
Immunity and Viral Infections: Modulating Antiviral Response via CRISPR–Cas Systems

 , , , , ,
, , , , ,  and
and 
Abstract
:1. Introduction
2. The Role of Innate Immunity in Restricting Viral Replication
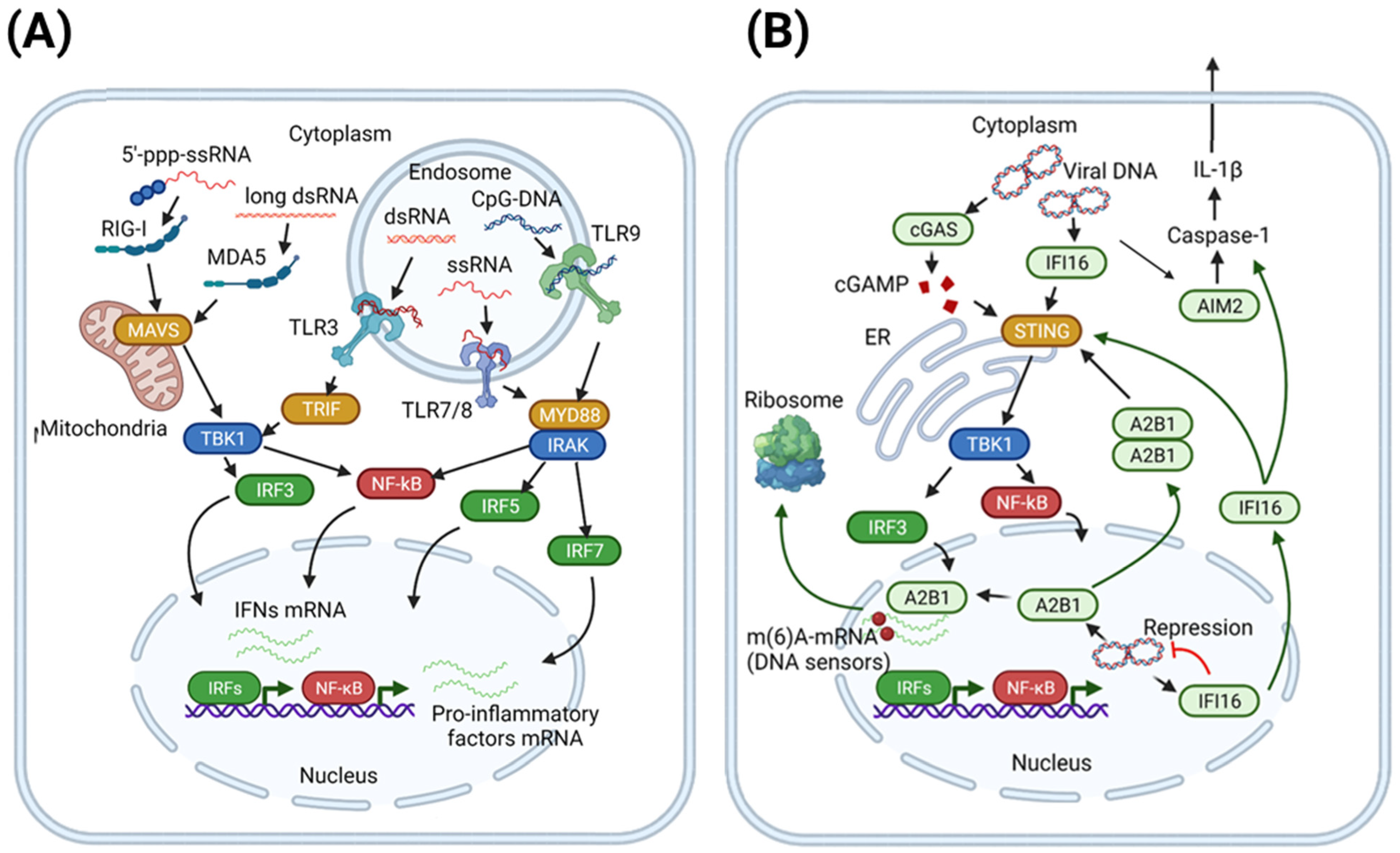
2.1. Pattern Recognition Receptors (PRR)
2.1.1. Viral Sensing by TLR and RLR: Concise Overview
2.1.2. Foreign DNA Recognition by Cytosolic and Nuclear DNA Sensors
2.2. Restriction of Viral Replication by Interferon-Stimulated Genes (ISGs)
2.2.1. Restriction of Viral Entry
2.2.2. Restriction of Protein Translation
2.2.3. Restriction of Viral Replication
2.2.4. Induction of Inflammatory Response
2.3. Host Factors Targeted by Viruses for Immune Evasion

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | Virus | Viral Protein | Mechanism |
|---|---|---|---|
| RIG-I | Influenza virus | NS1 | Direct interaction, TRIM and RIPLET binding |
| Coxsackievirus B3 | 2Apro | Cleavage | |
| Epstein-Barr virus | LMP1 | Proteasomal degradation | |
| SARS-CoV | Nucleocapsid protein | Binding TRIM25 | |
| Respiratory syncytial virus | NS1 | Binding TRIM25 | |
| Human papilloma virus | E6 | Proteasomal degradation of TRIM25 | |
| WNV | NS1 | Proteasomal degradation of TRIM25 | |
| HCV | NS3-4A | Cleavage of RIPLET | |
| DENV/WNV | NS3 | Binding 14-3-3ε | |
| TLR9 | Vaccinia virus | A46R | MyD88 adaptor binding |
| TLR3 | Human cytomegalovirus (HCMV) | US7 | Resultant ubiquitination of TLR3 |
| HIV | No data | Inhibits phosphorylation of IRF3, IRF7, STAT1, STAT3 | |
| MDA5 | Measles virus | V | Prevention of MDA5 dephosphorylation |
| SARS-CoV-2 | NSP8 | Impairment of K63-linked polyubiquitination | |
| DENV/WNV | NS3 | Binding 14-3-3ε | |
| IFI16 | HSV1 | ICP0/Ul41 | Ubiquitinoylation/inhibition of expression |
| CMV | pUL83 | Direct interaction | |
| cGAS | HSV1 | Ul41/Ul37/Vp22 | Inhibition of expression or enzymatic activity |
| HCMV | UL31/pp65 | Direct interaction/enzymatic activity inhibition | |
| Kaposi’s sarcoma-associated herpesvirus (KSHV) | ORF52/LANA | Direct interaction/enzymatic activity inhibition | |
| ZIKV | NS1 | Stabilization of caspase1 | |
| TRIF | HCV | NS3-4A | Cleavage |
| MAVS | DENV | NS2B3 | Binding of MFN1 and MFN2 proteins |
| Rhinoviruses | 2A and 3C | Cleavage | |
| STING | Adenovirus | E1A | Binding of STING |
| HPV18 | E7 | Binding of STING | |
| KSHV | vIRF1 | Prevention of STING interaction with downstream factors | |
| HBV | Pol | Prevention of STING polyubiquitylation | |
| HCV | NS4B | Inhibition of downstream signaling | |
| HIV | Vpx | Antagonizes cGAS/STING-triggered NF-κB signaling [94] | |
| DENV | NS2B3 | Inhibition of downstream signaling | |
| Yellow fever virus | NS4B | Inhibition of downstream signaling | |
| IKKε | Lassa fever virus | Nucleoprotein | Inhibition of autocatalytic activity |
| EBOV | Vp53 | Direct binding | |
| TBK1 | HIV | Vpr and Vif | Direct binding |
| EBOV | Vp53 | Direct binding | |
| Human herpesvirus 8 | vIRF1 | Interaction with CBP/p300 | |
| HSV1 | ICP34.5 | Binding of TBK1 | |
| KSHV | ORF45 | Alternative substrate for TBK1 | |
| IRF7 | Enterovirus 68 | 3Cpro | Cleavage |
| STAT2 | DENV | NS5 | Ubiquitination |
| IRF3 | SARS-CoV | ORF3b, ORF6 and N | IRF-3 inhibition |
3. Emerging CRISPR–Cas Tools
3.1. Modulation of Gene Transcription by CRISPR
3.2. CRISPR-Based DNA- and RNA-Editing Tools
4. CRISPR–Cas Systems for Modulating Antiviral Responses
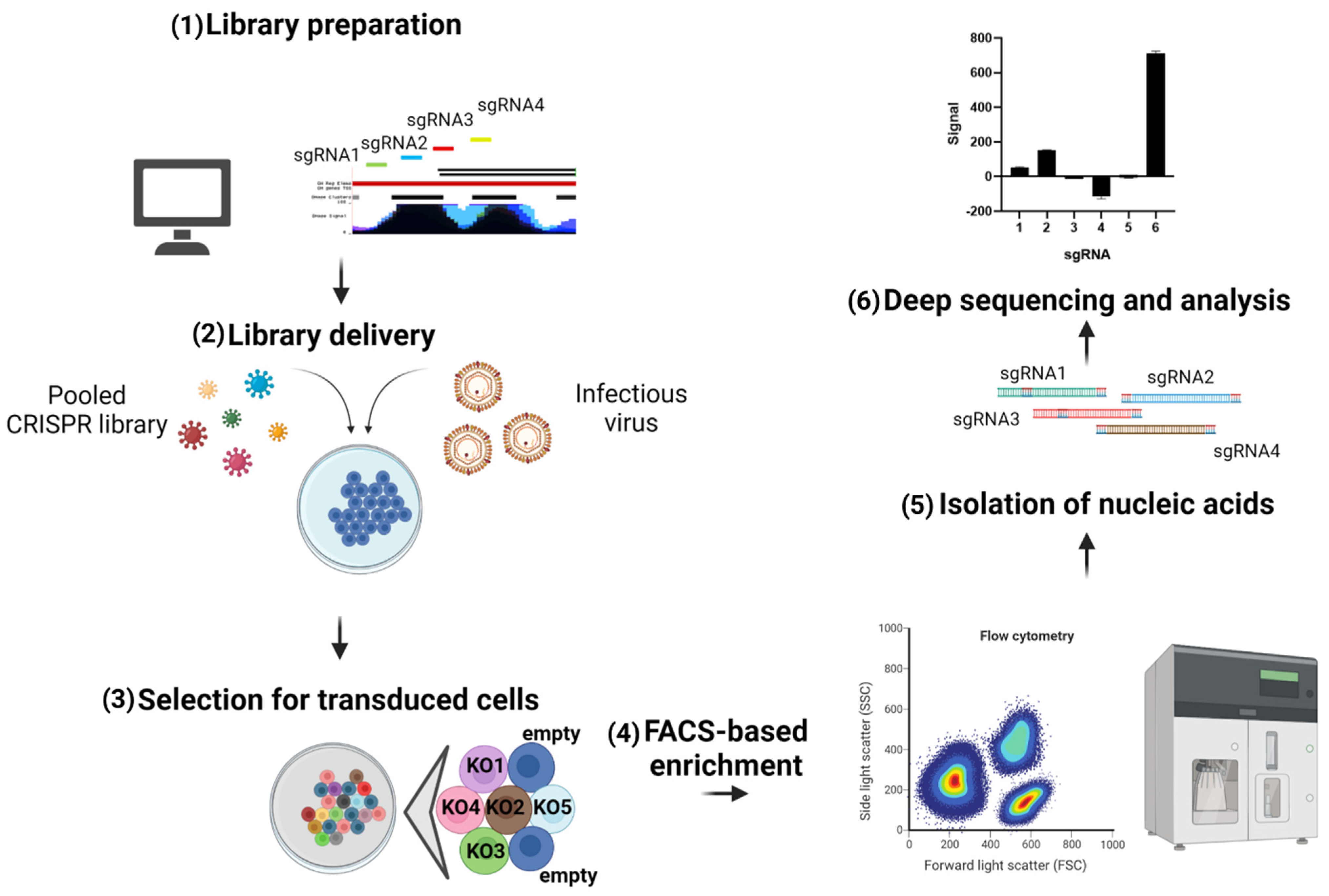
4.1. Screens to Identify Antiviral Genes
| CRISPR Screen Type | Model Virus | Most Potent Factors Identified | Ref. |
|---|---|---|---|
| CRISPR KO | Integrase-deficient MLV | NP220, HUSH complex | [185] |
| CRISPR KO | YFV and other flaviviruses | IFI6, HSPA5 | [77] |
| CRISPR KO | IAV | IFNAR2, TYK2, JAK1, IFNAR1, IRF9, RTF2 | [189] |
| CRISPR KO (ISGs only) | HIV | MxB, TRIM5α, IFITM1, tetherin, SAMD9L, UBE2L6 | [187] |
| CRISPR KO (ISGs only) | VSVg-pseudotyped viral-like particles | IFITM1, IFITM3 | [188] |
| CRISPRa | MNoV | TRIM7, GBP2, MX1 | [190] |
| CRISPRa | ZIKV | IFI6, IFNL2, ISG20, HELZ2 | [191] |
4.2. Investigation of Fundamental Mechanisms of Antiviral Immunity
4.3. Investigation of Signaling Pathways Involved in Antiviral Response
4.4. Regulation of Antiviral Immunity
4.5. Investigating Viral Immune Evasion Mechanisms
4.6. ISG Antiviral Action
| Purpose of Investigation | Model Virus | CRISPR Target Genes | Signaling Pathways | Ref. |
|---|---|---|---|---|
| Signaling pathways | CMV | cGAS, IFI16, STING | STING pathway | [198] |
| CMV, MVA, VSV | cGAS, IFI16, STING | STING pathway | [195] | |
| HSV-2 | STING | STING pathway | [197] | |
| CMV | STING | cGAS/STING | [196] | |
| WNV, IAV, VACV, SINV | OAS1, OAS2, OAS3, RNAse L | OAS signaling | [200] | |
| SeV, SVV | RIG-I | RIG-I/MAVS | [201] | |
| VSV, EMCV | IFNAR1, IFNAR2, STAT1, STAT2, IRF1, STAT3 | IFN receptor signaling | [204] | |
| IAV | IFNAR2 | IFN receptor signaling | [203] | |
| HCV | STAT1, STAT2, IRF9, STAT3, STAT6 | IFN receptor signaling | [205] | |
| VHSV | IRF9 | IFN receptor signaling | [206] | |
| CMV | IRF3 | ISG induction | [207] | |
| Signaling pathways, ISG antiviral activity | HSV-1 | IFI16, STING, cGAS, TRIM19 | STING pathway, | [199] |
| caspase-3 pathway, | ||||
| effector ISGs | ||||
| Signaling pathwaysViral evasion | CMV | ASC, CASP1, AIM2, IFI16, NLRP3, cGAS, STING, IL-1β, GSDMD | STING pathway, | [226] |
| IL-1β/caspase-1 pathway | ||||
| ZIKV | RIG-I, MDA5, IFNAR | RIG-I/MDA5/MAVS | [202] | |
| IFN receptor signaling | ||||
| Immunity regulation | VSV, HSV-1, SeV | DUB family enzymes | RIG-I/MAVS | [209] |
| STING pathway | ||||
| SeV | TTLL12 | RIG-I/MAVS | [212] | |
| WNV, SINV-EEEV, VSV, HSV-1 | Banf, cGAS, STING, Irf3, and other STING pathway genes | STING pathway | [215] | |
| HSV-1 | IFI16, STING | STING pathway | [210] | |
| RV, CHIKV, IAV, VSV | STAG2, STAT1, STING | cGAS/STING | [216] | |
| HIV-1 | TREX1, cGAS | cGAS/STING | [217] | |
| HSV-1 | TRIM41 | cGAS/STING | [211] | |
| VSV | 4E-BP1 | TLR signaling IFN receptor signaling | [213] | |
| Viral evasion | ZIKV, DENV, SINV | OAS3, RNAseL, STAT1, STAT2, MAVS | OAS signaling | [218] |
| IFN receptor signaling | ||||
| RIG-I/MAVS | ||||
| DENV | MyD88 | TLR signaling | [219] | |
| HIV-1 | USP18 | IFN receptor signaling | [220] | |
| CMV | ISG15, IKKα, IKKβ | NF-κB pathway | [222] | |
| HIV-1 | miR-146a precursor sequence | NF-κB pathway | [223] | |
| HIV-2 | BST-2 | Effector ISGs | [224] | |
| HIV-1 | USP18 | Effector ISGs | [221] | |
| IAV | p53 | Effector ISGs | [239] | |
| RRV, KSHV | TRIM19, SP100, DAXX, ATRX | Effector ISGs | [225] | |
| ISG antiviral activityViral evasion | HIV-1 | STAT1, MxB and 53 other ISGs | ISG induction | [227] |
| ISG antiviral activity | HIV-1 | SAMHD1 | Effector ISGs | [228] |
| RV | eIF4A, eIF4E, eIF4G | Effector ISGs | [229] | |
| HSV-1 | LXRα, LXRβ, CH25H, SULT2B1 | Effector ISGs | [230] | |
| SARS-CoV-2 | CH25H | Effector ISGs | [233] | |
| VHSV | Mx1, ISG15 | Effector ISGs | [231] | |
| HBV | APOBEC3A, APOBEC3B | Effector ISGs | [232] | |
| HBV | MOV10 | Effector ISGs | [240] | |
| HPV16 | SAMHD1 | Effector ISGs | [241] | |
| HIV-1 | SERINC5 (KO/iHA Knock-in) | Effector ISGs | [234] | |
| SHIV | IFITM1, IFITM3, IFITM3A | Effector ISGs | [242] | |
| IAV, ZIKV, VEEV, YFV, ONNV, WNV, VSV, DENV | IFITM1, IFITM2, IFITM3 | Effector ISGs | [243] | |
| VACV mutant | SAMD9, WDR6 | Effector ISGs | [244] | |
| Poxviruses | mSAMD9L, hSAMD9L | Effector ISGs | [245] | |
| HSV-1 | IFI16 | Effector ISGs | [246] | |
| ZIKV | Viperin | Effector ISGs | [247] | |
| Drug antiviral mechanisms | CHIKV, VEEV, SINV | IRF3, STAT1, MAVS, STING | STING pathway | [237] |
| JAK/STAT | ||||
| RIG-I/MAVS | ||||
| ZIKV | Viperin | TLR signaling | [238] | |
| Effector ISGs |
4.7. Developing Antiviral Approaches
5. Modulating Epitranscriptomics in Viral Infections
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| HIV | Human immunodeficiency virus |
| HBV | Hepatitis B virus |
| HCV | Hepatitis C virus |
| HDV | Hepatitis D virus |
| NK | Natural killer cells |
| IFN | Interferon |
| ISGs | Interferon-stimulated genes |
| JAK/STAT | Janus kinase/signal transducers and activators of transcription |
| DSB | Double-strand breaks |
| PAMP | Pathogen-associated molecular patterns |
| PRR | Pattern recognition receptors |
| TLR | Toll-like receptors |
| ssRNA | Single-stranded RNA |
| dsRNA | Double-stranded RNA |
| RIG-I | Retinoic acid-inducible gene I |
| cGAMP | Cyclic GMP-AMP |
| cGAS | Cyclic GMP-AMP synthase |
| AIM2 | Absent in melanoma 2 |
| TIR | Toll/IL-1 receptor |
| TRIF | TIR domain-containing adaptor-inducer interferon-β |
| MAVS | Mitochondrial antiviral-signaling protein |
| ssDNA | Single-stranded DNA |
| STING | Stimulator of interferon genes |
| ALRs | AIM2-like receptors |
| ASC | Apoptosis-associated speck-like protein containing a CARD |
| 25HC | 25-hydrocholesterol |
| VSV | Vesicular stomatitis virus |
| HSV | Herpes simplex virus |
| EBOV | Ebola virus |
| ZIKV | Zika virus |
| NCOA7 | Nuclear receptor coactivator 7 |
| IAV | Influenza A virus |
| DENV | Dengue virus |
| WNV | West Nile virus |
| PKR | Protein kinase R |
| SLFN11 | Schlafen 11 |
| APOBEC | Apolipoprotein B mRNA-editing enzyme catalytic polypeptide-like |
| IFI16 | Gamma-interferon-inducible protein 16 |
| IFITM1 | Interferon-induced transmembrane protein 1 |
| OAS | Oligoadenylate synthetase |
| VACV | Vaccinia virus |
| HCMV | Human cytomegalovirus |
| KSHV | Kaposi’s sarcoma-associated herpesvirus |
| MV | Measles virus |
| LMP1 | Latent membrane protein 1 |
| YFV | Yellow fever virus |
| HDAC | Histone deacetylase |
| SINV | Sin Nombre virus |
| ONNV | o’nyong’nyong virus |
| SINV | Sindbis virus |
| SVV | Seneca Valley virus |
| VEEV | Venezuelan encephalitis virus |
| CHIKV | Chikungunya virus |
| RV | Rotavirus |
| MVA | Modified vaccinia virus Ankara |
| CMV | Cytomegalovirus |
| SINV-EEEV | chimeric eastern equine encephalitis virus |
| SeV | Sendai virus |
| m6A | methyl-6-adenosine |
| TTLL12 | Tubulin Tyrosine Ligase Like 12 |
References
- Sayed, A.; Peng, B. Pandemics and income inequality: A historical review. SN Bus. Econ. 2021, 1, 1–17. [Google Scholar] [CrossRef]
- World Health Organization. Global Hepatitis Report; World Health Organization: Geneva, Switzerland, 2017. [Google Scholar]
- GBD 2016 Mortality Collaborators. Global, regional, and national under-5 mortality, adult mortality, age-specific mortality, and life expectancy, 1970–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet 2017, 390, 1084–1150. [Google Scholar] [CrossRef] [Green Version]
