
Regulation of Viral Restriction by Post-Translational Modifications

 ,
,  , and
, and {kind=link}
{kind=link}
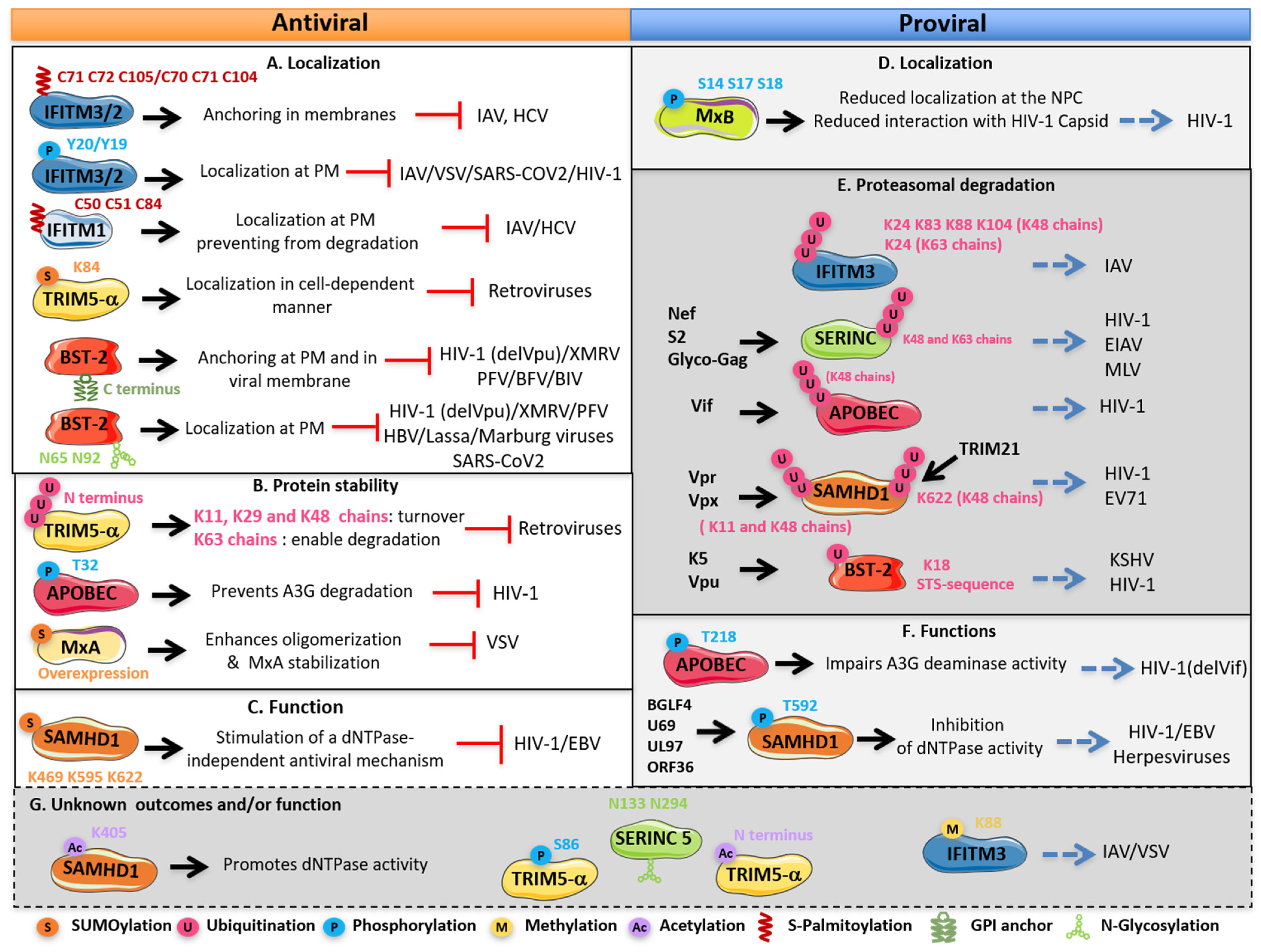{kind=link}
Abstract
1. Introduction
2. Post-Translational Modifications (PTMs)
2.1. PTMs Based on the Addition of Polypeptides: Ubiquitination and SUMOylation
2.2. PTMs Based on the Addition of Functional Groups: Glycosylation and S-Palmitoylation
2.3. Protein Chemical Changes: Phosphorylation, Acetylation and Methylation
3. Antiviral Factors Are Highly Regulated by PTMs
3.1. IFITM Proteins
3.1.1. S-Palmitoylation of IFITM Is Required for Its Localization and Antiviral Activity
3.1.2. Phosphorylation of IFITM3 Regulates Its Cell Trafficking and Promotes Its Antiviral Activity
3.1.3. Ubiquitination of IFITM3 Promotes Its Degradation and Reduces Its Antiviral Activity
3.1.4. Methylation of IFITM Impairs Its Antiviral Effect
3.2. Ubiquitination of SERINC Proteins Lead to Its Degradation upon Infection
3.3. TRIM5α
3.3.1. TRIM5α Exerts Its Antiviral Activity through Auto-Polyubiquitination
3.3.2. SUMOylation of TRIM5α Regulates Its Antiviral Activity and Affects Its Subcellular Localization in a Cell-Dependent Manner
3.4. APOBEC3s Proteins
3.4.1. Ubiquitination of APOBEC3 Is Useful for Viruses
3.4.2. Phosphorylation of APOBEC3: Divergent Actions Affecting Its Antiviral Activity
3.5. SAMHD1
3.5.1. Inactivation of SAMHD1 through Phosphorylation
3.5.2. A Potential Role of Acetylation and SUMOylation in the Regulation of SAMHD1 Functions
3.5.3. Lentiviral Antagonism by Ubiquitination of SAMHD1
3.6. MxA and MxB
3.6.1. SUMOylation Regulates MxA Antiviral Activity
3.6.2. Phosphorylation Regulates MxB Antiviral Activity
3.7. BST2
3.7.1. BST2 Glycosylation Modulates Its Activity
3.7.2. Viral Antagonisms of BST2 by Its Ubiquitination and Degradation
4. Recent Identified Restriction Factors and Their PTMs
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Doyle, T.; Goujon, C.; Malim, M.H. HIV-1 and Interferons: Who’s Interfering with Whom? Nat. Rev. Microbiol. 2015, 13, 403–413. [Google Scholar] [CrossRef] [PubMed]
- Hotter, D.; Kirchhoff, F. Interferons and beyond: Induction of Antiretroviral Restriction Factors. J. Leukoc. Biol. 2018, 103, 465–477. [Google Scholar] [CrossRef]
- Lindenmann, J. Resistance of Mice to Mouse-Adapted Influenza A Virus. Virology 1962, 16, 203–204. [Google Scholar] [CrossRef]
- Lilly, F. Fv-2: Identification and Location of a Second Gene Governing the Spleen Focus Response to Friend Leukemia Virus in Mice. J. Natl. Cancer Inst. 1970, 45, 163–169. [Google Scholar] [PubMed]
- Stremlau, M.; Owens, C.M.; Perron, M.J.; Kiessling, M.; Autissier, P.; Sodroski, J. The Cytoplasmic Body Component TRIM5a Restricts HIV-1 Infection in Old World Monkeys. Nature 2004, 427, 8. [Google Scholar] [CrossRef]
- Pertel, T.; Hausmann, S.; Morger, D.; Züger, S.; Guerra, J.; Lascano, J.; Reinhard, C.; Santoni, F.A.; Uchil, P.D.; Chatel, L.; et al. TRIM5 Is an Innate Immune Sensor for the Retrovirus Capsid Lattice. Nature 2011, 472, 361–365. [Google Scholar] [CrossRef] [PubMed]
- Sheehy, A.M.; Gaddis, N.C.; Choi, J.D.; Malim, M.H. Isolation of a Human Gene That Inhibits HIV-1 Infection and Is Suppressed by the Viral Vif Protein. Nature 2002, 418, 646–650. [Google Scholar] [CrossRef]
- Mangeat, B.; Turelli, P.; Caron, G.; Friedli, M.; Perrin, L.; Trono, D. Broad Antiretroviral Defence by Human APOBEC3G through Lethal Editing of Nascent Reverse Transcripts. Nature 2003, 424, 99–103. [Google Scholar] [CrossRef]
- Zhang, H.; Yang, B.; Pomerantz, R.J.; Zhang, C.; Arunachalam, S.C.; Gao, L. The Cytidine Deaminase CEM15 Induces Hypermutation in Newly Synthesized HIV-1 DNA. Nature 2003, 424, 94–98. [Google Scholar] [CrossRef]
