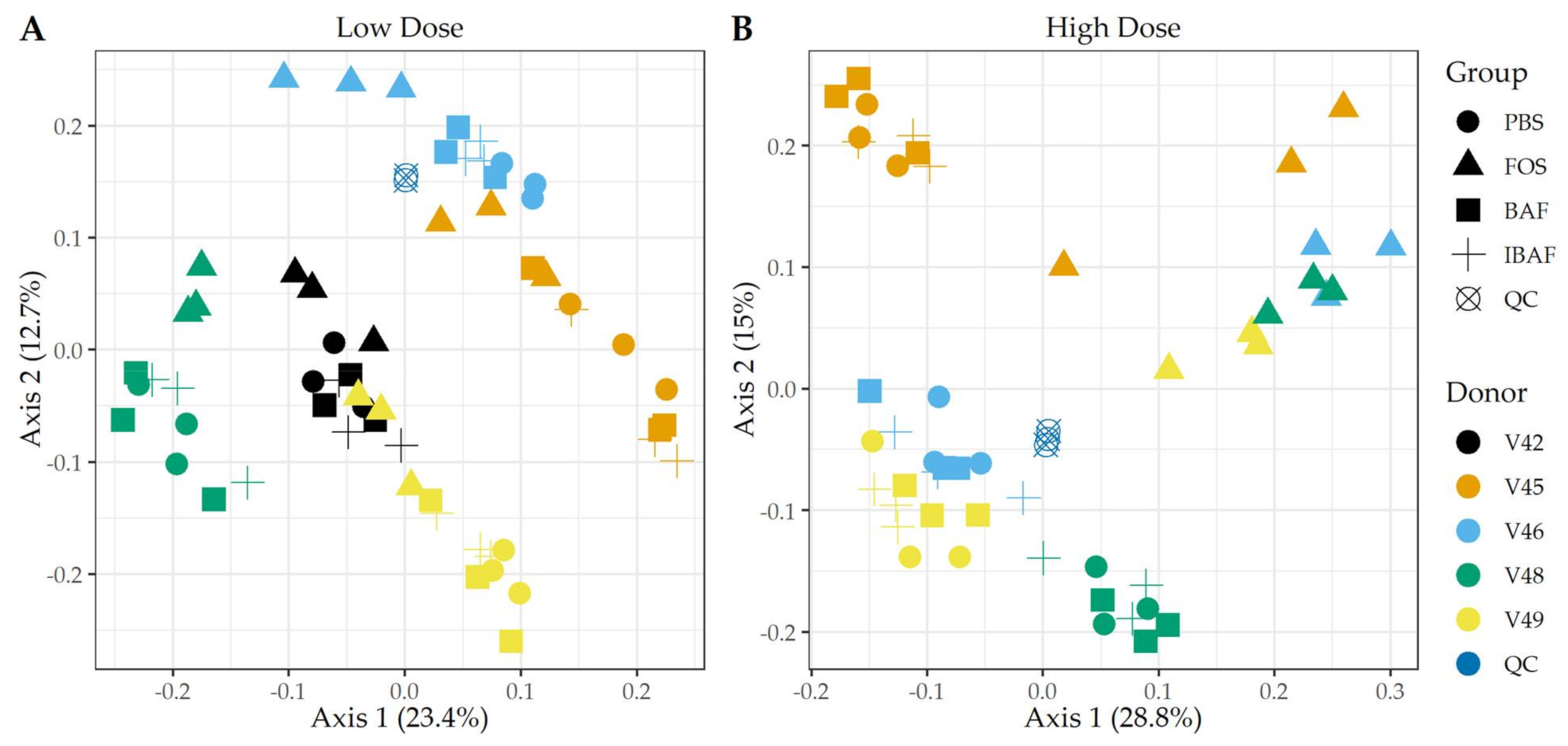
3.3.1. RapidAIM and Sequencing Library Reproducibility
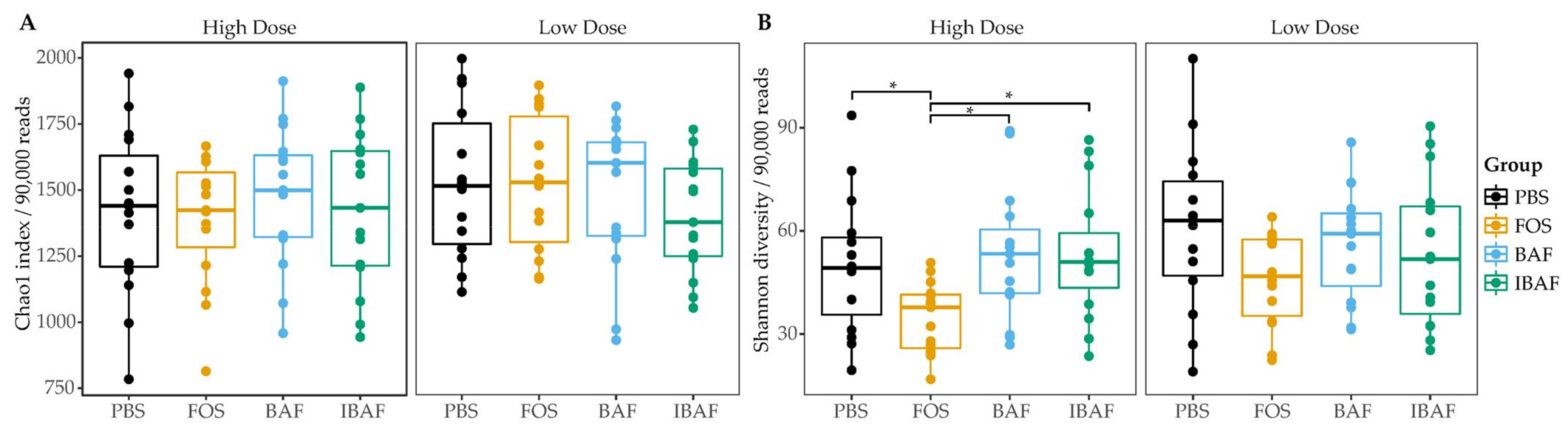
3.3.2. Microbial Diversity Analyses
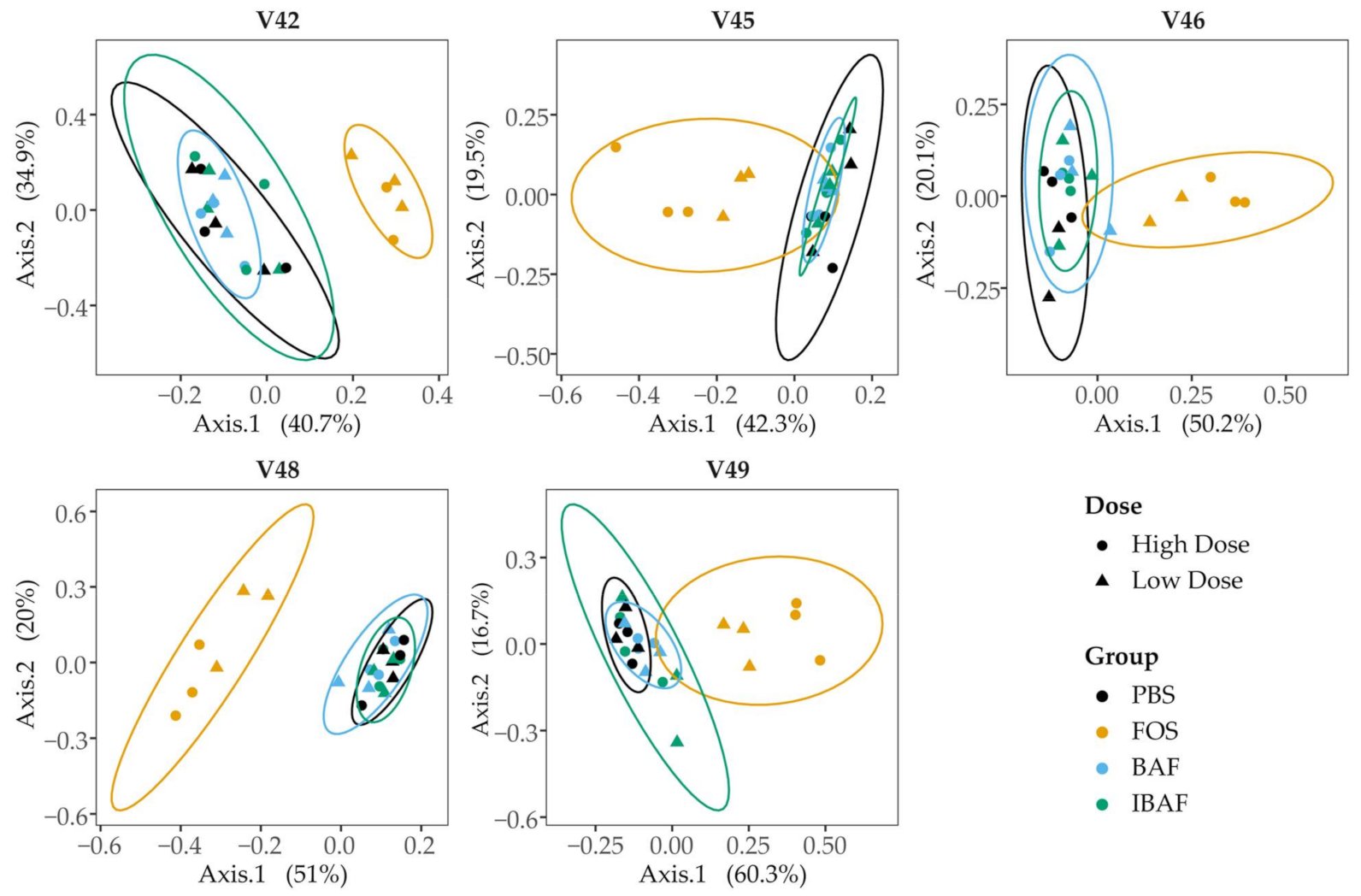
3.3.3. Compositional Differences
3.4. Functional Analyses
3.4.1. Metaproteomic Assessed Responses to BAFASAL®
3.4.2. Functional Responses to BAFASAL®
3.5. Metaproteomic Comparative Taxonomic Analyses
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- CDC Newsroom. 8 Zoonotic Diseases Shared Between Animals and People of Most Concern in the U.S. Centers for Disease Prevention and Control. 2019. Available online: https://www.cdc.gov/media/releases/2019/s0506-zoonotic-diseases-shared.html (accessed on 10 October 2020).
- World Health Organization. Food Safety. 2020. Available online: https://www.who.int/health-topics/food-safety/ (accessed on 10 October 2020).