- Malmgaard, L. Induction and regulation of IFNs during viral infections. J. Interf. Cytokine Res. 2004, 24, 439–454. [Google Scholar] [CrossRef]
- Biron, C.A.; Nguyen, K.B.; Pien, G.C.; Cousens, L.P.; Salazar-Mather, T.P. Natural killer cells in antiviral defense: Function and regulation by innate cytokines. Annu. Rev. Immunol. 1999, 17, 189–220. [Google Scholar] [CrossRef]
- Uzhachenko, R.V.; Shanker, A. CD8+ T lymphocyte and NK cell network: Circuitry in the cytotoxic domain of immunity. Front. Immunol. 2019, 10, 1906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waggoner, S.N.; Reighard, S.D.; Gyurova, I.E.; Cranert, S.A.; Mahl, S.E.; Karmele, E.P.; McNally, J.P.; Moran, M.T.; Brooks, T.R.; Yaqoob, F.; et al. Roles of natural killer cells in antiviral immunity. Curr. Opin. Virol. 2016, 16, 15–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen recognition and innate immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Platanias, L.C. Mechanisms of type-I-and type-II-interferon-mediated signalling. Nat. Rev. Immunol. 2005, 5, 375–386. [Google Scholar] [CrossRef] [PubMed]
- Antonelli, G.; Scagnolari, C.; Moschella, F.; Proietti, E. Twenty-five years of type I interferon-based treatment: A critical analysis of its therapeutic use. Cytokine Growth Factor Rev. 2015, 26, 121–131. [Google Scholar] [CrossRef]
- Park, A.; Iwasaki, A. Type I and type III interferons–induction, signaling, evasion, and application to combat COVID-19. Cell Host Microbe 2020, 27, 870–878. [Google Scholar] [CrossRef] [PubMed]
- Isorce, N.; Lucifora, J.; Zoulim, F.; Durantel, D. Immune-modulators to combat hepatitis B virus infection: From IFN- D to novel investigational immunotherapeutic strategies. Antiviral Res. 2015, 122, 69–81. [Google Scholar] [CrossRef]
- Durantel, D.; Zoulim, F. Review New antiviral targets for innovative treatment concepts for hepatitis B virus and hepatitis delta virus. J. Hepatol. 2016, 64, S117–S131. [Google Scholar] [CrossRef] [PubMed]
- Martinsen, J.T.; Gunst, J.D.; Højen, J.F.; Tolstrup, M.; Søgaard, O.S. The use of Toll-like receptor agonists in HIV-1 cure strategies. Front. Immunol. 2020, 11, 1112. [Google Scholar] [CrossRef] [PubMed]
- Fanning, G.C.; Zoulim, F.; Hou, J.; Bertoletti, A. Therapeutic strategies for hepatitis B virus infection: Towards a cure. Nat. Rev. Drug Discov. 2019, 18, 827–844. [Google Scholar] [CrossRef] [PubMed]
- Zhen, A.; Carrillo, M.A.; Kitchen, S.G. Chimeric antigen receptor engineered stem cells: A novel HIV therapy. Immunotherapy 2017, 9, 401–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nyakatura, E.K.; Soare, A.Y.; Lai, J.R. Bispecific antibodies for viral immunotherapy. Hum. Vaccin. Immunother. 2017, 13, 836–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finlay, B.B.; McFadden, G. Anti-immunology: Evasion of the host immune system by bacterial and viral pathogens. Cell 2006, 124, 767–782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowie, A.G.; Unterholzner, L. Viral evasion and subversion of pattern-recognition receptor signalling. Nat. Rev. Immunol. 2008, 8, 911–922. [Google Scholar] [CrossRef]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A Programmable Dual-RNA–Guided DNA Endonuclease in Adaptive Bacterial Immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef]
- Brezgin, S.; Kostyusheva, A.; Kostyushev, D.; Chulanov, V. Dead Cas Systems: Types, Principles, and Applications. Int. J. Mol. Sci. 2019, 20, 6041. [Google Scholar] [CrossRef] [Green Version]
- Barrangou, R.; Doudna, J.A. Applications of CRISPR technologies in research and beyond. Nat. Biotechnol. 2016, 34, 933–941. [Google Scholar] [CrossRef]
- Thompson, M.R.; Kaminski, J.J.; Kurt-Jones, E.A.; Fitzgerald, K.A. Pattern recognition receptors and the innate immune response to viral infection. Viruses 2011, 3, 920–940. [Google Scholar] [CrossRef] [Green Version]
- Feng, J.; Wickenhagen, A.; Turnbull, M.L.; Rezelj, V.V.; Kreher, F.; Tilston-Lunel, N.L.; Slack, G.S.; Brennan, B.; Koudriakova, E.; Shaw, A.E.; et al. Interferon-Stimulated Gene (ISG)-Expression Screening Reveals the Specific Antibunyaviral Activity of ISG20. J. Virol. 2018, 92, e02140-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burleigh, K.; Maltbaek, J.H.; Cambier, S.; Green, R.; Gale, M., Jr.; James, R.C.; Stetson, D.B. Human DNA-PK activates a STING-independent DNA sensing pathway. Sci. Immunol. 2020, 5, eaba4219. [Google Scholar] [CrossRef] [Green Version]
- Akira, S.; Takeda, K. Toll-like receptor signalling. Nat. Rev. Immunol. 2004, 4, 499–511. [Google Scholar] [CrossRef] [PubMed]
- Barral, P.M.; Sarkar, D.; Su, Z.-Z.; Barber, G.N.; DeSalle, R.; Racanello, V.R.; Fisher, P.B. Functions of the cytoplasmic RNA sensors RIG-I and MDA-5: Key regulators of innate immunity. Pharmacol. Ther. 2009, 124, 219–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burckstummer, T.; Baumann, C.; Blüml, S.; Dixit, E.; Dürnberger, G.; Jahn, H.; Planyavsky, M.; Bilban, M.; Colinge, J.; Bennet, K.L.; et al. An orthogonal proteomic-genomic screen identifies AIM2 as a cytoplasmic DNA sensor for the inflammasome. Nat. Immunol. 2009, 10, 266–272. [Google Scholar] [CrossRef] [PubMed]
- Yoneyama, M.; Kikuchi, M.; Natsukawa, T.; Shinobu, N.; Imaizumi, T.; Miyagushi, M.; Taira, K.; Akira, S.; Fujita, T. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat. Immunol. 2004, 5, 730–737. [Google Scholar] [CrossRef]
- Sun, L.; Wu, J.; Du, F.; Chen, X.; Chen, Z.J. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 2013, 339, 786–791. [Google Scholar] [CrossRef] [Green Version]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef]
- Sato, S.; Sugiyama, M.; Yamamoto, M.; Watanabe, Y.; Kawai, T.; Takeda, K.; Akira, S. Toll/IL-1 receptor domain-containing adaptor inducing IFN-beta (TRIF) associates with TNF receptor-associated factor 6 and TANK-binding kinase 1, and activates two distinct transcription factors, NF-kappa B and IFN-regulatory factor-3, in the Toll-like receptor signaling. J. Immunol. 2003, 171, 4304–4310. [Google Scholar]
- Lin, S.-C.; Lo, Y.-C.; Wu, H. Helical assembly in the MyD88-IRAK4-IRAK2 complex in TLR/IL-1R signalling. Nature 2010, 465, 885–890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawai, T.; Sato, S.; Ishii, K.J.; Coban, C.; Hemmi, H.; Yamamoto, M.; Terai, K.; Matsuda, M.; Inoue, J.-i.; Uematsu, S.; et al. Interferon-alpha induction through Toll-like receptors involves a direct interaction of IRF7 with MyD88 and TRAF6. Nat. Immunol. 2004, 5, 1061–1068. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Takahashi, K.; Sato, S.; Coban, C.; Kumar, H.; Kato, H.; Ishii, K.J.; Takeuchi, O.; Akira, S. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat. Immunol. 2005, 6, 981–988. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y.; Moore, C.B.; Leisman, R.M.; O’Connor, B.P.; Berhstralh, D.T.; Chen, Z.J.; Pickles, R.J.; Ting, J.P.-Y. MAVS-mediated apoptosis and its inhibition by viral proteins. PLoS ONE 2009, 4, e5466. [Google Scholar] [CrossRef]
- Hornung, V.; Ellegast, J.; Kim, S.; Brzózka, K.; Jung, A.; Kato, H.; Poeck, H.; Akira, S.; Conzelmann, K.-K.; Schlee, M.; et al. 5′-Triphosphate RNA is the ligand for RIG-I. Science 2006, 314, 994–997. [Google Scholar] [CrossRef] [Green Version]
- Jiang, X.; Kinch, L.N.; Brautigam, C.A.; Chen, X.; Du, F.; Grishin, N.V.; Chen, Z.J. Ubiquitin-induced oligomerization of the RNA sensors RIG-I and MDA5 activates antiviral innate immune response. Immunity 2012, 36, 959–973. [Google Scholar] [CrossRef] [Green Version]
- Hou, F.; Sun, L.; Zheng, H.; Skaug, B.; Jiang, Q.-X.; Chen, Z.J. MAVS forms functional prion-like aggregates to activate and propagate antiviral innate immune response. Cell 2011, 146, 448–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Chen, J.; Cai, X.; Wu, J.; Chen, X.; Wu, Y.-T.; Sun, L.; Chen, Z.J. MAVS recruits multiple ubiquitin E3 ligases to activate antiviral signaling cascades. Elife 2013, 2, e00785. [Google Scholar] [CrossRef]
- Kato, H.; Takeuchi, O.; Sato, S.; Yoneyama, M.; Yamamoto, M.; Matsui, K.; Uematsu, S.; Jung, A.; Kawai, T.; Ishii, K.J.; et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 2006, 441, 101–105. [Google Scholar] [CrossRef]
- Saito, T.; Owen, D.M.; Jiang, F.; Marcotrigiano, J.; Gale, M., Jr. Innate immunity induced by composition-dependent RIG-I recognition of hepatitis C virus RNA. Nature 2008, 454, 523–527. [Google Scholar] [CrossRef] [Green Version]
- Sato, S.; Li, K.; Kameyama, T.; Hayashi, T.; Ishida, Y.; Murakami, S.; Watanabe, T.; Lijima, S.; Sakurai, Y.; Watashi, K.; et al. The RNA Sensor RIG-I Dually Functions as an Innate Sensor and Direct Antiviral Factor for Hepatitis B Virus Article The RNA Sensor RIG-I Dually Functions as an Innate Sensor and Direct Antiviral Factor for Hepatitis B Virus. Immunity 2015, 42, 123–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, Z.; Zhang, J.; Han, Q.; Su, C.; Qu, J.; Xu, D.; Zhang, C.; Tian, Z. Hepatitis B virus inhibits intrinsic RIG-I and RIG-G immune signaling via inducing miR146a. Sci. Rep. 2016, 6, 26150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Wang, X.; Li, J.; Zhou, Y.; Ho, W. RIG-I activation inhibits HIV replication in macrophages. J. Leukoc. Biol. 2013, 94, 337–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solis, M.; Nakhaei, P.; Jalalirad, M.; Lacoste, J.; Douville, R.; Arguello, M.; Zhao, T.; Laughrea, M.; Wainberg, M.A.; Hiscott, J. RIG-I-mediated antiviral signaling is inhibited in HIV-1 infection by a protease-mediated sequestration of RIG-I. J. Virol. 2011, 85, 1224–1236. [Google Scholar] [CrossRef] [Green Version]
- Civril, F.; Deimling, T.; de Oliviera Mann, C.C.; Ablasser, A.; Moldt, M.; Witte, G.; Hornung, V.; Hopfner, K.-P. Structural mechanism of cytosolic DNA sensing by cGAS. Nature 2013, 498, 332–337. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Wu, J.; Du, F.; Xu, H.; Sun, L.; Chen, Z.; Brautigam, C.A.; Zhang, X.; Chen, Z.J. The cytosolic DNA sensor cGAS forms an oligomeric complex with DNA and undergoes switch-like conformational changes in the activation loop. Cell Rep. 2014, 6, 421–430. [Google Scholar] [CrossRef] [Green Version]
- Gao, P.; Ascano, M.; Zillinger, T.; Wang, W.; Dai, P.; Serganov, A.A.; Gaffney, B.L.; Shuman, S.; Jones, R.A.; Deng, L.; et al. Structure-function analysis of STING activation by c[G(2′,5′)pA(3′,5′)p] and targeting by antiviral DMXAA. Cell 2013, 154, 748–762. [Google Scholar] [CrossRef] [Green Version]
- Ishikawa, H.; Barber, G.N. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 2008, 455, 674–678. [Google Scholar] [CrossRef]