- Laguette, N.; Sobhian, B.; Casartelli, N.; Ringeard, M.; Chable-Bessia, C.; Ségéral, E.; Yatim, A.; Emiliani, S.; Schwartz, O.; Benkirane, M. SAMHD1 Is the Dendritic- and Myeloid-Cell-Specific HIV-1 Restriction Factor Counteracted by Vpx. Nature 2011, 474, 654–657. [Google Scholar] [CrossRef]
- Lahouassa, H.; Daddacha, W.; Hofmann, H.; Ayinde, D.; Logue, E.C.; Dragin, L.; Bloch, N.; Maudet, C.; Bertrand, M.; Gramberg, T.; et al. SAMHD1 Restricts the Replication of Human Immunodeficiency Virus Type 1 by Depleting the Intracellular Pool of Deoxynucleoside Triphosphates. Nat. Immunol. 2012, 13, 223–228. [Google Scholar] [CrossRef] [PubMed]
- Goujon, C.; Moncorgé, O.; Bauby, H.; Doyle, T.; Ward, C.C.; Schaller, T.; Hué, S.; Barclay, W.S.; Schulz, R.; Malim, M.H. Human MX2 Is an Interferon-Induced Post-Entry Inhibitor of HIV-1 Infection. Nature 2013, 502, 559–562. [Google Scholar] [CrossRef] [PubMed]
- Kane, M.; Yadav, S.S.; Bitzegeio, J.; Kutluay, S.B.; Zang, T.; Wilson, S.J.; Schoggins, J.W.; Rice, C.M.; Yamashita, M.; Hatziioannou, T.; et al. MX2 Is an Interferon-Induced Inhibitor of HIV-1 Infection. Nature 2013, 502, 563–566. [Google Scholar] [CrossRef]
- Liu, Z.; Pan, Q.; Ding, S.; Qian, J.; Xu, F.; Zhou, J.; Cen, S.; Guo, F.; Liang, C. The Interferon-Inducible MxB Protein Inhibits HIV-1 Infection. Cell Host Microbe 2013, 14, 398–410. [Google Scholar] [CrossRef]
- Neil, S.J.D.; Zang, T.; Bieniasz, P.D. Tetherin Inhibits Retrovirus Release and Is Antagonized by HIV-1 Vpu. Nature 2008, 451, 425–430. [Google Scholar] [CrossRef]
- Van Damme, N.; Goff, D.; Katsura, C.; Jorgenson, R.L.; Mitchell, R.; Johnson, M.C.; Stephens, E.B.; Guatelli, J. The Interferon-Induced Protein BST-2 Restricts HIV-1 Release and Is Downregulated from the Cell Surface by the Viral Vpu Protein. Cell Host Microbe 2008, 3, 245–252. [Google Scholar] [CrossRef]
- Usami, Y.; Wu, Y.; Göttlinger, H.G. SERINC3 and SERINC5 Restrict HIV-1 Infectivity and Are Counteracted by Nef. Nature 2015, 526, 218–223. [Google Scholar] [CrossRef]
- Rosa, A.; Chande, A.; Ziglio, S.; De Sanctis, V.; Bertorelli, R.; Goh, S.L.; McCauley, S.M.; Nowosielska, A.; Antonarakis, S.E.; Luban, J.; et al. HIV-1 Nef Promotes Infection by Excluding SERINC5 from Virion Incorporation. Nature 2015, 526, 212–217. [Google Scholar] [CrossRef]
- Brass, A.L.; Huang, I.-C.; Benita, Y.; John, S.P.; Krishnan, M.N.; Feeley, E.M.; Ryan, B.J.; Weyer, J.L.; van der Weyden, L.; Fikrig, E.; et al. The IFITM Proteins Mediate Cellular Resistance to Influenza A H1N1 Virus, West Nile Virus, and Dengue Virus. Cell 2009, 139, 1243–1254. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-C.; Peterson, S.E.; Loring, J.F. Protein Post-Translational Modifications and Regulation of Pluripotency in Human Stem Cells. Cell Res. 2014, 24, 143–160. [Google Scholar] [CrossRef]
- Spoel, S.H. Orchestrating the Proteome with Post-Translational Modifications. J. Exp. Bot. 2018, 69, 4499–4503. [Google Scholar] [CrossRef] [PubMed]
- Orford, K.; Crockett, C.; Jensen, J.P.; Weissman, A.M.; Byers, S.W. Serine Phosphorylation-Regulated Ubiquitination and Degradation of Beta-Catenin. J. Biol. Chem. 1997, 272, 24735–24738. [Google Scholar] [CrossRef]
- Waby, J.S.; Bingle, C.D.; Corfe, B.M. Post-Translational Control of Sp-Family Transcription Factors. Curr. Genomics 2008, 9, 301–311. [Google Scholar] [CrossRef] [PubMed]
- Hannoun, Z.; Maarifi, G.; Chelbi-Alix, M.K. The Implication of SUMO in Intrinsic and Innate Immunity. Cytokine Growth Factor Rev. 2016, 29, 3–16. [Google Scholar] [CrossRef]
- Swatek, K.N.; Komander, D. Ubiquitin Modifications. Cell Res. 2016, 26, 399–422. [Google Scholar] [CrossRef]