- van Boeckel, T.P.; Pires, J.; Silvester, R.; Zhao, C.; Song, J.; Criscuolo, N.G.; Gilbert, M.; Bonhoeffer, S.; Laxminarayan, R. Global trends in antimicrobial resistance in animals in low- and middle-income countries. Science 2019, 365, eaaw1944. [Google Scholar] [CrossRef]
- Patel, S.J.; Wellington, M.; Shah, R.M.; Ferreira, M.J. Antibiotic Stewardship in Food-producsing Animals: Challenges, Progress, and Opportunities. Clin. Ther. 2020, 42, 1649–1658. [Google Scholar] [CrossRef]
- Stanton, I.C.; Bethel, A.; Leonard, A.F.; Gaze, W.H.; Garside, R. What is the research evidence for antibiotic resistance exposure and transmission to humans from the environment? A systematic map protocol. Environ. Evid. 2020, 9, 12. [Google Scholar] [CrossRef] [PubMed]
- Wallinga, D. Don’t ignore another disease threat. Nature 2020, 586, S64. [Google Scholar] [CrossRef]
- Salmond, G.P.; Fineran, P.C. A century of the phage: Past, present and future. Nat. Rev. Microbiol. 2015, 13, 777–786. [Google Scholar] [CrossRef] [PubMed]
- Keen, E.C. A century of phage research: Bacteriophages and the shaping of modern biology. Bioessays 2015, 37, 6–9. [Google Scholar] [CrossRef]
- Sarhan, W.A.; Azzazy, H.M. Phage approved in food, why not as a therapeutic? Expert. Rev. Anti-Infect. Ther. 2015, 13, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Aleshkin, A.V.; Rubalskii, E.O.; Volozhantsev, N.V.; Verevkin, V.V.; Svetoch, E.A.; Kiseleva, I.A.; Bochkareva, S.S.; Borisova, O.Y.; Popova, A.V.; Bogun, A.G.; et al. A small-scale experiment of using phage-based probiotic dietary supplement for prevention of E. coli traveler’s diarrhea. Bacteriophage 2015, 5, e1074329. [Google Scholar] [CrossRef][Green Version]
- Doss, J.; Culbertson, K.; Hahn, D.; Camacho, J.; Barekzi, N. A Review of Phage Therapy against Bacterial Pathogens of Aquatic and Terrestrial Organisms. Viruses 2017, 9, 50. [Google Scholar] [CrossRef]
- Wójcik, E.A.; Stańczyk, M.; Wojtasik, A.; Kowalska, J.D.; Nowakowska, M.; Łukasiak, M.; Bartnicka, M.; Kazimierczak, J.; Dastych, J. Comprehensive Evaluation of the Safety and Efficacy of BAFASAL(R) Bacteriophage Preparation for the Reduction of Salmonella in the Food Chain. Viruses 2020, 12, 742. [Google Scholar] [CrossRef]
- Sobel, J.; Hirshfeld, A.B.; Mctigue, K.; Burnett, C.L.; Altekruse, S.; Brenner, F.; Malcolm, G.; Mottice, S.L.; Swerdlow, C.R.N.L. The pandemic of Salmonella enteritidis phage type 4 reaches Utah: A complex investigation confirms the need for continuing rigorous control measures. Epidemiol. Infect. 2000, 125, 1–8. [Google Scholar] [CrossRef]
- Caenepeel, C.; Tabib, N.S.S.; Vieira-Silva, S.; Vermeire, S. Review article: How the intestinal microbiota may reflect disease activity and influence therapeutic outcome in inflammatory bowel disease. Aliment. Pharmacol. Ther. 2020, 52, 1453–1468. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.H.W.; Bäckhed, F.; Landmesser, U.; Hazen, S.L. Intestinal Microbiota in Cardiovascular Health and Disease: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2019, 73, 2089–2105. [Google Scholar] [CrossRef] [PubMed]