- Tschopp, J.; Martinon, F.; Burns, K. NALPs: A novel protein family involved in inflammation. Nat. Rev. Mol. Cell Biol. 2003, 4, 95–104. [Google Scholar] [CrossRef]
- Fernandes-Alnemri, T.; Yu, J.-W.; Datta, P.; Wu, J.; Alnemri, E.S. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature 2009, 458, 509–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamkanfi, M.; Dixit, V.M. Mechanisms and functions of inflammasomes. Cell 2014, 157, 1013–1022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, F.; Yuan, Y.; Ma, F. Function and regulation of nuclear DNA sensors during viral infection and tumorigenesis. Front. Immunol. 2020, 11, 624556. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.-Y.; Aliyari, R.; Chikere, K.; Li, G.; Marsden, M.D.; Smith, J.K.; Pernet, O.; Guo, H.; Nusbaum, R.; Zack, J.A.; et al. Interferon-inducible cholesterol-25-hydroxylase broadly inhibits viral entry by production of 25-hydroxycholesterol. Immunity 2013, 38, 92–105. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Deng, Y.-Q.; Wang, S.; Ma, F.; Aliyari, R.; Huang, X-Y.; Zhang, N-N.; Watanabe, M.; Dong, H-L.; Liu, P.; et al. 25-Hydroxycholesterol protects host against Zika virus infection and its associated microcephaly in a mouse model. Immunity 2017, 46, 446–456. [Google Scholar] [CrossRef] [Green Version]
- Romero-Brey, I.; Romero-Brey, I.; Berger, C.; Colpitts, C.C.; Boldanova, T.; Engelmann, M.; Todt, D.; Perin, P.M.; Behrendt, P.; Vondran, F.W.R.; et al. Interferon-inducible cholesterol-25-hydroxylase restricts hepatitis C virus replication through blockage of membranous web formation. Hepatology 2015, 62, 702–714. [Google Scholar]
- Doyle, T.; Moncorgé, O.; Bonaventure, B.; Pollpeter, D.; Lussignol, M.; Tauziet, M.; Apolonia, L.; Catanese, M.-T.; Goujon, C.; Malim, M.H. The interferon-inducible isoform of NCOA7 inhibits endosome-mediated viral entry. Nat. Microbiol. 2018, 3, 1369–1376. [Google Scholar] [CrossRef] [PubMed]
- Kühnl, A.; Musiol, A.; Heitzig, N.; Johnson, D.; Ehrhardt, C.; Grewal, T.; Gerke, V.; Ludwig, S.; Rescher, U. Late endosomal/lysosomal cholesterol accumulation is a host cell-protective mechanism inhibiting endosomal escape of influenza A virus. MBio 2018, 9, e01345-18. [Google Scholar] [CrossRef] [Green Version]
- John, S.P.; Chin, C.R.; Perreira, J.M.; Feeley, E.M.; Aker, A.M.; Savidis, G.; Smith, S.E.; Elia, A.E.H.; Everitt, A.R.; Vora, M.; et al. The CD225 domain of IFITM3 is required for both IFITM protein association and inhibition of influenza A virus and dengue virus replication. J. Virol. 2013, 87, 7837–7852. [Google Scholar] [CrossRef] [Green Version]
- Tartour, K.; Nguyen, X.-N.; Approurchaux, R.; Assil, S.; Barateau, V.; Bloyet, L-M.; Gaillard, J.B.; Confort, M-P.; Escudero-Perez, B.; Gruffat, H.; et al. Interference with the production of infectious viral particles and bimodal inhibition of replication are broadly conserved antiviral properties of IFITMs. PLoS Pathog. 2017, 13, e1006610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wrensch, F.; Karsten, C.B.; Gnirß, K.; Hoffmann, M.; Lu, K.; Takada, A.; Winkler, M.; Simmons, G.; Pöhlmann, S. Interferon-Induced Transmembrane Protein–Mediated Inhibition of Host Cell Entry of Ebolaviruses. J. Infect. Dis. 2015, 212, S210–S218. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Sehgal, M.; Hou, Z.; Cheng, J.; Shu, S.; Wu, S.; Guo, F.; Le Marchand, S.J.; Lin, H.; Chang, J.; et al. Identification of residues controlling restriction versus enhancing activities of IFITM proteins on entry of human coronaviruses. J. Virol. 2018, 92, e01535-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, G.; Kenney, A.D.; Kudryashova, E.; Zani, A.; Zhang, L.; Lai, K.K.; Hall-Stoodley, L.; Robinson, R.; Kudryashov, D.S.; Compton, A.A.; et al. Opposing activities of IFITM proteins in SARS-CoV-2 infection. EMBO J. 2021, 40, e106501. [Google Scholar] [CrossRef]
- Weidner, J.M.; Jiang, D.; Pan, X.B.; Chang, J.; Block, T.M.; Guo, J.T. Interferon-induced cell membrane proteins, IFITM3 and tetherin, inhibit vesicular stomatitis virus infection via distinct mechanisms. J. Virol. 2010, 84, 12646–12657. [Google Scholar] [CrossRef] [Green Version]
- Mudhasani, R.; Tran, J.P.; Retterer, C.; Radoshitzky, S.R.; Kota, K.P.; Altamura, L.A.; Smith, J.M.; Packard, B.Z.; Luhn, J.H.; Constantino, J.; et al. IFITM-2 and IFITM-3 but not IFITM-1 restrict Rift Valley fever virus. J. Virol. 2013, 87, 8451–8464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narayana, S.K.; Helbig, K.J.; McCartney, E.M.; Eyre, N.S.; Bull, R.A.; Eltahla, A.; Lloyd, A.R.; Beard, M.R. The interferon-induced transmembrane proteins, IFITM1, IFITM2, and IFITM3 inhibit hepatitis C virus entry. J. Biol. Chem. 2015, 290, 25946–25959. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Pan, Q.; Rong, L.; Liu, S.-L.; Liang, C. The IFITM proteins inhibit HIV-1 infection. J. Virol. 2011, 85, 2126–2137. [Google Scholar] [CrossRef] [Green Version]
- Pfaender, S.; Mar, K.B.; Michailidis, E.; Kratzel, A.; Boys, I.N.; V’kovski, P.; Fan, W.; Kelly, J.N.; Hirt, D.; Ebert, N.; et al. LY6E impairs coronavirus fusion and confers immune control of viral disease. Nat. Microbiol. 2020, 5, 1330–1339. [Google Scholar] [CrossRef] [PubMed]
- Pindel, A.; Sadler, A. The role of protein kinase R in the interferon response. J. Interf. cytokine Res. Off. J. Int. Soc. Interf. Cytokine Res. 2011, 31, 59–70. [Google Scholar] [CrossRef]
- Vladimer, G.I.; Górna, M.W.; Superti-Furga, G. IFITs: Emerging Roles as Key Anti-Viral Proteins. Front. Immunol. 2014, 5, 94. [Google Scholar] [CrossRef] [Green Version]
- Durfee, L.A.; Lyon, N.; Seo, K.; Huibregtse, J.M. The ISG15 conjugation system broadly targets newly synthesized proteins: Implications for the antiviral function of ISG15. Mol. Cell 2010, 38, 722–732. [Google Scholar] [CrossRef]
- Li, M.; Kao, E.; Gao, X.; Sandig, H.; Limmer, K.; Pavon-Eternod, M.; Jones, T.E.; Landry, S.; Pan, T.; Weitzman, M.D.; et al. Codon-usage-based inhibition of HIV protein synthesis by human schlafen 11. Nature 2012, 491, 125–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schoggins, J.W.; Wilson, S.J.; Panis, M.; Murphy, M.Y.; Jones, C.T.; Beiniasz, P.; Rice, C.M.; et al. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 2011, 472, 481–485. [Google Scholar] [CrossRef] [PubMed]
- Helbig, K.J.; Beard, M.R. The role of viperin in the innate antiviral response. J. Mol. Biol. 2014, 426, 1210–1219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malim, M.H.; Bieniasz, P.D. HIV Restriction Factors and Mechanisms of Evasion. Cold Spring Harb. Perspect. Med. 2012, 2, a006940. [Google Scholar] [CrossRef]
- Richardson, R.B.; Ohlson, M.B.; Eitson, J.L.; Kumar, A.; McDougal, M.B.; Boys, I.N.; Mar, K.B.; De La Cruz-Rivera, P.C.; Douglas, C.; Konopka, G.; et al. A CRISPR screen identifies IFI6 as an ER-resident interferon effector that blocks flavivirus replication. Nat. Microbiol. 2018, 3, 1214–1223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batra, J.; Hultquist, J.F.; Liu, D.; Shtanko, O.; Dollen, J.V.; Satkamp, L.; Dollen, J.V.; Satkamp, L.; Jang, G.M.; Luthra, P.; et al. Protein Interaction Mapping Identifies RBBP6 as a Negative Regulator of Ebola Virus Replication. Cell 2018, 175, 1917–1930. [Google Scholar] [CrossRef] [Green Version]
- Chakrabarti, A.; Jha, B.K.; Silverman, R.H. New insights into the role of RNase L in innate immunity. J. Interf. Cytokine Res. Off. J. Int. Soc. Interf. Cytokine Res. 2011, 31, 49–57. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.; Ma, J.; Sun, J.; Gao, G. The zinc-finger antiviral protein recruits the RNA processing exosome to degrade the target mRNA. Proc. Natl. Acad. Sci. USA 2007, 104, 151–156. [Google Scholar] [CrossRef] [Green Version]
- Lucifora, J.; Xia, Y.; Reisinger, F.; Zhang, K.; Stadler, D.; Cheng, X.; Sprinzl, M.F.; Koppemsteiner, H.; Makowska, Z.; Volz, T.; et al. Specific and nonhepatotoxic degradation of nuclear hepatitis B virus cccDNA. Science 2014, 343, 1221–1228. [Google Scholar] [CrossRef]
- Qiao, Y.; Han, X.; Guan, G.; Wu, N.; Sun, J.; Pak, V.; Liang, G. TGF-beta triggers HBV cccDNA degradation through AID-dependent deamination. FEBS Lett. 2016, 590, 419–427. [Google Scholar] [CrossRef]
- Espert, L.; Degols, G.; Gongora, C.; Blondel, D.; Williams, B.R.; Silverman, R.H.; Mechti, N. ISG20, a new interferon-induced RNase specific for single-stranded RNA, defines an alternative antiviral pathway against RNA genomic viruses. J. Biol. Chem. 2003, 278, 16151–16158. [Google Scholar] [CrossRef] [Green Version]
- Wu, N.; Nguyen, X.-N.; Wang, L.; Approurchaux, R.; Zhang, C.; Panthu, B.; Gruffat, H.; Journo, C.A.; lais, S.; Qin, J.; et al. The interferon stimulated gene 20 protein (ISG20) is an innate defense antiviral factor that discriminates self versus non-self translation. PLoS Pathog. 2019, 15, e1008093. [Google Scholar] [CrossRef] [PubMed]
- Stadler, D.; Kächele, M.; Jones, A.N.; Hess, J.; Urban, C.; Schneider, J.; Xia, Y.; Oswald, A.; Nebioglu, F.; Bester, R.; et al. Interferon-induced degradation of the persistent hepatitis B virus cccDNA form depends on ISG20. EMBO Rep. 2021, 22, e49568. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. TLR Signaling. In Seminars in Immunology; Elsevier: Amsterdam, The Netherlands, 2007; Volume 19, pp. 24–32. [Google Scholar]
- Könner, A.C.; Brüning, J.C. Toll-like receptors: Linking inflammation to metabolism. Trends Endocrinol. Metab. 2011, 22, 16–23. [Google Scholar] [CrossRef]
- Oka, T.; Hikoso, S.; Yamaguchi, O.; Taneike, M.; Takeda, T.; Tamai, T.; Oyabu, J.; Murakawa, T.; Nakayama, H.; Nishida, K.; et al. Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature 2012, 485, 251–255. [Google Scholar] [CrossRef] [PubMed]
- Vogl, T.; Tenbrock, K.; Ludwig, S.; Leukert, N.; Ehrhardt, C.; van Zoelen, M.A.D.; Nacken, W.; Foell, D.; van der Poll, T.; Sorg, C.; et al. Mrp8 and Mrp14 are endogenous activators of Toll-like receptor 4, promoting lethal, endotoxin-induced shock. Nat. Med. 2007, 13, 1042–1049. [Google Scholar] [CrossRef]
- Kalamvoki, M.; Roizman, B. HSV-1 degrades, stabilizes, requires, or is stung by STING depending on ICP0, the US3 protein kinase, and cell derivation. Proc. Natl. Acad. Sci. USA 2014, 111, E611–E617. [Google Scholar] [CrossRef] [Green Version]
- Park, A.; Ra, E.A.; Lee, T.A.; Choi, H.J.; Lee, E.; Kang, S.; Seo, J.-Y.; Lee, S.; Parl, B. HCMV-encoded US7 and US8 act as antagonists of innate immunity by distinctively targeting TLR-signaling pathways. Nat. Commun. 2019, 10, 4670. [Google Scholar] [CrossRef]