- Morreale, F.E.; Walden, H. Types of Ubiquitin Ligases. Cell 2016, 165, 248–248.e1. [Google Scholar] [CrossRef] [PubMed]
- Mevissen, T.E.T.; Komander, D. Mechanisms of Deubiquitinase Specificity and Regulation. Annu. Rev. Biochem. 2017, 86, 159–192. [Google Scholar] [CrossRef]
- Husnjak, K.; Dikic, I. Ubiquitin-Binding Proteins: Decoders of Ubiquitin-Mediated Cellular Functions. Annu. Rev. Biochem. 2012, 81, 291–322. [Google Scholar] [CrossRef]
- Haglund, K.; Dikic, I. Ubiquitylation and Cell Signaling. EMBO J. 2005, 24, 3353–3359. [Google Scholar] [CrossRef] [PubMed]
- Ciechanover, A.; Stanhill, A. The Complexity of Recognition of Ubiquitinated Substrates by the 26S Proteasome. Biochim. Biophys. Acta 2014, 1843, 86–96. [Google Scholar] [CrossRef]
- Li, S.; Ahmad, I.; Shi, J.; Wang, B.; Yu, C.; Zhang, L.; Zheng, Y.-H. Murine Leukemia Virus Glycosylated Gag Reduces Murine SERINC5 Protein Expression at Steady-State Levels via the Endosome/Lysosome Pathway to Counteract SERINC5 Antiretroviral Activity. J. Virol. 2018, 93, e01651-18. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, I.; Li, S.; Li, R.; Chai, Q.; Zhang, L.; Wang, B.; Yu, C.; Zheng, Y.-H. The Retroviral Accessory Proteins S2, Nef, and GlycoMA Use Similar Mechanisms for Antagonizing the Host Restriction Factor SERINC5. J. Biol. Chem. 2019, 294, 7013–7024. [Google Scholar] [CrossRef]
- Pardieu, C.; Vigan, R.; Wilson, S.J.; Calvi, A.; Zang, T.; Bieniasz, P.; Kellam, P.; Towers, G.J.; Neil, S.J.D. The RING-CH Ligase K5 Antagonizes Restriction of KSHV and HIV-1 Particle Release by Mediating Ubiquitin-Dependent Endosomal Degradation of Tetherin. PLoS Pathog. 2010, 6, e1000843. [Google Scholar] [CrossRef] [PubMed]
- Roy, N.; Pacini, G.; Berlioz-Torrent, C.; Janvier, K. Characterization of E3 Ligases Involved in Lysosomal Sorting of the HIV-1 Restriction Factor BST2. J. Cell Sci. 2017, 130, 1596–1611. [Google Scholar] [CrossRef]
- Sheehy, A.M.; Gaddis, N.C.; Malim, M.H. The Antiretroviral Enzyme APOBEC3G Is Degraded by the Proteasome in Response to HIV-1 Vif. Nat. Med. 2003, 9, 1404–1407. [Google Scholar] [CrossRef] [PubMed]
- Hrecka, K.; Hao, C.; Gierszewska, M.; Swanson, S.K.; Kesik-Brodacka, M.; Srivastava, S.; Florens, L.; Washburn, M.P.; Skowronski, J. Vpx Relieves Inhibition of HIV-1 Infection of Macrophages Mediated by the SAMHD1 Protein. Nature 2011, 474, 658–661. [Google Scholar] [CrossRef]
- Ciechanover, A.; Ben-Saadon, R. N-Terminal Ubiquitination: More Protein Substrates Join In. Trends Cell Biol. 2004, 14, 103–106. [Google Scholar] [CrossRef]
- Celen, A.B.; Sahin, U. Sumoylation on Its 25th Anniversary: Mechanisms, Pathology, and Emerging Concepts. FEBS J. 2020, 287, 3110–3140. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; He, Y.; Wang, X.; Liang, Z.; He, G.; Zhang, P.; Zhu, H.; Xu, N.; Liang, S. Protein SUMOylation Modification and Its Associations with Disease. Open Biol. 2017, 7, 170167. [Google Scholar] [CrossRef]
- Boulanger, M.; Chakraborty, M.; Tempé, D.; Piechaczyk, M.; Bossis, G. SUMO and Transcriptional Regulation: The Lessons of Large-Scale Proteomic, Modifomic and Genomic Studies. Molecules 2021, 26, 828. [Google Scholar] [CrossRef]
- Hendriks, I.A.; Vertegaal, A.C.O. A Comprehensive Compilation of SUMO Proteomics. Nat. Rev. Mol. Cell Biol. 2016, 17, 581–595. [Google Scholar] [CrossRef]
- Hendriks, I.A.; Lyon, D.; Su, D.; Skotte, N.H.; Daniel, J.A.; Jensen, L.J.; Nielsen, M.L. Site-Specific Characterization of Endogenous SUMOylation across Species and Organs. Nat. Commun. 2018, 9, 2456. [Google Scholar] [CrossRef]