- Miaoa, S.; Kaib, M.; Jiec, W.; Guangxiand, W.; Changliangd, Z.; Qic, L.; Xiaofenga, B.; Huia, W. A Review of the Brain-Gut-Microbiome Axis and the Potential Role of Microbiota in Alzheimer’s Disease. J. Alzheimer’s Dis. 2020, 73, 849–865. [Google Scholar]
- Malkki, H. Parkinson disease: Could gut microbiota influence severity of Parkinson disease? Nat. Rev. Neurol. 2017, 13, 66–67. [Google Scholar] [CrossRef] [PubMed]
- Simpson, C.A.; Mu, A.; Haslam, N.; Schwartz, O.S.; Simmons, J.G. Feeling down? A systematic review of the gut microbiota in anxiety/depression and irritable bowel syndrome. J. Affect. Disord. 2020, 266, 429–446. [Google Scholar] [CrossRef]
- Divya Ganeshan, S.; Hosseinidoust, Z. Phage Therapy with a Focus on the Human Microbiota. Antibiotics 2019, 8, 131. [Google Scholar] [CrossRef]
- Li, L.; Ning, Z.; Zhang, X.; Mayne, J.; Cheng, K.; Stintzi, A.; Figeys, D. RapidAIM: A culture- and metaproteomics-based Rapid Assay of Individual Microbiome responses to drugs. Microbiome 2020, 8, 33. [Google Scholar] [CrossRef]
- Li, L.; Abou-Samra, E.; Ning, Z.; Zhang, X.; Mayne, J.; Wang, J.; Cheng, K.; Walker, K.; Stintzi, A.; Figeys, D. An in vitro model maintaining taxon-specific functional activities of the gut microbiome. Nat. Commun. 2019, 10, 4146. [Google Scholar] [CrossRef]
- Mao, B.; Li, D.; Zhao, J.; Liu, X.; Gu, Z.; Chen, Y.Q.; Zhang, H.; Chen, W. Metagenomic insights into the effects of fructo-oligosaccharides (FOS) on the composition of fecal microbiota in mice. J. Agric. Food Chem. 2015, 63, 856–863. [Google Scholar] [CrossRef]
- Gu, J.; Mao, B.; Cui, S.; Liu, X.; Zhang, H.; Zhao, J.; Chen, W. Metagenomic Insights into the Effects of Fructooligosaccharides (FOS) on the Composition of Luminal and Mucosal Microbiota in C57BL/6J Mice, Especially the Bifidobacterium Composition. Nutrients 2019, 11, 2431. [Google Scholar] [CrossRef]
- Tandon, D.; Haque, M.M.; Gote, M.; Jain, M.; Bhaduri, A.; Dubey, A.K.; Mande, S.S. A prospective randomized, double-blind, placebo-controlled, dose-response relationship study to investigate efficacy of fructo-oligosaccharides (FOS) on human gut microflora. Sci. Rep. 2019, 9, 5473. [Google Scholar] [CrossRef]
- Liu, F.; Li, P.; Chen, M.; Luo, Y.; Prabhakar, M.; Zheng, H.; He, Y.; Qi, Q.; Long, H.; Zhang, Y.; et al. Fructooligosaccharide (FOS) and Galactooligosaccharide (GOS) Increase Bifidobacterium but Reduce Butyrate Producing Bacteria with Adverse Glycemic Metabolism in healthy young population. Sci. Rep. 2017, 7, 11789. [Google Scholar] [CrossRef]
- Sivieri, K.; Morales, M.L.V.; Saad, S.M.I.; Adorno, M.A.T.; Sakamoto, I.K.; Rossi, E.A. Prebiotic effect of fructooligosaccharide in the simulator of the human intestinal microbial ecosystem (SHIME(R) model). J. Med. 2014, 17, 894–901. [Google Scholar] [CrossRef]
- Li, L.; Zhang, X.; Ning, Z.; Mayne, J.; Moore, J.I.; Butcher, J.; Chiang, C.; Mack, D.; Stintzi, A.; Figeys, D. Evaluating in Vitro Culture Medium of Gut Microbiome with Orthogonal Experimental Design and a Metaproteomics Approach. J. Proteome Res. 2018, 17, 154–163. [Google Scholar] [CrossRef]