- Bussey, K.A.; Reimer, E.; Todt, H.; Denker, B.; Gallo, A.; Konrad, A.; Ottinger, M.; Adler, H.; Stürzl, M.; Brune, W.; et al. The gammaherpesviruses Kaposi’s sarcoma-associated herpesvirus and murine gammaherpesvirus 68 modulate the Toll-like receptor-induced proinflammatory cytokine response. J. Virol. 2014, 88, 9245–9259. [Google Scholar] [CrossRef] [Green Version]
- Vincent, I.E.; Zannnetti, C.; Lucifora, J.; Norder, H.; Protzer, U.L.; Hainaut, P.; Zoulim, F.; Tommasino, M.; Trépo, C.; Hasan, U.; et al. Hepatitis B virus impairs TLR9 expression and function in plasmacytoid dendritic cells. PLoS ONE 2011, 6, e26315. [Google Scholar] [CrossRef] [Green Version]
- Su, J.; Rui, Y.; Lou, M.; Yin, L.; Xiong, H.; Zhou, Z.; Shen, S.; Chen, T.; Zhang, Z.; Zhao, N.; et al. HIV-2/SIV Vpx targets a novel functional domain of STING to selectively inhibit cGAS–STING-mediated NF-κB signalling. Nat. Microbiol. 2019, 4, 2552–2564. [Google Scholar] [CrossRef]
- Feng, Q.; Langereis, M.A.; Lork, M.; Nguyen, M.; Hato, S.V.; Lanke, K.; Emdad, L.; Bhoopathi, P.; Fisher, P.B.; Lloyd, R.E.; et al. Enterovirus 2Apro targets MDA5 and MAVS in infected cells. J. Virol. 2014, 88, 3369–3378. [Google Scholar] [CrossRef] [Green Version]
- Davis, M.E.; Wang, M.K.; Rennick, L.J.; Full, F.; Gableske, S.; Mesman, A.W.; Gringhuis, S.I.; Geijtenbeek, T.B.H.; Duprex, W.P.; Gack, M.U. Antagonism of the phosphatase PP1 by the measles virus V protein is required for innate immune escape of MDA5. Cell Host Microbe 2014, 16, 19–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, C.; Sun, L.; Liu, W.; Duan, Z. Latent Membrane Protein 1 of Epstein-Barr Virus Promotes RIG-I Degradation Mediated by Proteasome Pathway. Front. Immunol. 2018, 9, 1446. [Google Scholar] [CrossRef]
- Jureka, A.S.; Kleinpeter, A.B.; Cornilescu, G.; Cornilescu, C.C.; Petit, C.M. Structural Basis for a Novel Interaction between the NS1 Protein Derived from the 1918 Influenza Virus and RIG-I. Structure 2015, 23, 2001–2010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.; Zhang, X.; Wang, F.; Wang, P.; Kuang, E.; Li, X. Suppression of MDA5-mediated antiviral immune responses by NSP8 of SARS-CoV-2. bioRxiv 2020, 2020.08.12.247767. [Google Scholar] [CrossRef]
- Rajsbaum, R.; Albrecht, R.A.; Wang, M.K.; Maharaj, N.P.; Versteeg, G.A.; Nistal-Villán, E.; García-Sastre, A.; Gack, M.U. Species-specific inhibition of RIG-I ubiquitination and IFN induction by the influenza A virus NS1 protein. PLoS Pathog. 2012, 8, e1003059. [Google Scholar] [CrossRef]
- Hu, Y.; Li, W.; Gao, T.; Cui, Y.; Jin, Y.; Ma, Q.; Liu, X.; Cao, C. SARS coronavirus nucleocapsid inhibits type I interferon production by interfering with TRIM25-mediated RIG-I ubiquitination. J. Virol. 2017, 91, e02143-16. [Google Scholar] [CrossRef] [Green Version]
- Ban, J.; Lee, N.-R.; Lee, N.-J.; Lee, J.K.; Quan, F.-S.; Inn, K.-S. Human Respiratory Syncytial Virus NS 1 Targets TRIM25 to Suppress RIG-I Ubiquitination and Subsequent RIG-I-Mediated Antiviral Signaling. Viruses 2018, 10, 716. [Google Scholar] [CrossRef] [Green Version]
- Chiang, C.; Pauli, E.-K.; Biryukov, J.; Feister, K.F.; Meng, M.; White, E.A.; Münger, K.; Howley, P.M.; Meyers, C.; Gack, M.U. The human papillomavirus E6 oncoprotein targets USP15 and TRIM25 to suppress RIG-I-mediated innate immune signaling. J. Virol. 2018, 92, e01737-17. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.-L.; Ye, H.-Q.; Liu, S.-Q.; Deng, C.-L.; Li, X.-D.; Shi, P.-Y.; Zhang, B. West Nile Virus NS1 Antagonizes Interferon Beta Production by Targeting RIG-I and MDA5. J. Virol. 2017, 91, e02396-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oshiumi, H.; Miyashita, M.; Matsumoto, M.; Seya, T. A distinct role of Riplet-mediated K63-Linked polyubiquitination of the RIG-I repressor domain in human antiviral innate immune responses. PLoS Pathog. 2013, 9, e1003533. [Google Scholar] [CrossRef] [Green Version]
- Chan, Y.K.; Gack, M.U. A phosphomimetic-based mechanism of dengue virus to antagonize innate immunity. Nat. Immunol. 2016, 17, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Kerur, N.; Veettil, M.V.; Sharma-Walia, N.; Bottero, V.; Sadagopan, S.; Otageri, P.; Chandran, B. IFI16 acts as a nuclear pathogen sensor to induce the inflammasome in response to Kaposi Sarcoma-associated herpesvirus infection. Cell Host Microbe 2011, 9, 363–375. [Google Scholar] [CrossRef] [Green Version]
- Orzalli, M.H.; Broekema, N.M.; Diner, B.A.; Hancks, D.C.; Elde, N.C.; Cristea, I.M.; Knipe, D.M. cGAS-mediated stabilization of IFI16 promotes innate signaling during herpes simplex virus infection. Proc. Natl. Acad. Sci. USA 2015, 112, E1773–E1781. [Google Scholar] [CrossRef] [Green Version]
- Orzalli, M.H.; DeLuca, N.A.; Knipe, D.M. Nuclear IFI16 induction of IRF-3 signaling during herpesviral infection and degradation of IFI16 by the viral ICP0 protein. Proc. Natl. Acad. Sci. USA 2012, 109, E3008–E3017. [Google Scholar] [CrossRef] [Green Version]
- Cristea, I.M.; Moorman, N.J.; Terhune, S.S.; Cuevas, C.D.; O’Keefe, E.S.; Rout, M.P.; Chait, B.T.; Shenk, T. Human cytomegalovirus pUL83 stimulates activity of the viral immediate-early promoter through its interaction with the cellular IFI16 protein. J. Virol. 2010, 84, 7803–7814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orzalli, M.H.; Broekema, N.M.; Knipe, D.M. Relative Contributions of Herpes Simplex Virus 1 ICP0 and vhs to Loss of Cellular IFI16 Vary in Different Human Cell Types. J. Virol. 2016, 90, 8351–8359. [Google Scholar] [CrossRef] [Green Version]
- Su, C.; Zheng, C. Herpes Simplex Virus 1 Abrogates the cGAS/STING-Mediated Cytosolic DNA-Sensing Pathway via Its Virion Host Shutoff Protein, UL41. J. Virol. 2017, 91, e02414-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.J.; Li, W.; Shao, Y.; Avey, D.; Fu, B.; Gillen, J.; Hand, T.; Ma, S.; Liu, X.; Miley, W.; et al. Inhibition of cGAS DNA Sensing by a Herpesvirus Virion Protein. Cell Host Microbe 2015, 18, 333–344. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.; Liu, Q.; Wu, Y.; Ma, L.; Zhang, Z.; Liu, T.; Jin, S.; She, Y.; Li, Y.-P.; Cui, J. Zika virus elicits inflammation to evade antiviral response by cleaving cGAS via NS1-caspase-1 axis. EMBO J. 2018, 37, e99347. [Google Scholar] [CrossRef]
- Zhang, J.; Zhao, J.; Xu, J.; He, S.; Zeng, Y.; Xie, L.; Xie, N.; Liu, T.; Lee, K.; et al. Species-Specific Deamidation of cGAS by Herpes Simplex Virus UL37 Protein Facilitates Viral Replication. Cell Host Microbe 2018, 24, 234–248. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; You, H.; Su, C.; Li, Y.; Chen, S.; Zheng, C. Herpes Simplex Virus 1 Tegument Protein VP22 Abrogates cGAS/STING-Mediated Antiviral Innate Immunity. J. Virol. 2018, 92, e00841-18. [Google Scholar] [CrossRef] [Green Version]
- Biolatti, M.; Dell’Oste, V.; Pautasso, S.; Gugliesi, F.; von Eineem, J.; Krapp, C.; Jakobsen, M.R.; Borgogna, C.; Gariglio, M.; De Andrea, M.; et al. Human Cytomegalovirus Tegument Protein pp65 (pUL83) Dampens Type I Interferon Production by Inactivating the DNA Sensor cGAS without Affecting STING. J. Virol. 2018, 92, e01774-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Z.-F.; Zou, H.-M.; Liao, B.-W.; Zhang, H.-Y.; Yang, Y.; Fu, Y.-Z.; Wang, S.-Y.; Luo, M.-H.; Wang, Y.-Y. Human Cytomegalovirus Protein UL31 Inhibits DNA Sensing of cGAS to Mediate Immune Evasion. Cell Host Microbe 2018, 24, 69–80. [Google Scholar] [CrossRef] [Green Version]
- Zhang, G.; Chan, B.; Samarina, N.; Abere, B.; Weidner-Glunde, M.; Buch, A.; Pich, A.; Brinkmann, M.M.; Schulz, T.F. Cytoplasmic isoforms of Kaposi sarcoma herpesvirus LANA recruit and antagonize the innate immune DNA sensor cGAS. Proc. Natl. Acad. Sci. USA 2016, 113, E1034–E1043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meylan, E.; Joseph, C.; Hofmann, K.; Moradpour, D.; Binder, M.; Bartenschlager, R.; Tschopp, J. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature 2005, 437, 1167–1172. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Foy, E.; Ferreon, J.C.; Nakamura, M.; Ferreon, A.C.M.; Ikeda, M.; Ray, S.C.; Gale Jr, M.; Lemon, S.M. Immune evasion by hepatitis C virus NS3/4A protease-mediated cleavage of the Toll-like receptor 3 adaptor protein TRIF. Proc. Natl. Acad. Sci. USA 2005, 102, 2992–2997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, C.-Y.; Liang, J.-J.; Li, J.-K.; Lee, Y.-L.; Chang, B.-L.; Su, C.-I.; Huang, W.-J.; Lai, M.M.C.; Lin, Y.-L. Dengue Virus Impairs Mitochondrial Fusion by Cleaving Mitofusins. PLoS Pathog. 2015, 11, e1005350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drahos, J.; Racaniello, V.R. Cleavage of IPS-1 in cells infected with human rhinovirus. J. Virol. 2009, 83, 11581–11587. [Google Scholar] [CrossRef] [Green Version]
- Lau, L.; Gray, E.E.; Brunette, R.L.; Stetson, D.B. DNA tumor virus oncogenes antagonize the cGAS-STING DNA-sensing pathway. Science 2015, 350, 568–571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Z.; Jacobs, S.R.; West, J.A.; Stopford, C.; Zhang, Z.; Davis, Z.; Barber, G.N.; Glaunsinger, B.A.; Dittmer, D.P.; Damania, B. Modulation of the cGAS-STING DNA sensing pathway by gammaherpesviruses. Proc. Natl. Acad. Sci. USA 2015, 112, E4306–E4315. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Li, J.; Chen, J.; Li, Y.; Wang, W.; Du, X.; Song, W.; Zhang, W.; Lin, L.; Yuan, Z. Hepatitis B Virus Polymerase Disrupts K63-Linked Ubiquitination of STING To Block Innate Cytosolic DNA-Sensing Pathways. J. Virol. 2015, 89, 2287–2300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aguirre, S.; Maestre, A.M.; Pagni, S.; Patel, J.R.; Savage, T.; Gutman, D.; Maringer, K.; Bernal-Rubio, D.; Shabman, R.S.; Simon, V.; et al. DENV inhibits type I IFN production in infected cells by cleaving human STING. PLoS Pathog. 2012, 8, e1002934. [Google Scholar] [CrossRef] [Green Version]
- Pythoud, C.; Shanaka I Rodrigo, W.W.; Pasqual, G.; Rothenberger, S.; Martínez-Sobrido, L.; Calros de la Torre, J.; Kunz, S. Arenavirus nucleoprotein targets interferon regulatory factor-activating kinase IKKε. J. Virol. 2012, 86, 7728–7738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harman, A.N.; Nasr, N.; Feetham, A.; Galoyan, A.; Alshehri, A.A.; Rambukwelle, D.; Botting, R.A.; Heiner, B.M.; Diefenbach, E.; Diefenbach, R.J.; et al. HIV Blocks Interferon Induction in Human Dendritic Cells and Macrophages by Dysregulation of TBK1. J. Virol. 2015, 89, 6575–6584. [Google Scholar] [CrossRef] [Green Version]
- Prins, K.C.; Cárdenas, W.B.; Basler, C.F. Ebola virus protein VP35 impairs the function of interferon regulatory factor-activating kinases IKKepsilon and TBK-1. J. Virol. 2009, 83, 3069–3077. [Google Scholar] [CrossRef] [Green Version]