- Maarifi, G.; Fernandez, J.; Portilho, D.M.; Boulay, A.; Dutrieux, J.; Oddos, S.; Butler-Browne, G.; Nisole, S.; Arhel, N.J. RanBP2 Regulates the Anti-Retroviral Activity of TRIM5α by SUMOylation at a Predicted Phosphorylated SUMOylation Motif. Commun. Biol. 2018, 1, 193. [Google Scholar] [CrossRef] [PubMed]
- De la Cruz-Herrera, C.F.; Campagna, M.; García, M.A.; Marcos-Villar, L.; Lang, V.; Baz-Martínez, M.; Gutiérrez, S.; Vidal, A.; Rodríguez, M.S.; Esteban, M.; et al. Activation of the Double-Stranded RNA-Dependent Protein Kinase PKR by Small Ubiquitin-like Modifier (SUMO). J. Biol. Chem. 2014, 289, 26357–26367. [Google Scholar] [CrossRef] [PubMed]
- Brantis-de-Carvalho, C.E.; Maarifi, G.; Gonçalves Boldrin, P.E.; Zanelli, C.F.; Nisole, S.; Chelbi-Alix, M.K.; Valentini, S.R. MxA Interacts with and Is Modified by the SUMOylation Machinery. Exp. Cell Res. 2015, 330, 151–163. [Google Scholar] [CrossRef] [PubMed]
- Dutrieux, J.; Portilho, D.M.; Arhel, N.J.; Hazan, U.; Nisole, S. TRIM5α Is a SUMO Substrate. Retrovirology 2015, 12, 28. [Google Scholar] [CrossRef]
- Portilho, D.M.; Fernandez, J.; Ringeard, M.; Machado, A.K.; Boulay, A.; Mayer, M.; Müller-Trutwin, M.; Beignon, A.-S.; Kirchhoff, F.; Nisole, S.; et al. Endogenous TRIM5α Function Is Regulated by SUMOylation and Nuclear Sequestration for Efficient Innate Sensing in Dendritic Cells. Cell Rep. 2016, 14, 355–369. [Google Scholar] [CrossRef]
- Maarifi, G.; Hannoun, Z.; Geoffroy, M.C.; El Asmi, F.; Zarrouk, K.; Nisole, S.; Blondel, D.; Chelbi-Alix, M.K. MxA Mediates SUMO-Induced Resistance to Vesicular Stomatitis Virus. J. Virol. 2016, 90, 6598–6610. [Google Scholar] [CrossRef]
- Maarifi, G.; El Asmi, F.; Maroui, M.A.; Dianoux, L.; Chelbi-Alix, M.K. Differential Effects of SUMO1 and SUMO3 on PKR Activation and Stability. Sci. Rep. 2018, 8, 1277. [Google Scholar] [CrossRef]
- Martinat, C.; Cormier, A.; Tobaly-Tapiero, J.; Palmic, N.; Casartelli, N.; Mahboubi, B.; Coggins, S.A.; Buchrieser, J.; Persaud, M.; Diaz-Griffero, F.; et al. SUMOylation of SAMHD1 at Lysine 595 Is Required for HIV-1 Restriction in Non-Cycling Cells. Nat. Commun. 2021, 12, 4582. [Google Scholar] [CrossRef]
- Arriagada, G.; Muntean, L.N.; Goff, S.P. SUMO-Interacting Motifs of Human TRIM5α Are Important for Antiviral Activity. PLoS Pathog. 2011, 7, e1002019. [Google Scholar] [CrossRef] [PubMed]
- Lukic, Z.; Goff, S.P.; Campbell, E.M.; Arriagada, G. Role of SUMO-1 and SUMO Interacting Motifs in Rhesus TRIM5α-Mediated Restriction. Retrovirology 2013, 10, 10. [Google Scholar] [CrossRef]
- Maillet, S.; Fernandez, J.; Decourcelle, M.; El Koulali, K.; Blanchet, F.P.; Arhel, N.J.; Maarifi, G.; Nisole, S. Daxx Inhibits HIV-1 Reverse Transcription and Uncoating in a SUMO-Dependent Manner. Viruses 2020, 12, 636. [Google Scholar] [CrossRef] [PubMed]
- Colomer-Lluch, M.; Castro-Gonzalez, S.; Serra-Moreno, R. Ubiquitination and SUMOylation in HIV Infection: Friends and Foes. Curr. Issues Mol. Biol. 2020, 35, 159–194. [Google Scholar] [CrossRef]
- Moremen, K.W.; Tiemeyer, M.; Nairn, A.V. Vertebrate Protein Glycosylation: Diversity, Synthesis and Function. Nat. Rev. Mol. Cell Biol. 2012, 13, 448–462. [Google Scholar] [CrossRef]
- Clerc, F.; Reiding, K.R.; Jansen, B.C.; Kammeijer, G.S.M.; Bondt, A.; Wuhrer, M. Human Plasma Protein N-Glycosylation. Glycoconj. J. 2016, 33, 309–343. [Google Scholar] [CrossRef]
- Kinoshita, T.; Fujita, M. Biosynthesis of GPI-Anchored Proteins: Special Emphasis on GPI Lipid Remodeling. J. Lipid Res. 2016, 57, 6–24. [Google Scholar] [CrossRef]
- Sharma, S.; Lewinski, M.K.; Guatelli, J. An N-Glycosylated Form of SERINC5 Is Specifically Incorporated into HIV-1 Virions. J. Virol. 2018, 92, e00753-18. [Google Scholar] [CrossRef] [PubMed]