- Zhang, X.; Ning, Z.; Mayne, J.; Moore, J.I.; Li, J.; Butcher, J.; Deeke, S.A.; Chen, R.; Chiang, C.; Wen, M.; et al. MetaPro-IQ: A universal metaproteomic approach to studying human and mouse gut microbiota. Microbiome 2016, 4, 31. [Google Scholar] [CrossRef]
- Cheng, K.; Ning, Z.; Zhang, X.; Li, L.; Liao, B.; Mayne, J.; Stintzi, A.; Figeys, D. MetaLab: An automated pipeline for metaproteomic data analysis. Microbiome 2017, 5, 157. [Google Scholar] [CrossRef]
- Butcher, J.; Unger, S.; Li, J.; Bando, N.; Romain, G.; Francis, J.; Mottawea, W.; Mack, D.; Stintzi, A.; O’Connor, D.L. Independent of Birth Mode or Gestational Age, Very-Low-Birth-Weight Infants Fed Their Mothers’ Milk Rapidly Develop Personalized Microbiotas Low in Bifidobacterium. J. Nutr. 2018, 148, 326–335. [Google Scholar] [CrossRef]
- Mottawea, W.; Chiang, C.; Mühlbauer, M.; Starr, A.E.; Butcher, J.; Abujamel, T.; Deeke, S.A.; Brandel, A.; Zhou, H.; Shokralla, S.; et al. Altered intestinal microbiota-host mitochondria crosstalk in new onset Crohn’s disease. Nat. Commun. 2016, 7, 13419. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Lin, H.; Jing, Y.; Wang, J. Broad-host-range Salmonella bacteriophage STP4-a and its potential application evaluation in poultry industry. Poult. Sci. 2020, 99, 3643–3654. [Google Scholar] [CrossRef]
- Ramirez, K.; Cazarez-Montoya, C.; Lopez-Moreno, H.S.; Campo, N.C. Bacteriophage cocktail for biocontrol of Escherichia coli O157:H7: Stability and potential allergenicity study. PLoS ONE 2018, 13, e0195023. [Google Scholar] [CrossRef]
- Chijiiwa, R.; Hosokawa, M.; Kogawa, M.; Nishikawa, Y.; Ide, K.; Sakanashi, C.; Takahashi, K.; Takeyama, H. Single-cell genomics of uncultured bacteria reveals dietary fiber responders in the mouse gut microbiota. Microbiome 2020, 8, 5. [Google Scholar] [CrossRef] [PubMed]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mayne, J.; Zhang, X.; Butcher, J.; Walker, K.; Ning, Z.; Wójcik, E.; Dastych, J.; Stintzi, A.; Figeys, D. Examining the Effects of an Anti-Salmonella Bacteriophage Preparation, BAFASAL®, on Ex-Vivo Human Gut Microbiome Composition and Function Using a Multi-Omics Approach. Viruses 2021, 13, 1734. https://doi.org/10.3390/v13091734
Mayne J, Zhang X, Butcher J, Walker K, Ning Z, Wójcik E, Dastych J, Stintzi A, Figeys D. Examining the Effects of an Anti-Salmonella Bacteriophage Preparation, BAFASAL®, on Ex-Vivo Human Gut Microbiome Composition and Function Using a Multi-Omics Approach. Viruses. 2021; 13(9):1734. https://doi.org/10.3390/v13091734
Chicago/Turabian StyleMayne, Janice, Xu Zhang, James Butcher, Krystal Walker, Zhibin Ning, Ewelina Wójcik, Jarosław Dastych, Alain Stintzi, and Daniel Figeys. 2021. "Examining the Effects of an Anti-Salmonella Bacteriophage Preparation, BAFASAL®, on Ex-Vivo Human Gut Microbiome Composition and Function Using a Multi-Omics Approach" Viruses 13, no. 9: 1734. https://doi.org/10.3390/v13091734
APA StyleMayne, J., Zhang, X., Butcher, J., Walker, K., Ning, Z., Wójcik, E., Dastych, J., Stintzi, A., & Figeys, D. (2021). Examining the Effects of an Anti-Salmonella Bacteriophage Preparation, BAFASAL®, on Ex-Vivo Human Gut Microbiome Composition and Function Using a Multi-Omics Approach. Viruses, 13(9), 1734. https://doi.org/10.3390/v13091734