- Verpooten, D.; Ma, Y.; Hou, S.; Yan, Z.; He, B. Control of TANK-binding kinase 1-mediated signaling by the gamma(1)34.5 protein of herpes simplex virus 1. J. Biol. Chem. 2009, 284, 1097–1105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, R.; Genin, P.; Mamane, Y.; Sgarbanti, M.; Battistini, A.; Harrington, W.J., Jr.; Barber, G.N.; Hiscott, J. HHV-8 encoded vIRF-1 represses the interferon antiviral response by blocking IRF-3 recruitment of the CBP/p300 coactivators. Oncogene 2001, 20, 800–811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, Q.; Fu, B.; Wu, F.; Li, X.; Yuan, Y.; Zhu, F. ORF45 of Kaposi’s sarcoma-associated herpesvirus inhibits phosphorylation of interferon regulatory factor 7 by IKKε and TBK1 as an alternative substrate. J. Virol. 2012, 86, 10162–10172. [Google Scholar] [CrossRef] [Green Version]
- Xiang, Z.; Liu, L.; Lei, X.; Zhou, Z.; He, B.; Wang, J. 3C Protease of Enterovirus D68 Inhibits Cellular Defense Mediated by Interferon Regulatory Factor 7. J. Virol. 2015, 90, 1613–1621. [Google Scholar] [CrossRef] [Green Version]
- Watters, K.; Palmenberg, A.C. Differential processing of nuclear pore complex proteins by rhinovirus 2A proteases from different species and serotypes. J. Virol. 2011, 85, 10874–10883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrison, J.; Laurent-Rolle, M.; Maestre, A.M.; Rajsbaum, R.; Pisanelli, G.; Simon, V.; Mulder, L.C.F.; Fernandez-Sesma, A.; García-Sastre, A. Dengue virus co-opts UBR4 to degrade STAT2 and antagonize type I interferon signaling. PLoS Pathog. 2013, 9, e1003265. [Google Scholar] [CrossRef] [Green Version]
- Ashour, J.; Laurent-Rolle, M.; Shi, P.-Y.; Garcia-Sastre, A. NS5 of Dengue Virus Mediates STAT2 Binding and Degradation. J. Virol. 2009, 83, 5408–5418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kopecky-Bromberg, S.A.; Martínez-Sobrido, L.; Frieman, M.; Baric, R.A.; Palese, P. Severe acute respiratory syndrome coronavirus open reading frame (ORF) 3b, ORF 6, and nucleocapsid proteins function as interferon antagonists. J. Virol. 2007, 81, 548–557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, H.; Leo, H.; Shan, C.; Muruato, A.E.; Nunes, B.T.D.; Medeiros, D.B.A.; Zou, J.; Xie, X.; Giraldo, M.I.; Vasconcelos, P.F.C.; et al. An evolutionary NS1 mutation enhances Zika virus evasion of host interferon induction. Nat. Commun. 2018, 9, 414. [Google Scholar] [CrossRef]
- Fros, J.J.; Pijlman, G.P. Alphavirus Infection: Host Cell Shut-Off and Inhibition of Antiviral Responses. Viruses 2016, 8, 166. [Google Scholar] [CrossRef]
- White, L.K.; Sali, T.; Alvarado, D.; Gatti, E.; Pierre, P.; Streblow, D.; Defilippis, V.R. Chikungunya virus induces IPS-1-dependent innate immune activation and protein kinase R-independent translational shutoff. J. Virol. 2011, 85, 606–620. [Google Scholar] [CrossRef] [Green Version]
- Lamphear, B.J.; Yan, R.; Yang, F.; Waters, D.; Liebig, H.D.; Klump, H.; Kuechler, E.; Skern, T.; Rhoads, R.E. Mapping the cleavage site in protein synthesis initiation factor eIF-4 gamma of the 2A proteases from human Coxsackievirus and rhinovirus. J. Biol. Chem. 1993, 268, 19200–19203. [Google Scholar] [CrossRef]
- Walker, E.J.; Younessi, P.; Fulcher, A.J.; McCuaig, R.; Thomas, B.J.; Bardin, P.G.; Jans, D.A.; Ghildyal, R. Rhinovirus 3C protease facilitates specific nucleoporin cleavage and mislocalisation of nuclear proteins in infected host cells. PLoS ONE 2013, 8, e71316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conticello, S.G.; Harris, R.S.; Neuberger, M.S. The Vif protein of HIV triggers degradation of the human antiretroviral DNA deaminase APOBEC3G. Curr. Biol. 2003, 13, 2009–2013. [Google Scholar] [CrossRef] [Green Version]
- Neil, S.J.D.; Zang, T.; Bieniasz, P.D. Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature 2008, 451, 425–430. [Google Scholar] [CrossRef] [Green Version]
- Kostyushev, D.; Kostyusheva, A.; Brezgin, S.; Zarifyan, D.; Utkina, A.; Goptar, I.; Chulanov, V. Suppressing the NHEJ pathway by DNA-PKcs inhibitor NU7026 prevents degradation of HBV cccDNA cleaved by CRISPR/Cas9. Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Chang, H.H.Y.; Pannunzio, N.R.; Adachi, N.; Lieber, M.R. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat. Rev. Mol. Cell Biol. 2017, 18, 495–506. [Google Scholar] [CrossRef]
- Brandsma, I.; van Gent, D.C. Pathway choice in DNA double strand break repair: Observations of a balancing act. Genome Integr. 2012, 3, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Overbeek, M.; Capurso, D.; Carter, M.M.; Thhompson, M.S.; Frias, E.; Russ, C.; Reece-Hoyes, J.S.; Nye, C.; Gradia, S.; Vidal, B.; et al. DNA repair profiling reveals nonrandom outcomes at Cas9-mediated breaks. Mol. Cell 2016, 63, 633–646. [Google Scholar] [CrossRef] [Green Version]
- Ihry, R.J.; Worringer, K.A.; Salick, M.R.; Frias, E.; Ho, D.; Theriault, K.; Kommineni, S.; Chen, J.; Sondey, M.; Ye, C.; et al. p53 inhibits CRISPR-Cas9 engineering in human pluripotent stem cells. Nat. Med. 2018, 24, 939. [Google Scholar] [CrossRef]
- Zuccaro, M.V.; Xu, J.; Mitchell, C.; Marin, D.; Zimmerman, R.; Rana, B.; Weinstein, E.; King, R.T.; Palmerola, K.L.; Smith, M.E.; et al. Allele-specific chromosome removal after Cas9 cleavage in human embryos. Cell 2020, 183, 1650–1664. [Google Scholar] [CrossRef] [PubMed]
- Kosicki, M.; Tomberg, K.; Bradley, A. Repair of double-strand breaks induced by CRISPR–Cas9 leads to large deletions and complex rearrangements. Nat. Biotechnol. 2018, 36, 765–771. [Google Scholar] [CrossRef]
- Leibowitz, M.L.; Papathanasiou, S.; Doerfler, P.A.; Blaine, L.; Sun, L.; Yao, Y.; Zhang, C.-Z.; Weiss, M.J.; Pellman, D. Chromothripsis as an on-target consequence of CRISPR–Cas9 genome editing. Nat. Genet. 2021, 53, 895–905. [Google Scholar] [CrossRef]
- Liu, M.; Rehman, S.; Tang, X.; Gu, K.; Fan, Q.; Chen, D.; Ma, W. Methodologies for improving HDR efficiency. Front. Genet. 2019, 9, 691. [Google Scholar] [CrossRef]
- Kim, S.; Koo, T.; Cho, H.-Y.; Lee, G.; Lim, D.-G.; Shin, H.S.; Kim, J.-S. CRISPR RNAs trigger innate immune responses in human cells. Genome Res. 2018, 28, 367–373. [Google Scholar] [CrossRef] [Green Version]
- Bae, S.; Kweon, J.; Kim, H.S.; Kim, J.-S. Microhomology-based choice of Cas9 nuclease target sites. Nat. Methods 2014, 11, 705–706. [Google Scholar] [CrossRef] [PubMed]
- Qi, L.S.; Larson, M.H.; Gilbert, L.A.; Doudna, J.A.; Weissman, J.S.; Arkin, A.P.; Lim, W.A. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 2013, 152, 1173–1183. [Google Scholar] [CrossRef] [Green Version]
- Cheng, A.W.; Wang, H.; Yang, H.; Shi, L.; Katz, Y.; Theunissen, T.W.; Rangarajan, S.; Shivalia, C.S.; Dadon, D.B.; Jaenisch, R. Multiplexed activation of endogenous genes by CRISPR-on, an RNA-guided transcriptional activator system. Cell Res. 2013, 23, 1163–1171. [Google Scholar] [CrossRef]
- Baeumler, T.A.; Ahmed, A.A.; Fulga, T.A. Engineering Synthetic Signaling Pathways with Programmable dCas9-Based Chimeric Receptors. Cell Rep. 2017, 20, 2639–2653. [Google Scholar] [CrossRef] [Green Version]
- Hilton, I.B.; D’ippolito, A.M.; Vockley, C.M.; Thakore, P.I.; Crawford, G.E.; Reddy, T.E.; Gersbach, C.A. Epigenome editing by a CRISPR-Cas9-based acetyltransferase activates genes from promoters and enhancers. Nat. Biotechnol. 2015, 33, 510–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez-Pinera, P.; Kocak, D.D.; Vockley, C.M.; Adler, A.F.; Kabadi, A.M.; Polstein, L.R.; Thakore, P.I.; Glass, K.A.; Ousterout, D.G.; Leong, K.W.; et al. RNA-guided gene activation by CRISPR-Cas9-based transcription factors. Nat. Methods 2013, 10, 973. [Google Scholar] [CrossRef] [Green Version]
- Fu, Y.; Rocha, P.P.; Luo, V.M.; Raviram, R.; Deng, Y.; Mazzoni, E.O.; Skok, J.A. CRISPR-dCas9 and sgRNA scaffolds enable dual-colour live imaging of satellite sequences and repeat-enriched individual loci. Nat. Commun. 2016, 7, 1–8. [Google Scholar] [CrossRef]
- Chavez, A.; Scheiman, J.; Vora, S.; Pruitt, B.W.; Tuttle, M.; Lyer, E.P.R.; Lin, S.; Kiani, S.; Guzman, C.D.; Wiegand, D.J.; et al. Highly efficient Cas9-mediated transcriptional programming. Nat. Methods 2015, 12, 326–328. [Google Scholar] [CrossRef] [Green Version]
- Pflueger, C.; Tan, D.; Swain, T.; Nguyen, T.; Pflueger, J.; Nefzger, C.; Polo, J.M.; Ford, E.; Lister, R. A modular dCas9-SunTag DNMT3A epigenome editing system overcomes pervasive off-target activity of direct fusion dCas9-DNMT3A constructs. Genome Res. 2018, 28, 1193–1206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konermann, S.; Brigham, M.D.; Trevino, A.E.; Joung, J.; Abudayyeh, O.O.; Barcena, C.; Hsu, P.D.; Habib, N.; Gootenberg, J.S.; Nishimasu, H.; et al. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature 2015, 517, 583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sajwan, S.; Mannervik, M. Gene activation by dCas9-CBP and the SAM system differ in target preference. Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, H.; Liu, J.; Zhou, C.; Gao, N.; Rao, Z.; Li, H.; Hu, X.; Li, C.; Yao, X.; Shen, X.; et al. In vivo simultaneous transcriptional activation of multiple genes in the brain using CRISPR-dCas9-activator transgenic mice. Nat. Neurosci. 2018, 21, 440. [Google Scholar] [CrossRef]
- Thakore, P.I.; D’Ippolito, A.M.; Song, L.; Safi, A.; Shivakumar, N.K.; Kabadi, A.M.; Reddy, T.E.; Crawford, G.E.; Gersbach, C.A. Highly specific epigenome editing by CRISPR-Cas9 repressors for silencing of distal regulatory elements. Nat. Methods 2015, 1–9. [Google Scholar] [CrossRef] [Green Version]
- O’Geen, H.; Bates, S.L.; Carter, S.S.; Nisson, K.A.; Halmai, J.; Fink, K.D.; Rhie, S.K.; Farnham, P.J.; Segal, D.J. Ezh2-dCas9 and KRAB-dCas9 enable engineering of epigenetic memory in a context-dependent manner. Epigenetics Chromatin 2019, 12, 26. [Google Scholar] [CrossRef]
- Rees, H.A.; Liu, D.R. Base editing: Precision chemistry on the genome and transcriptome of living cells. Nat. Rev. Genet. 2018, 19, 770–788. [Google Scholar] [CrossRef]
- Abbott, T.R.; Dhamdhere, G.; Liu, Y.; Lin, X.; Goudy, L.; Zeng, L.; Chemparathy, A.; Chmura, S.; Heaton, N.S.; Debs, R.; et al. Development of CRISPR as an antiviral strategy to combat SARS-CoV-2 and influenza. Cell 2020, 181, 865–876. [Google Scholar] [CrossRef]
- Freije, C.A.; Myhrvold, C.; Boehm, C.K.; Lin, A.E.; Welch, N.L.; Carter, A.; Metsky, H.C.; Luo, C.Y.; Abudayyeh, O.O.; Gootenberg, J.S.; et al. Programmable inhibition and detection of RNA viruses using Cas13. Mol. Cell 2019, 76, 826–837. [Google Scholar] [CrossRef] [Green Version]