- Kupzig, S.; Korolchuk, V.; Rollason, R.; Sugden, A.; Wilde, A.; Banting, G. Bst-2/HM1.24 Is a Raft-Associated Apical Membrane Protein with an Unusual Topology: A Raft Protein with an Unusual Topology. Traffic 2003, 4, 694–709. [Google Scholar] [CrossRef]
- Sobocińska, J.; Roszczenko-Jasińska, P.; Ciesielska, A.; Kwiatkowska, K. Protein Palmitoylation and Its Role in Bacterial and Viral Infections. Front. Immunol. 2018, 8, 2003. [Google Scholar] [CrossRef]
- Yount, J.S.; Karssemeijer, R.A.; Hang, H.C. S -Palmitoylation and Ubiquitination Differentially Regulate Interferon-Induced Transmembrane Protein 3 (IFITM3)-Mediated Resistance to Influenza Virus. J. Biol. Chem. 2012, 287, 19631–19641. [Google Scholar] [CrossRef]
- Narayana, S.K.; Helbig, K.J.; McCartney, E.M.; Eyre, N.S.; Bull, R.A.; Eltahla, A.; Lloyd, A.R.; Beard, M.R. The Interferon-Induced Transmembrane Proteins, IFITM1, IFITM2, and IFITM3 Inhibit Hepatitis C Virus Entry. J. Biol. Chem. 2015, 290, 25946–25959. [Google Scholar] [CrossRef]
- Sällman Almén, M.; Bringeland, N.; Fredriksson, R.; Schiöth, H.B. The Dispanins: A Novel Gene Family of Ancient Origin That Contains 14 Human Members. PLoS ONE 2012, 7, e31961. [Google Scholar] [CrossRef]
- Roskoski, R. ERK1/2 MAP Kinases: Structure, Function, and Regulation. Pharmacol. Res. 2012, 66, 105–143. [Google Scholar] [CrossRef]
- Li, X.; Wilmanns, M.; Thornton, J.; Köhn, M. Elucidating Human Phosphatase-Substrate Networks. Sci. Signal. 2013, 6, rs10. [Google Scholar] [CrossRef]
- Ardito, F.; Giuliani, M.; Perrone, D.; Troiano, G.; Lo Muzio, L. The Crucial Role of Protein Phosphorylation in Cell Signaling and Its Use as Targeted Therapy. Int. J. Mol. Med. 2017, 40, 271–280. [Google Scholar] [CrossRef]
- Bononi, A.; Agnoletto, C.; De Marchi, E.; Marchi, S.; Patergnani, S.; Bonora, M.; Giorgi, C.; Missiroli, S.; Poletti, F.; Rimessi, A.; et al. Protein Kinases and Phosphatases in the Control of Cell Fate. Enzyme Res. 2011, 2011, 329098. [Google Scholar] [CrossRef]
- Jia, R.; Pan, Q.; Ding, S.; Rong, L.; Liu, S.-L.; Geng, Y.; Qiao, W.; Liang, C. The N-Terminal Region of IFITM3 Modulates Its Antiviral Activity by Regulating IFITM3 Cellular Localization. J. Virol. 2012, 86, 13697–13707. [Google Scholar] [CrossRef]
- Foster, T.L.; Wilson, H.; Iyer, S.S.; Coss, K.; Doores, K.; Smith, S.; Kellam, P.; Finzi, A.; Borrow, P.; Hahn, B.H.; et al. Resistance of Transmitted Founder HIV-1 to IFITM-Mediated Restriction. Cell Host Microbe 2016, 20, 429–442. [Google Scholar] [CrossRef]
- Winstone, H.; Lista, M.J.; Reid, A.C.; Bouton, C.; Pickering, S.; Galao, R.P.; Kerridge, C.; Doores, K.J.; Swanson, C.M.; Neil, S.J.D. The Polybasic Cleavage Site in SARS-CoV-2 Spike Modulates Viral Sensitivity to Type I Interferon and IFITM2. J. Virol. 2021, 95, 9. [Google Scholar] [CrossRef]
- Shirakawa, K.; Takaori-Kondo, A.; Yokoyama, M.; Izumi, T.; Matsui, M.; Io, K.; Sato, T.; Sato, H.; Uchiyama, T. Phosphorylation of APOBEC3G by Protein Kinase A Regulates Its Interaction with HIV-1 Vif. Nat. Struct. Mol. Biol. 2008, 15, 1184–1191. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Demorest, Z.L.; Li, M.; Harris, R.S. Phosphorylation Directly Regulates the Intrinsic DNA Cytidine Deaminase Activity of Activation-Induced Deaminase and APOBEC3G Protein. J. Biol. Chem. 2011, 286, 26568–26575. [Google Scholar] [CrossRef]
- Matsumoto, T.; Shirakawa, K.; Yokoyama, M.; Fukuda, H.; Sarca, A.D.; Koyabu, S.; Yamazaki, H.; Kazuma, Y.; Matsui, H.; Maruyama, W.; et al. Protein Kinase A Inhibits Tumor Mutator APOBEC3B through Phosphorylation. Sci. Rep. 2019, 9, 8307. [Google Scholar] [CrossRef]
- Cribier, A.; Descours, B.; Valadão, A.L.C.; Laguette, N.; Benkirane, M. Phosphorylation of SAMHD1 by Cyclin A2/CDK1 Regulates Its Restriction Activity toward HIV-1. Cell Rep. 2013, 3, 1036–1043. [Google Scholar] [CrossRef]
- Welbourn, S.; Dutta, S.M.; Semmes, O.J.; Strebel, K. Restriction of Virus Infection but Not Catalytic DNTPase Activity Is Regulated by Phosphorylation of SAMHD1. J. Virol. 2013, 87, 11516–11524. [Google Scholar] [CrossRef]
- White, T.E.; Brandariz-Nuñez, A.; Valle-Casuso, J.C.; Amie, S.; Nguyen, L.A.; Kim, B.; Tuzova, M.; Diaz-Griffero, F. The Retroviral Restriction Ability of SAMHD1, but Not Its Deoxynucleotide Triphosphohydrolase Activity, Is Regulated by Phosphorylation. Cell Host Microbe 2013, 13, 441–451. [Google Scholar] [CrossRef] [PubMed]
- St. Gelais, C.; de Silva, S.; Hach, J.C.; White, T.E.; Diaz-Griffero, F.; Yount, J.S.; Wu, L. Identification of Cellular Proteins Interacting with the Retroviral Restriction Factor SAMHD1. J. Virol. 2014, 88, 5834–5844. [Google Scholar] [CrossRef]
- Drazic, A.; Myklebust, L.M.; Ree, R.; Arnesen, T. The World of Protein Acetylation. Biochim. Biophys. Acta 2016, 1864, 1372–1401. [Google Scholar] [CrossRef]
- Choudhary, C.; Kumar, C.; Gnad, F.; Nielsen, M.L.; Rehman, M.; Walther, T.C.; Olsen, J.V.; Mann, M. Lysine Acetylation Targets Protein Complexes and Co-Regulates Major Cellular Functions. Science 2009, 325, 834–840. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.J.; Seo, J.H.; Park, J.-H.; Vo, T.T.L.; An, S.; Bae, S.-J.; Le, H.; Lee, H.S.; Wee, H.-J.; Lee, D.; et al. SAMHD1 Acetylation Enhances Its Deoxynucleotide Triphosphohydrolase Activity and Promotes Cancer Cell Proliferation. Oncotarget 2017, 8, 68517–68529. [Google Scholar] [CrossRef]
- Fletcher, A.J.; Christensen, D.E.; Nelson, C.; Tan, C.P.; Schaller, T.; Lehner, P.J.; Sundquist, W.I.; Towers, G.J. TRIM 5α Requires Ube2W to Anchor Lys63-linked Ubiquitin Chains and Restrict Reverse Transcription. EMBO J. 2015, 34, 2078–2095. [Google Scholar] [CrossRef]
- Fletcher, A.J.; Vaysburd, M.; Maslen, S.; Zeng, J.; Skehel, J.M.; Towers, G.J.; James, L.C. Trivalent RING Assembly on Retroviral Capsids Activates TRIM5 Ubiquitination and Innate Immune Signaling. Cell Host Microbe 2018, 24, 761–775. [Google Scholar] [CrossRef]
- Murn, J.; Shi, Y. The Winding Path of Protein Methylation Research: Milestones and New Frontiers. Nat. Rev. Mol. Cell Biol. 2017, 18, 517–527. [Google Scholar] [CrossRef]
- Ravichandran, M.; Jurkowska, R.Z.; Jurkowski, T.P. Target Specificity of Mammalian DNA Methylation and Demethylation Machinery. Org. Biomol. Chem. 2018, 16, 1419–1435. [Google Scholar] [CrossRef]
- Weber, M.; Schübeler, D. Genomic Patterns of DNA Methylation: Targets and Function of an Epigenetic Mark. Curr. Opin. Cell Biol. 2007, 19, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Ringeard, M.; Marchand, V.; Decroly, E.; Motorin, Y.; Bennasser, Y. FTSJ3 Is an RNA 2’-O-Methyltransferase Recruited by HIV to Avoid Innate Immune Sensing. Nature 2019, 565, 500–504. [Google Scholar] [CrossRef] [PubMed]
- Kikkert, M. Innate Immune Evasion by Human Respiratory RNA Viruses. J. Innate Immun. 2020, 12, 4–20. [Google Scholar] [CrossRef] [PubMed]
- Milavetz, B.I.; Balakrishnan, L. Viral Epigenetics. Methods Mol. Biol. 2015, 1238, 569–596. [Google Scholar] [CrossRef]
- Chen, Y.; Guo, D. Molecular Mechanisms of Coronavirus RNA Capping and Methylation. Virol. Sin. 2016, 31, 3–11. [Google Scholar] [CrossRef]
- Shan, Z.; Han, Q.; Nie, J.; Cao, X.; Chen, Z.; Yin, S.; Gao, Y.; Lin, F.; Zhou, X.; Xu, K.; et al. Negative Regulation of Interferon-Induced Transmembrane Protein 3 by SET7-Mediated Lysine Monomethylation. J. Biol. Chem. 2013, 288, 35093–35103. [Google Scholar] [CrossRef]