- Cox, D.B.T.; Gootenberg, J.S.; Abudayyeh, O.O.; Franklin, B.; Kellner, M.J.; Joung, J.; Zhang, F. RNA editing with CRISPR-Cas13. Science 2017, 358, 1019–1027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abudayyeh, O.O.; Gootenberg, J.S.; Franklin, B.; Koob, J.; Kellner, M.J.; Ladha, A.; Joung, J.; Kirchgatterer, P.; Cox, D.B.T.; Zhang, F. A cytosine deaminase for programmable single-base RNA editing. Science 2019, 365, 382–386. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Lv, J.; Li, Y.; Mao, S.; Li, Z.; Jing, Z.; Sun, Y.; Zhang, X.; Shen, S.; Wang, X.; et al. Programmable C-to-U RNA editing using the human APOBEC 3A deaminase. EMBO J. 2020, 39, e104741. [Google Scholar] [CrossRef]
- Xu, C.; Zhou, Y.; Xiao, Q.; He, B.; Geng, G.; Wang, Z.; Cao, B.; Dong, X.; Bai, W.; Wang, Y.; et al. Programmable RNA editing with compact CRISPR–Cas13 systems from uncultivated microbes. Nat. Methods 2021, 18, 499–506. [Google Scholar] [CrossRef] [PubMed]
- Yeo, N.C.; Chavez, A.; Lance-Byrne, A.; Chan, Y.; Menn, D.; Milanova, D.; Kuo, C.-C.; Guo, X.; Sharma, S.; Tung, A.; et al. An enhanced CRISPR repressor for targeted mammalian gene regulation. Nat. Methods 2018, 15, 611. [Google Scholar] [CrossRef]
- Komor, A.C.; Kim, Y.B.; Packer, M.S.; Zuris, J.A.; Liu, D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 2016, 533, 420. [Google Scholar] [CrossRef] [Green Version]
- Komor, A.C.; Zhao, K.T.; Packer, M.S.; Gaudelli, N.M.; Waterbury, A.L.; Koblan, L.W.; Kim, Y.B.; Badran, A.H.; Liu, D.R. Improved base excision repair inhibition and bacteriophage Mu Gam protein yields C: G-to-T: A base editors with higher efficiency and product purity. Sci. Adv. 2017, 3, eaao4774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Li, J.; Wang, Y.; Yang, B.; Weo, J.; Wu, J.; Wang, R.; Huang, X.; Chen, J.; Yang, L. Efficient base editing in methylated regions with a human APOBEC3A-Cas9 fusion. Nat. Biotechnol. 2018, 36, 946. [Google Scholar] [CrossRef]
- Jiang, W.; Feng, S.; Huang, S.; Yu, W.; Li, G.; Yang, G.; Liu, Y.; Zhang, Y.; Zhang, L.; Hou, Y.; et al. BE-PLUS: A new base editing tool with broadened editing window and enhanced fidelity. CELL Res. 2018, 28, 855–861. [Google Scholar] [CrossRef] [Green Version]
- Hess, G.T.; Frésard, L.; Han, K.; Lee, C.H.; Li, A.; Cimprich, K.A.; Montgomery, S.B.; Bassik, M.C. Directed evolution using dCas9-targeted somatic hypermutation in mammalian cells. Nat. Methods 2016, 13, 1036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaudelli, N.M.; Komor, A.C.; Rees, H.A.; Packer, M.S.; Badran, A.H.; Bryson, D.I.; Liu, D.R. Programmable base editing of A• T to G• C in genomic DNA without DNA cleavage. Nature 2017, 551, 464. [Google Scholar] [CrossRef]
- Dominguez, A.A.; Lim, W.A.; Qi, L.S. Beyond editing: Repurposing CRISPR–Cas9 for precision genome regulation and interrogation. Nat. Rev. Mol. Cell Biol. 2016, 17, 5. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Wang, G.Z.; Cingöz, O.; Goff, S.P. NP220 mediates silencing of unintegrated retroviral DNA. Nature 2018, 564, 278–282. [Google Scholar] [CrossRef]
- Chia, B.S.; Li, B.; Cui, A.; Eisenhaure, T.; Raychowdhury, R.; Lieb, D.; Hacohen, N. Loss of the nuclear protein RTF2 enhances influenza virus replication. J. Virol. 2020, 94, e00319-20. [Google Scholar] [CrossRef] [PubMed]
- OhAinle, M.; Helms, L.; Vermeire, J.; Roesch, F.; Humes, D.; Bason, R.; Delrow, J.J.; Overbaugh, J.; Emerman, M. A virus-packageable CRISPR screen identifies host factors mediating interferon inhibition of HIV. Elife 2018, 7, e39823. [Google Scholar] [CrossRef] [PubMed]
- Roesch, F.; OhAinle, M.; Emerman, M. A CRISPR screen for factors regulating SAMHD1 degradation identifies IFITMs as potent inhibitors of lentiviral particle delivery. Retrovirology 2018, 15, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, J.; Perez, J.T.; Chen, C.; Li, Y.; Benitez, A.; Kandasamy, M.; Lee, Y.; Andrade, J.; tenOever, B.; Manicassamy, B. Genome-wide CRISPR/Cas9 Screen Identifies Host Factors Essential for Influenza Virus Replication. Cell Rep. 2018, 23, 596–607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orchard, R.C.; Sullender, M.E.; Dunlap, B.F.; Balce, D.R.; Doench, J.G.; Virgin, H.W. Identification of antinorovirus genes in human cells using Genome-Wide CRISPR activation screening. J. Virol. 2019, 93, e01324-18. [Google Scholar] [CrossRef] [Green Version]
- Dukhovny, A.; Lamkiewicz, K.; Chen, Q.; Fricke, M.; Jabrane-Ferrat, N.; Marz, M.; Jung, J.U.; Sklan, E.H. A CRISPR Activation Screen Identifies Genes That Protect against Zika Virus Infection. J. Virol. 2019, 93, e00211-19. [Google Scholar] [CrossRef] [Green Version]
- LaFleur, M.W.; Nguyen, T.H.; Coxe, M.A.; Yates, K.B.; Trombley, J.D.; Weiss, S.A.; Brown, F.D.; Gillis, J.E.; Coxe, D.J.; Doench, J.G.; et al. A CRISPR-Cas9 delivery system for in vivo screening of genes in the immune system. Nat. Commun. 2019, 10, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Honda, K.; Takaoka, A.; Taniguchi, T. Type I inteferon gene induction by the interferon regulatory factor family of transcription factors. Immunity 2006, 25, 349–360. [Google Scholar] [CrossRef] [Green Version]
- Lugrin, J.; Martinon, F. The AIM 2 inflammasome: Sensor of pathogens and cellular perturbations. Immunol. Rev. 2018, 281, 99–114. [Google Scholar] [CrossRef] [PubMed]
- Paijo, J.; Döring, M.; Spanier, J.; Grabski, E.; Nooruzzaman, M.; Schmidt, T.; Witte, G.; Messerle, M.; Hornung, V.; Kaever; et al. cGAS senses human cytomegalovirus and induces type I interferon responses in human monocyte-derived cells. PLoS Pathog. 2016, 12, e1005546. [Google Scholar] [CrossRef] [PubMed]
- Lio, C.-W. J.; McDonald, B.; Takahashi, M.; Dhanwani, R.; Sharma, N.; Huang, J.; Pham, E.; Benedict, C.A.; Sharma, S. cGAS-STING signaling regulates initial innate control of cytomegalovirus infection. J. Virol. 2016, 90, 7789–7797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sui, H.; Zhou, M.; Imamichi, H.; Jiao, X.; Sherman, B.T.; Lane, H.C.; Imamichi, T. STING is an essential mediator of the Ku70-mediated production of IFN-lambda 1 in response to exogenous DNA. Sci. Signal. 2017, 10, eaah5054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gray, E.E.; Winship, D.; Snyder, J.M.; Chuld, S.J.; Geballe, A.P.; Stetson, D.B. The AIM2-like receptors are dispensable for the interferon response to intracellular DNA. Immunity 2016, 45, 255–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diner, B.A.; Lum, K.K.; Toettcher, J.E.; Cristea, I.M. Viral DNA sensors IFI16 and cyclic GMP-AMP synthase possess distinct functions in regulating viral gene expression, immune defenses, and apoptotic responses during herpesvirus infection. MBio 2016, 7, e01553-16. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Banerjee, S.; Wang, Y.; Goldstein, S.A.; Dong, B.; Gaughan, C.; Silverman, R.H.; Weiss, S.R. Activation of RNase L is dependent on OAS3 expression during infection with diverse human viruses. Proc. Natl. Acad. Sci. USA 2016, 113, 2241–2246. [Google Scholar] [CrossRef] [Green Version]
- Li, P.; Zhang, X.; Cao, W.; Yang, F.; Du, X.; Shi, Z.; Zhang, M.; Liu, X.; Zhu, Z.; Zheng, H. RIG-I is responsible for activation of type I interferon pathway in Seneca Valley virus-infected porcine cells to suppress viral replication. Virol. J. 2018, 15, 162. [Google Scholar] [CrossRef] [Green Version]
- Schilling, M.; Bridgeman, A.; Gray, N.; Hertzog, J.; Hubiltz, P.; Kohl, A.; Rehwinkel, J. RIG-I plays a dominant role in the induction of transcriptional changes in Zika virus-infected cells, which protect from virus-induced cell death. Cells 2020, 9, 1476. [Google Scholar] [CrossRef]
- Zhang, Q.; Zeng, L.-P.; Zhou, P.; Irving, A.T.; Li, S.; Shi, Z-L.; Wang, L-F. IFNAR2-dependent gene expression profile induced by IFN-α in Pteropus alecto bat cells and impact of IFNAR2 knockout on virus infection. PLoS ONE 2017, 12, e0182866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urin, V.; Shemesh, M.; Schreiber, G. CRISPR/Cas9-based Knockout Strategy Elucidates Components Essential for Type 1 Interferon Signaling in Human HeLa Cells. J. Mol. Biol. 2019, 431, 3324–3338. [Google Scholar] [CrossRef]
- Yamauchi, S.; Takeuchi, K.; Chihara, K.; Honjoh, C.; Kato, Y.; Yoshiki, H.; Hotta, H.; Sada, K. STAT1 is essential for the inhibition of hepatitis C virus replication by interferon-λ but not by interferon-α. Sci. Rep. 2016, 6, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.S.; Shin, M.J.; Kim, K.H. Increase of viral hemorrhagic septicemia virus growth by knockout of IRF9 gene in Epithelioma papulosum cyprini cells. Fish Shellfish Immunol. 2018, 83, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Ashley, C.L.; Abendroth, A.; McSharry, B.P.; Slobedman, B. Interferon-independent upregulation of interferon-stimulated genes during human cytomegalovirus infection is dependent on IRF3 expression. Viruses 2019, 11, 246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, W.M.; Chevillotte, M.D.; Rice, C.M. Interferon-stimulated genes: A complex web of host defenses. Annu. Rev. Immunol. 2014, 32, 513–545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Q.; Wu, Y.; Qin, Y.; Hu, J.; Xie, W.; Qin, F.X.-F.; Cui, J. Broad and diverse mechanisms used by deubiquitinase family members in regulating the type I interferon signaling pathway during antiviral responses. Sci. Adv. 2018, 4, eaar2824. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Wu, R.; Guo, W.; Xie, L.; Qiao, Z.; Chen, S.; Zhu, J.; Huang, C.; Huang, J.; Chen, B.; et al. STING-Mediated IFI16 Degradation Negatively Controls Type I Interferon Production. Cell Rep. 2019, 29, 1249–1260. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.-S.; Zhang, Z.-Y.; Cai, H.; Zhao, M.; Mao, J.; Dai, J.; Xia, T.; Zhang, X-M.; Li, T. RINCK-mediated monoubiquitination of cGAS promotes antiviral innate immune responses. Cell Biosci. 2018, 8, 35. [Google Scholar] [CrossRef] [Green Version]
- Ju, L.-G.; Zhu, Y.; Lei, P.-J.; Yan, D.; Zhu, K.; Wang, X.; Li, Q.-L.; Li, X.-J.; Chen, J.-W.; Li, L.-Y.; et al. TTLL12 inhibits the activation of cellular antiviral signaling through interaction with VISA/MAVS. J. Immunol. 2017, 198, 1274–1284. [Google Scholar] [CrossRef] [PubMed]
- Ramírez-Carvajal, L.; Singh, N.; de los Santos, T.; Rodríguez, L.L.; Long, C.R. Depletion of elongation initiation factor 4E binding proteins by CRISPR/Cas9 enhances the antiviral response in porcine cells. Antiviral Res. 2016, 125, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Atianand, M.K.; Fitzgerald, K.A. Molecular basis of DNA recognition in the immune system. J. Immunol. 2013, 190, 1911–1918. [Google Scholar] [CrossRef]