- Chesarino, N.M.; Compton, A.A.; McMichael, T.M.; Kenney, A.D.; Zhang, L.; Soewarna, V.; Davis, M.; Schwartz, O.; Yount, J.S. IFITM 3 Requires an Amphipathic Helix for Antiviral Activity. EMBO Rep. 2017, 18, 1740–1751. [Google Scholar] [CrossRef]
- Rahman, K.; Coomer, C.A.; Majdoul, S.; Ding, S.Y.; Padilla-Parra, S.; Compton, A.A. Homology-Guided Identification of a Conserved Motif Linking the Antiviral Functions of IFITM3 to Its Oligomeric State. eLife 2020, 9, e58537. [Google Scholar] [CrossRef]
- Spence, J.S.; He, R.; Hoffmann, H.-H.; Das, T.; Thinon, E.; Rice, C.M.; Peng, T.; Chandran, K.; Hang, H.C. IFITM3 Directly Engages and Shuttles Incoming Virus Particles to Lysosomes. Nat. Chem. Biol. 2019, 15, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Bailey, C.C.; Zhong, G.; Huang, I.-C.; Farzan, M. IFITM-Family Proteins: The Cell’s First Line of Antiviral Defense. Annu. Rev. Virol. 2014, 1, 261–283. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.-F.; Nisole, S. West Nile Virus Restriction in Mosquito and Human Cells: A Virus under Confinement. Vaccines 2020, 8, 256. [Google Scholar] [CrossRef]
- Marziali, F.; Cimarelli, A. Membrane Interference Against HIV-1 by Intrinsic Antiviral Factors: The Case of IFITMs. Cells 2021, 10, 1171. [Google Scholar] [CrossRef]
- Shi, G.; Kenney, A.D.; Kudryashova, E.; Zani, A.; Zhang, L.; Lai, K.K.; Hall-Stoodley, L.; Robinson, R.T.; Kudryashov, D.S.; Compton, A.A.; et al. Opposing Activities of IFITM Proteins in SARS-CoV-2 Infection. EMBO J. 2020, 40, e106501. [Google Scholar] [PubMed]
- Zhao, X.; Guo, F.; Liu, F.; Cuconati, A.; Chang, J.; Block, T.M.; Guo, J.-T. Interferon Induction of IFITM Proteins Promotes Infection by Human Coronavirus OC43. Proc. Natl. Acad. Sci. USA 2014, 111, 6756–6761. [Google Scholar] [CrossRef] [PubMed]
- Prelli Bozzo, C.; Nchioua, R.; Volcic, M.; Koepke, L.; Krüger, J.; Schütz, D.; Heller, S.; Stürzel, C.M.; Kmiec, D.; Conzelmann, C.; et al. IFITM Proteins Promote SARS-CoV-2 Infection and Are Targets for Virus Inhibition in Vitro. Nat. Commun. 2021, 12, 4584. [Google Scholar] [CrossRef]
- Gea-Mallorquí, E. Does a Host Restriction Factor Facilitate Entry of SARS-CoV-2? Nat. Rev. Immunol. 2020, 20, 648. [Google Scholar] [CrossRef]
- Yount, J.S.; Moltedo, B.; Yang, Y.-Y.; Charron, G.; Moran, T.M.; López, C.B.; Hang, H.C. Palmitoylome Profiling Reveals S-Palmitoylation–Dependent Antiviral Activity of IFITM3. Nat. Chem. Biol. 2010, 6, 610–614. [Google Scholar] [CrossRef] [PubMed]
- Percher, A.; Ramakrishnan, S.; Thinon, E.; Yuan, X.; Yount, J.S.; Hang, H.C. Mass-Tag Labeling Reveals Site-Specific and Endogenous Levels of Protein S-Fatty Acylation. Proc. Natl. Acad. Sci. USA 2016, 113, 4302–4307. [Google Scholar] [CrossRef] [PubMed]
- John, S.P.; Chin, C.R.; Perreira, J.M.; Feeley, E.M.; Aker, A.M.; Savidis, G.; Smith, S.E.; Elia, A.E.H.; Everitt, A.R.; Vora, M.; et al. The CD225 Domain of IFITM3 Is Required for Both IFITM Protein Association and Inhibition of Influenza A Virus and Dengue Virus Replication. J. Virol. 2013, 87, 7837–7852. [Google Scholar] [CrossRef]
- McMichael, T.M.; Zhang, L.; Chemudupati, M.; Hach, J.C.; Kenney, A.D.; Hang, H.C.; Yount, J.S. The Palmitoyltransferase ZDHHC20 Enhances Interferon-Induced Transmembrane Protein 3 (IFITM3) Palmitoylation and Antiviral Activity. J. Biol. Chem. 2017, 292, 21517–21526. [Google Scholar] [CrossRef]
- Hach, J.C.; McMichael, T.; Chesarino, N.M.; Yount, J.S. Palmitoylation on Conserved and Nonconserved Cysteines of Murine IFITM1 Regulates Its Stability and Anti-Influenza A Virus Activity. J. Virol. 2013, 87, 9923–9927. [Google Scholar] [CrossRef] [PubMed]