- Ma, H.; Qian, W.; Bamouskova, M.; Collins, P.L.; Porter, S.I.; Byrum, A.K.; Zhang, R.; Artyomov, M.; Oltz, E.M.; Mosammaparast, N.; et al. Barrier-to-Autointegration Factor 1 Protects against a Basal cGAS-STING Response. MBio 2020, 11, e00136-20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, S.; Diep, J.; Feng, N.; Ren, L.; Li, B.; Ooi, Y.S.; Wang, X.; Brulois, K.F.; Yasukawa, L.L.; Li, X.; et al. STAG2 deficiency induces interferon responses via cGAS-STING pathway and restricts virus infection. Nat. Commun. 2018, 9, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Morrison, J.H.; Dingli, D.; Poeschla, E. HIV-1 activation of innate immunity depends strongly on the intracellular level of TREX1 and sensing of incomplete reverse transcription products. J. Virol. 2018, 92, e00001-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whelan, J.N.; Li, Y.; Silverman, R.H.; Weiss, S.R. Zika virus production is resistant to RNase L antiviral activity. J. Virol. 2019, 93, e00313-19. [Google Scholar] [CrossRef] [Green Version]
- Schafer, A.R.M.; Smith, J.L.; Pryke, K.M.; DeFilippis, V.R.; Hirsch, A.J. The E3 Ubiquitin Ligase SIAH1 Targets MyD88 for Proteasomal Degradation During Dengue Virus Infection. Front. Microbiol. 2020, 11, 24. [Google Scholar] [CrossRef] [Green Version]
- Taylor, J.P.; Cash, M.N.; Santostefano, K.E.; Nakanishi, M.; Terada, N.; Wallet, M.A. CRISPR/Cas9 knockout of USP18 enhances type I IFN responsiveness and restricts HIV-1 infection in macrophages. J. Leukoc. Biol. 2018, 103, 1225–1240. [Google Scholar] [CrossRef] [PubMed]
- Kuffour, E.O.; Schott, K.; Vasudevan, A.A.J.; Holler, J.; Schulz, W.A.; Lang, P.A.; Lang, K.S.; Kim, B.; Häussinger, D.; König, R.; et al. USP18 (UBP43) abrogates p21-mediated inhibition of HIV-1. J. Virol. 2018, 92, e00592-18. [Google Scholar] [CrossRef] [Green Version]
- Goodwin, C.M.; Schafer, X.; Munger, J. UL26 Attenuates IKKβ-Mediated Induction of Interferon-Stimulated Gene (ISG) Expression and Enhanced Protein ISGylation during Human Cytomegalovirus Infection. J. Virol. 2019, 93, e01052-19. [Google Scholar] [CrossRef]
- Teng, Y.; Luo, M.; Yu, T.; Chen, L.; Huang, Q.; Chen, S.; Xie, L.; Zeng, Y.; Luo, F.; Xiong, H.; et al. CRISPR/Cas9-mediated deletion of miR-146a enhances antiviral response in HIV-1 infected cells. Genes Immun. 2019, 20, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Dufrasne, F.E.; Lombard, C.; Goubau, P.; Ruelle, J. Single amino acid substitution N659D in HIV-2 envelope glycoprotein (Env) impairs viral release and hampers BST-2 antagonism. Viruses 2016, 8, 285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hahn, A.S.; Großkopf, A.K.; Jungnickl, D.; Scholz, B.; Ensser, A. Viral FGARAT homolog ORF75 of rhesus monkey rhadinovirus effects proteasomal degradation of the ND10 components SP100 and PML. J. Virol. 2016, 90, 8013–8028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Botto, S.; Abraham, J.; Mizuno, N.; Pryke, K.; Gall, B.; Landais, I.; Streblow, D.N.; Fruh, K.J.; DeFilippis, V.R. Human cytomegalovirus immediate early 86-kDa protein blocks transcription and induces degradation of the immature interleukin-1β protein during virion-mediated activation of the AIM2 inflammasome. MBio 2019, 10, e02510-18. [Google Scholar] [CrossRef] [Green Version]
- Xu, B.; Pan, Q.; Liang, C. Role of MxB in alpha interferon-mediated inhibition of HIV-1 Infection. J. Virol. 2018, 92, e00422-18. [Google Scholar] [CrossRef] [Green Version]
- Bonifati, S.; Daly, M.B.; Gelais, C.St.; Kim, S.H.; Hollenbaugh, J.A.; Shepard, C.; Kennedy, E.M.; Kim, D.-H.; Schinazi, R.F.; Kim, B.; et al. SAMHD1 controls cell cycle status, apoptosis and HIV-1 infection in monocytic THP-1 cells. Virology 2016, 495, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Feng, C.; Fang, Y.; Zhou, X.; Xu, L.; Wang, W.; Kong, X.; Peppelenbosch, M.P.; Pan, Q.; Yin, Y. The eukaryotic translation initiation factor 4F complex restricts rotavirus infection via regulating the expression of IRF1 and IRF7. Int. J. Mol. Sci. 2019, 20, 1580. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Wei, Z.; Zhang, Y.; Ma, X.; Chen, Y.; Yu, M.; Ma, C.; Li, X.; Cao, Y.; Liu, J.; et al. Activation of liver X receptor plays a central role in antiviral actions of 25-hydroxycholesterol. J. Lipid Res. 2018, 59, 2287–2296. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.S.; Kim, K.H. Effect of CRISPR/Cas9-mediated knockout of either Mx1 or ISG15 gene in EPC cells on resistance against VHSV infection. Fish Shellfish Immunol. 2019, 93, 1041–1046. [Google Scholar] [CrossRef]
- Brezgin, S.; Kostyusheva, A.; Bayurova, E.; Gordeychuk, I.; Isaguliants, M.; Goptar, I.; Nikiforova, A.; Smirnov, V.; Volchkova, E.; Glebe, D.; et al. Replenishment of Hepatitis B Virus cccDNA Pool Is Restricted by Baseline Expression of Host Restriction Factors In Vitro. Microorganisms 2019, 7, 533. [Google Scholar] [CrossRef] [Green Version]
- Zang, R.; Case, J.B.; Yutuc, E.; Ma, X.; Shen, S.; Castro, M.F.G.; Liu, Z.; Zeng, Q.; Zhao, H.; Son, J.; et al. Cholesterol 25-hydroxylase suppresses SARS-CoV-2 replication by blocking membrane fusion. Proc. Natl. Acad. Sci. USA 2020, 117, 32105–32113. [Google Scholar] [CrossRef] [PubMed]
- Passos, V.; Zillinger, T.; Casartelli, N.; Wachs, A.S.; Xu, S.; Malassa, A.; Steppich, K.; Schilling, H.; Franz, S.; Todt, D.; et al. Characterization of endogenous SERINC5 protein as anti-HIV-1 factor. J. Virol. 2019, 93, e01221-19. [Google Scholar] [CrossRef] [Green Version]
- Wąchalska, M.; Graul, M.; Praest, P.; Luteijn, R.D.; Babnis, A.W.; Wiertz, E.J.H.J.; Bieńkowska-Szewczyk, K.; Lipińska, A.D. Fluorescent TAP as a Platform for Virus-Induced Degradation of the Antigenic Peptide Transporter. Cells 2019, 8, 1590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- den Haan, J.M.M.; Lehar, S.M.; Bevan, M.J. CD8+ but not CD8− dendritic cells cross-prime cytotoxic T cells in vivo. J. Exp. Med. 2000, 192, 1685–1696. [Google Scholar] [CrossRef] [PubMed]
- Sali, T.M.; Pryke, K.M.; Abraham, J.; Liu, A.; Archer, I.; Broeckel, R.; Staverosky, J.A.; Smith, J.L.; Al-Shammari, A.; Amsler, L.; et al. Characterization of a novel human-specific STING agonist that elicits antiviral activity against emerging alphaviruses. PLoS Pathog. 2015, 11, e1005324. [Google Scholar] [CrossRef]
- Vanwalscappel, B.; Tada, T.; Landau, N.R. Toll-like receptor agonist R848 blocks Zika virus replication by inducing the antiviral protein viperin. Virology 2018, 522, 199–208. [Google Scholar] [CrossRef]
- Wang, B.; Lam, T.H.; Soh, M.K.; Ye, Z.; Chen, J.; Ren, E.C. Influenza A virus facilitates its infectivity by activating p53 to inhibit the expression of interferon-induced transmembrane proteins. Front. Immunol. 2018, 9, 1193. [Google Scholar] [CrossRef] [PubMed]
- Puray-Chavez, M.N.; Farghali, M.H.; Yapo, V.; Huber, A.D.; Liu, D.; Ndongwe, T.P.; Casey, M.C.; Laughlin, T.G.; Hannik, M.; Tedbury, P.R.; et al. Effects of Moloney Leukemia Virus 10 Protein on Hepatitis B Virus Infection and Viral Replication. Viruses 2019, 11, 651. [Google Scholar] [CrossRef] [Green Version]
- James, C.D.; Prabhakar, A.T.; Otoa, R.; Evans, M.R.; Wang, X.; Bristol, M.L.; Zhang, K.; Li, R.; Morgan, I.M. SAMHD1 Regulates Human Papillomavirus 16-Induced Cell Proliferation and Viral Replication during Differentiation of Keratinocytes. mSphere 2019, 4, e00448-19. [Google Scholar] [CrossRef] [Green Version]
- Sharma, A.; McLaughlin, R.N., Jr.; Basom, R.S.; Kikawa, C.; OhAinle, M.; Yount, J.S.; Emerman, M.; Overbaugh, J. Macaque interferon-induced transmembrane proteins limit replication of SHIV strains in an Envelope-dependent manner. PLoS Pathog. 2019, 15, e1007925. [Google Scholar] [CrossRef]
- Spence, J.S.; He, R.; Hoffmann, H.-H.; Das, T.; Thinon, E.; Rice, C.M.; Peng, T.; Chandran, K.; Hang, H.C. IFITM3 directly engages and shuttles incoming virus particles to lysosomes. Nat. Chem. Biol. 2019, 15, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Sivan, G.; Ormanoglu, P.; Buehler, E.C.; Martin, S.E.; Moss, B. Identification of restriction factors by human genome-wide RNA interference screening of viral host range mutants exemplified by discovery of SAMD9 and WDR6 as inhibitors of the vaccinia virus K1L− C7L−mutant. MBio 2015, 6, e01122-15. [Google Scholar] [CrossRef] [Green Version]
- Meng, X.; Zhang, F.; Yan, B.; Si, C.; Honda, H.; Nagamachi, A.; Sun, L.Z.; Xian, Y. A paralogous pair of mammalian host restriction factors form a critical host barrier against poxvirus infection. PLoS Pathog. 2018, 14, e1006884. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.E.; Bottero, V.; Flaherty, S.; Dutta, S.; Singh, V.V.; Chandran, B. IFI16 restricts HSV-1 replication by accumulating on the hsv-1 genome, repressing HSV-1 gene expression, and directly or indirectly modulating histone modifications. PLoS Pathog. 2014, 10, e1004503. [Google Scholar] [CrossRef] [PubMed]
- Van der Hoek, K.H.; Eyre, N.S.; Shue, B.; Khantisitthiporn, O.; Glab-Ampi, K.; Carr, J.M.; Gartner, M.J.; Jolly, L.A.; Thomas, P.Q.; Adikusuma, F.; et al. Viperin is an important host restriction factor in control of Zika virus infection. Sci. Rep. 2017, 7, 1–14. [Google Scholar]
- Cacciarelli, T.V.; Martinez, O.M.; Gish, R.G.; Villanueva, J.C.; Krams, S.M. Immunoregulatory cytokines in chronic hepatitis C virus infection: Pre-and posttreatment with interferon alfa. Hepatology 1996, 24, 6–9. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Sheen, I.; Chien, R.; Chu, C.; Liaw, Y. Long-term beneficial effect of interferon therapy in patients with chronic hepatitis B virus infection. Hepatology 1999, 29, 971–975. [Google Scholar] [CrossRef] [PubMed]
- Kühl, U.; Pauschinger, M.; Schwimmbeck, P.L.; Seeberg, B.; Lober, C.; Noutsias, M.; Poller, W.; Schultheiss, H.P. Interferon-β treatment eliminates cardiotropic viruses and improves left ventricular function in patients with myocardial persistence of viral genomes and left ventricular dysfunction. Circulation 2003, 107, 2793–2798. [Google Scholar] [CrossRef] [Green Version]
- Bogerd, H.P.; Kornepati, A.V.R.; Marshall, J.B.; Kennedy, E.M.; Cullen, B.R. Specific induction of endogenous viral restriction factors using CRISPR/Cas-derived transcriptional activators. Proc. Natl. Acad. Sci. USA 2015, 112, E7249–E7256. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Ozono, S.; Yao, W.; Tobiume, M.; Yamaoka, S.; Kishigami, S.; Fujita, H.; Tokunaga, K. CRISPR-mediated activation of endogenous BST-2/tetherin expression inhibits wild-type HIV-1 production. Sci. Rep. 2019, 9, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Liang, G.; Liu, G.; Kitamura, K.; Wang, Z.; Chowdhury, S.; Monjurul, A.M.; Wakae, K.; Koura, M.; Shimadu, M.; Kinoshita, K.; et al. TGF-β suppression of HBV RNA through AID-dependent recruitment of an RNA exosome complex. PLoS Pathog 2015, 11, e1004780. [Google Scholar] [CrossRef]
- Brezgin, S.; Kostyusheva, A.; Ponomareva, N.; Volia, V.; Goptar, I.; Nikiforova, A.; Shilovskiy, I.; Smirnov, V.; Kostyushev, D.; Chulanov, V. Clearing of Foreign Episomal DNA from Human Cells by CRISPRa-Mediated Activation of Cytidine Deaminases. Int. J. Mol. Sci. 2020, 21, 6865. [Google Scholar] [CrossRef]
- Roberts, S.A.; Lawrence, M.S.; Klimczak, L.J.; Grimm, S.A.; Fargo, D.; Stojanov, P.; Kiezun, A.; Kryukov, G.V.; Carter, S.L.; Saksena, G.; et al. An APOBEC cytidine deaminase mutagenesis pattern is widespread in human cancers. Nat. Genet. 2013, 45, 1–8. [Google Scholar] [CrossRef]
- Schröfelbauer, B.; Chen, D.; Landau, N.R. A single amino acid of APOBEC3G controls its species-specific interaction with virion infectivity factor (Vif). Proc. Natl. Acad. Sci. USA 2004, 101, 3927–3932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pham, Q.T.; Bouchard, A.; Grütter, M.G.; Berthoux, L. Generation of human TRIM5 α mutants with high HIV-1 restriction activity. Gene Ther. 2010, 17, 859–871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dufour, C.; Claudel, A.; Joubarne, N.; Merindol, N.; Maisonnet, T.; Masroori, N.; Plourde, M.B.; Berthoux, L. Editing of the human TRIM5 gene to introduce mutations with the potential to inhibit HIV-1. PLoS ONE 2018, 13, e0191709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saayman, S.M.; Lazar, D.C.; Scott, T.A.; Hart, J.R.; Takahashi, M.; Burnett, J.C.; Planelles, V.; Morris, K.V.; Weinberg, M.S. Potent and Targeted Activation of Latent HIV-1 Using the CRISPR/dCas9 Activator Complex. Mol. Ther. 2016, 24, 488–498. [Google Scholar] [CrossRef] [Green Version]
- Limsirichai, P.; Gaj, T.; Schaffer, D.V. CRISPR-mediated Activation of Latent HIV-1 Expression. Mol. Ther. 2016, 24, 499–507. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Yin, C.; Zhang, T.; Li, F.; Yang, W.; Kaminski, R.; Fagan, P.R.; Putatunda, R.; Young, W.-B.; Khalili, K.; et al. CRISPR/gRNA-directed synergistic activation mediator (SAM) induces specific, persistent and robust reactivation of the HIV-1 latent reservoirs. Sci. Rep. 2015, 5, 1–14. [Google Scholar] [CrossRef]
- Ji, H.; Jiang, Z.; Lu, P.; Ma, L.; Li, C.; Pan, H.; Fu, Z.; Qu, X.; Wang, P.; Deng, J.; et al. Specific Reactivation of Latent HIV-1 by dCas9-SunTag-VP64-mediated Guide RNA Targeting the HIV-1 Promoter. Mol. Ther. 2016, 24, 508–521. [Google Scholar] [CrossRef] [Green Version]
- Linder, B.; Grozhik, A.V.; Olarerin-George, A.O.; Meydan, C.; Mason, C.E.; Jaffrey, S.R. Single-nucleotide-resolution mapping of m6A and m6Am throughout the transcriptome. Nat. Methods 2015, 12, 767–772. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Choe, J.; Park, O.H.; Kim, Y.K. Molecular mechanisms driving mRNA degradation by m6A modification. Trends Genet. 2020, 36, 177–188. [Google Scholar] [CrossRef] [Green Version]
- Meyer, K.D.; Jaffrey, S.R. Rethinking m6A readers, writers, and erasers. Annu. Rev. Cell Dev. Biol. 2017, 33, 319–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scarrow, M.; Chen, N.; Sun, G. Insights into the N6-methyladenosine mechanism and its functionality: Progress and questions. Crit. Rev. Biotechnol. 2020, 40, 639–652. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Li, H.; Wu, A.; Peng, Y.; Shu, G.; Yin, G. Functions of N6-methyladenosine and its role in cancer. Mol. Cancer 2019, 18, 176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, F.; et al. Association of N6-methyladenosine with viruses and related diseases. Virol. J. 2019, 16, 133. [Google Scholar] [CrossRef] [Green Version]
- Winkler, R.; Gillis, E.; Lasman, L.; Safra, M.; Geula, S.; Soyris, C.; Nachshon, A.; Tai-Schmeidel, J.; Freidman, N.; Le-Trilling, V.T.K.; et al. m 6 A modification controls the innate immune response to infection by targeting type I interferons. Nat. Immunol. 2019, 20, 173–182. [Google Scholar] [CrossRef]
- Wang, L.; Wen, M.; Cao, X. Nuclear hnRNPA2B1 initiates and amplifies the innate immune response to DNA viruses. Science 2019, 365, eaav0758. [Google Scholar] [CrossRef]
- Lu, M.; Zhang, Z.; Xue, M.; Zhao, B.S.; Harder, O.; Li, A.; Liang, X.; Gao, T.Z.; Xu, Y.; Zhou, J.; et al. N 6-methyladenosine modification enables viral RNA to escape recognition by RNA sensor RIG-I. Nat. Microbiol. 2020, 5, 584–598. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.G.; Chen, R.; Ahmad, S.; Verma, R.; Kasturi, S.P.; Amaya, L.; Broughton, J.P.; Kim, J.; Cadena, C.; Pulendran, B.; et al. N6-methyladenosine modification controls circular RNA immunity. Mol. Cell 2019, 76, 96–109. [Google Scholar] [CrossRef]
- Lewis, C.J.T.; Pan, T.; Kalsotra, A. RNA modifications and structures cooperate to guide RNA–protein interactions. Nat. Rev. Mol. Cell Biol. 2017, 18, 202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.M.; Zhou, J.; Mao, Y.H.; Ji, Q.Q.; Qian, S.B. Programmable RNA N-6-methyladenosine editing by CRISPR-Cas9 conjugates. Nat. Chem. Biol. 2019, 15, 865. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.; Chen, P.J.; Miao, Z.; Liu, D.R. Programmable m 6 A modification of cellular RNAs with a Cas13-directed methyltransferase. Nat. Biotechnol. 2020, 38, 1431–1440. [Google Scholar] [CrossRef] [PubMed]
- Tycko, J.; Myer, V.E.; Hsu, P.D. Methods for optimizing CRISPR-Cas9 genome editing specificity. Mol. Cell 2016, 63, 355–370. [Google Scholar] [CrossRef] [Green Version]
- Kocak, D.D.; Josephs, E.A.; Bhandarkar, V.; Adkar, S.S.; Kwon, J.B.; Gersbach, C.A. Increasing the specificity of CRISPR systems with engineered RNA secondary structures. Nat. Biotechnol. 2019, 37, 657. [Google Scholar] [CrossRef]
- Slaymaker, I.M.; Gao, L.; Zetscje, N.; Scott, D.A.; Yan, W.X.; Zhang, F. Rationally engineered Cas9 nucleases with improved specificity. Science 2016, 351, 84–88. [Google Scholar] [CrossRef] [Green Version]
- Kleinstiver, B.P.; Pattanayal, V.; Prew, M.S.; Tsai, S.Q.; Nguyen, N.T.; Zheng, Z.; Joung, J.K. High-fidelity CRISPR–Cas9 nucleases with no detectable genome-wide off-target effects. Nature 2016, 529, 490–495. [Google Scholar] [CrossRef] [Green Version]
- Agudelo, D.; Carter, S.; Velimirovic, M.; Duringer, A.; Rivest, J.-F.; Levesque, S.; Loehr, J.; Mouchiroud, M.; Cyr, D.; Waters, P.J.; et al. Versatile and robust genome editing with Streptococcus thermophilus CRISPR1-Cas9. Genome Res. 2020, 30, 107–117. [Google Scholar] [CrossRef]
- Kostyushev, D.; Brezgin, S.; Kostyusheva, A.; Zarifyan, D.; Goptar, I.; Chulanov, V. Orthologous CRISPR/Cas9 systems for specific and efficient degradation of covalently closed circular DNA of hepatitis B virus. Cell. Mol. LIFE Sci. 2019, 76, 1779–1794. [Google Scholar] [CrossRef]
- Charlesworth, C.T.; Deshpande, P.S.; Dever, D.P.; Camarena, J.; Lemgart, V.T.; Cromer, M.K.; Vakulskas, C.A.; Collingwood, M.A.; Zhang, L.; Bode, N.M.; et al. Identification of preexisting adaptive immunity to Cas9 proteins in humans. Nat. Med. 2019, 25, 249. [Google Scholar] [CrossRef]
- Ferdosi, S.R.; Ewaisha, R.; Moghadam, F.; Krishna, S.; Park, J.G.; Ebrahimkhani, M.R.; Kiani, S.; Anderson, K.S. Multifunctional CRISPR-Cas9 with engineered immunosilenced human T cell epitopes. Nat. Commun. 2019, 10, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kostyushev, D.; Kostyusheva, A.; Brezgin, S.; Smirnov, V.; Volchkova, E.; Lukashev, A.; Chulanov, V. Gene Editing by Extracellular Vesicles. Int. J. Mol. Sci. 2020, 21, 7362. [Google Scholar] [CrossRef]
- Gillmore, J.D.; Gane, E.; Taubel, J.; Kao, J.; Fontana, M.; Maitland, M.L.; Seitzer, J.; O’Connel, D.; Walsh, K.R.; Wood, K.; et al. CRISPR-Cas9 In Vivo Gene Editing for Transthyretin Amyloidosis. N. Engl. J. Med. 2021. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Wang, J.; Liu, Y.; Xie, L.; Su, B.; Mou, D.; Wang, L.; Liu, T.; Wang, X.; Zhao, L.; et al. CRISPR-edited stem cells in a patient with HIV and acute lymphocytic leukemia. N. Engl. J. Med. 2019, 381, 1240–1247. [Google Scholar] [CrossRef] [PubMed]
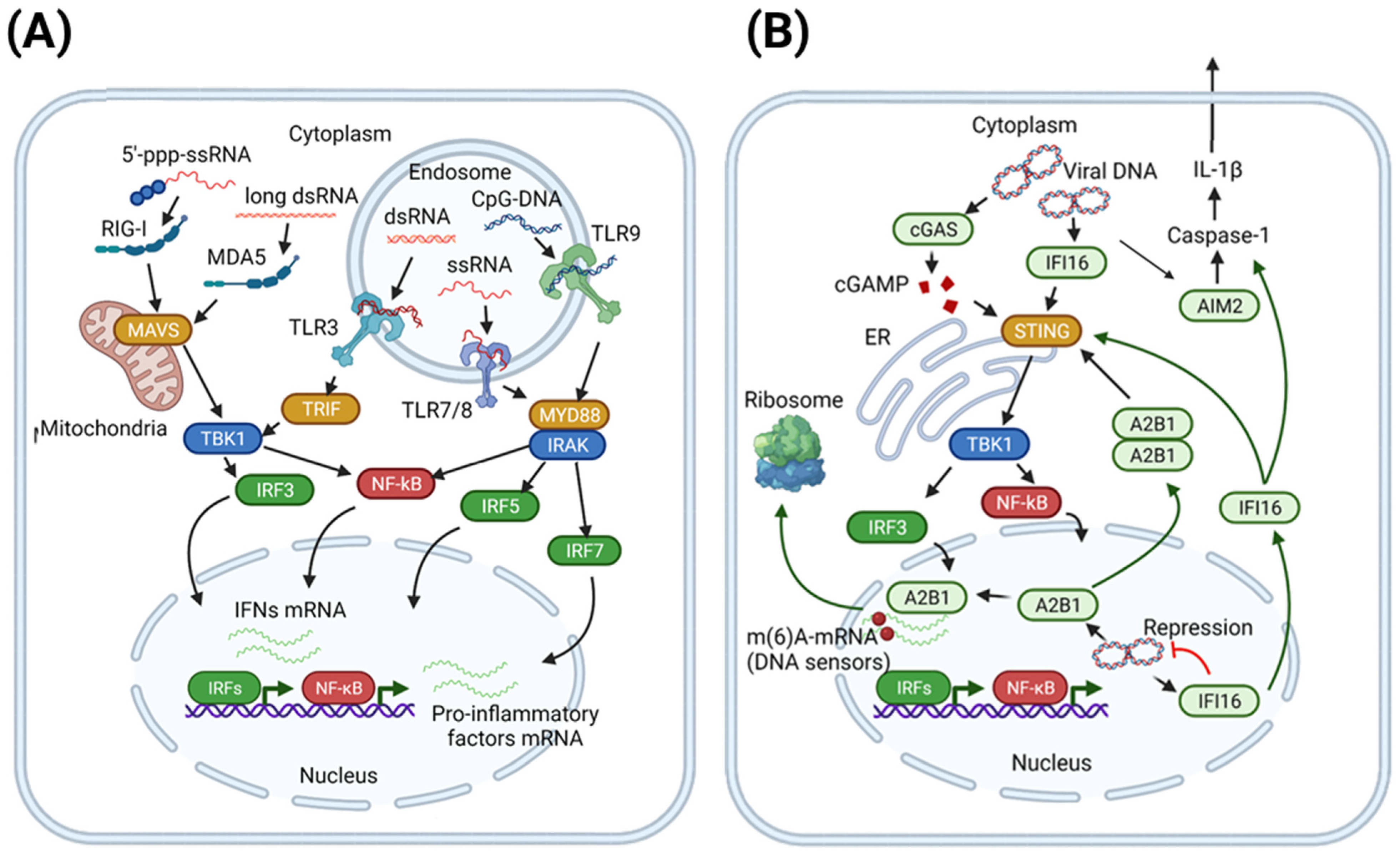
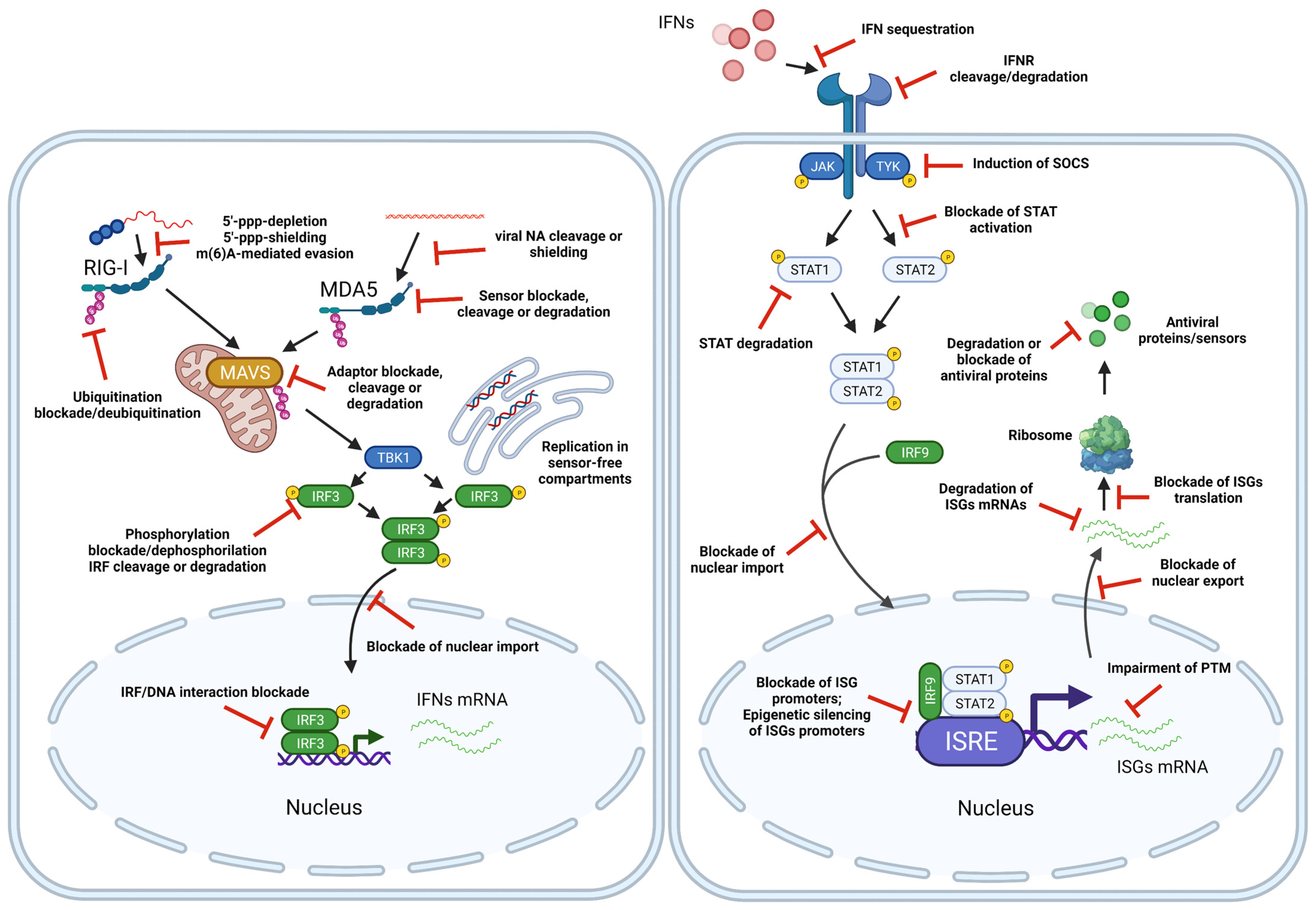

| System Effect | System | Number of Main Components | Relative Efficacy | Ref. | |
|---|---|---|---|---|---|
| Activation | dCas9–VP64 | 2 | ↑ | [161] | |
| dCas9–VP160/VP192 | 2 | ↑↑ | [158] | ||
| dCas9–p65/p65–HSF | 2 | ↑/↑↑ | [165] | ||
| dCas9–p300 | 2 | ↑↑ | [160] | ||
| dCas9–VPR | 2 | ↑↑↑ | [163] | ||
| dCas9–CBP | 2 | ↑↑↑ | [166] | ||
| Scaffold | 3 | ↑↑↑ | [162] | ||
| dCas9–SunTag–VP64 | 3 | ↑↑↑ | [164] | ||
| SPH (SunTag–p65–HSF1) | 3 | ↑↑↑↑ | [167] | ||
| SAM | 3 | ↑↑↑↑ | [165] | ||
| Repression | dCas9–KRAB | 2 | ↑↑ | [168] | |
| dCas9–KRAB–MeCP | 2 | ↑↑↑ | [177] | ||
| dCas9–EZH2 | 2 | ↑↑ | [169] | ||
| Base Editors | |||||
| System | Target | Base Change | Editing Window (nts from PAM) | Ref. | |
| APOBEC1–BE3 | DNA | C→T/G→A | 13-17 | [178] | |
| APOBEC1–BE4 | DNA | C→T/G→A | 13-17 | [179] | |
| APOBEC3A–BE3 | DNA | C→T/G→A | 13-17 | [180] | |
| BE–PLUS | DNA | C→T/G→A | 7-17 | [181] | |
| CRISPR-X | DNA | C→T/G→A | −50 to +50 from PAM | [182] | |
| ABE-7.10 | DNA | A→G/T→C | 14-17 | [183] | |
| REPAIR | RNA | A→I | - | [173] | |
| RESCUE | RNA | A→I/C→U | - | [174] | |
| CURE | RNA | C→U | - | [175] | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brezgin, S.; Kostyusheva, A.; Bayurova, E.; Volchkova, E.; Gegechkori, V.; Gordeychuk, I.; Glebe, D.; Kostyushev, D.; Chulanov, V. Immunity and Viral Infections: Modulating Antiviral Response via CRISPR–Cas Systems. Viruses 2021, 13, 1373. https://doi.org/10.3390/v13071373
Brezgin S, Kostyusheva A, Bayurova E, Volchkova E, Gegechkori V, Gordeychuk I, Glebe D, Kostyushev D, Chulanov V. Immunity and Viral Infections: Modulating Antiviral Response via CRISPR–Cas Systems. Viruses. 2021; 13(7):1373. https://doi.org/10.3390/v13071373
Chicago/Turabian StyleBrezgin, Sergey, Anastasiya Kostyusheva, Ekaterina Bayurova, Elena Volchkova, Vladimir Gegechkori, Ilya Gordeychuk, Dieter Glebe, Dmitry Kostyushev, and Vladimir Chulanov. 2021. "Immunity and Viral Infections: Modulating Antiviral Response via CRISPR–Cas Systems" Viruses 13, no. 7: 1373. https://doi.org/10.3390/v13071373
APA StyleBrezgin, S., Kostyusheva, A., Bayurova, E., Volchkova, E., Gegechkori, V., Gordeychuk, I., Glebe, D., Kostyushev, D., & Chulanov, V. (2021). Immunity and Viral Infections: Modulating Antiviral Response via CRISPR–Cas Systems. Viruses, 13(7), 1373. https://doi.org/10.3390/v13071373