- Jia, R.; Xu, F.; Qian, J.; Yao, Y.; Miao, C.; Zheng, Y.-M.; Liu, S.-L.; Guo, F.; Geng, Y.; Qiao, W.; et al. Identification of an Endocytic Signal Essential for the Antiviral Action of IFITM3: Endocytosis of IFITM3 and Its Antiviral Activity. Cell. Microbiol. 2014, 16, 1080–1093. [Google Scholar] [CrossRef]
- Chesarino, N.M.; McMichael, T.M.; Hach, J.C.; Yount, J.S. Phosphorylation of the Antiviral Protein Interferon-Inducible Transmembrane Protein 3 (IFITM3) Dually Regulates Its Endocytosis and Ubiquitination. J. Biol. Chem. 2014, 289, 11986–11992. [Google Scholar] [CrossRef]
- Perreira, J.M.; Chin, C.R.; Feeley, E.M.; Brass, A.L. IFITMs Restrict the Replication of Multiple Pathogenic Viruses. J. Mol. Biol. 2013, 425, 4937–4955. [Google Scholar] [CrossRef]
- Chesarino, N.M.; McMichael, T.M.; Yount, J.S. E3 Ubiquitin Ligase NEDD4 Promotes Influenza Virus Infection by Decreasing Levels of the Antiviral Protein IFITM3. PLoS Pathog. 2015, 11, e1005095. [Google Scholar] [CrossRef]
- Malakhova, O.A.; Zhang, D.-E. ISG15 Inhibits Nedd4 Ubiquitin E3 Activity and Enhances the Innate Antiviral Response. J. Biol. Chem. 2008, 283, 8783–8787. [Google Scholar] [CrossRef]
- Shan, J.; Zhao, B.; Shan, Z.; Nie, J.; Deng, R.; Xiong, R.; Tsun, A.; Pan, W.; Zhao, H.; Chen, L.; et al. Histone Demethylase LSD1 Restricts Influenza A Virus Infection by Erasing IFITM3-K88 Monomethylation. PLoS Pathog. 2017, 13, e1006773. [Google Scholar] [CrossRef]
- Matheson, N.J.; Sumner, J.; Wals, K.; Rapiteanu, R.; Weekes, M.P.; Vigan, R.; Weinelt, J.; Schindler, M.; Antrobus, R.; Costa, A.S.H.; et al. Cell Surface Proteomic Map of HIV Infection Reveals Antagonism of Amino Acid Metabolism by Vpu and Nef. Cell Host Microbe 2015, 18, 409–423. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wang, H.; Zhang, J.; Yang, J.; Bai, L.; Zheng, B.; Zheng, T.; Wang, Y.; Li, J.; Zhang, W. SERINC5 Inhibits the Secretion of Complete and Genome-Free Hepatitis B Virions Through Interfering With the Glycosylation of the HBV Envelope. Front. Microbiol. 2020, 11, 697. [Google Scholar] [CrossRef]
- Qiu, X.; Eke, I.E.; Johnson, S.F.; Ding, C.; Zheng, Y.-H. Proteasomal Degradation of Human SERINC4: A Potent Host Anti-HIV-1 Factor That Is Antagonized by Nef. Curr. Res. Virol. Sci. 2020, 1, 100002. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Ji, C.; Wang, L.; Cao, Y.; Dai, J.; Ye, X.; Zeng, L.; Dai, J.; Wu, Q.; Xie, Y.; et al. Cloning and Expression of a Novel Human C5orf12 Gene*, a Member of the TMS_TDE Family. Mol. Biol. Rep. 2003, 30, 47–52. [Google Scholar] [CrossRef]
- Sood, C.; Marin, M.; Chande, A.; Pizzato, M.; Melikyan, G.B. SERINC5 Protein Inhibits HIV-1 Fusion Pore Formation by Promoting Functional Inactivation of Envelope Glycoproteins. J. Biol. Chem. 2017, 292, 6014–6026. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhou, T.; Yang, J.; Lin, Y.; Shi, J.; Zhang, X.; Frabutt, D.A.; Zeng, X.; Li, S.; Venta, P.J.; et al. Identification of SERINC5-001 as the Predominant Spliced Isoform for HIV-1 Restriction. J. Virol. 2017, 91, e00137-17. [Google Scholar] [CrossRef]
- Chande, A.; Cuccurullo, E.C.; Rosa, A.; Ziglio, S.; Carpenter, S.; Pizzato, M. S2 from Equine Infectious Anemia Virus Is an Infectivity Factor Which Counteracts the Retroviral Inhibitors SERINC5 and SERINC3. Proc. Natl. Acad. Sci. USA 2016, 113, 13197–13202. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Xiong, R.; Zhou, T.; Su, P.; Zhang, X.; Qiu, X.; Li, H.; Li, S.; Yu, C.; Wang, B.; et al. HIV-1 Nef Antagonizes SERINC5 Restriction by Downregulation of SERINC5 via the Endosome/Lysosome System. J. Virol. 2018, 92, e00196-18. [Google Scholar] [CrossRef]
- Ganser-Pornillos, B.K.; Pornillos, O. Restriction of HIV-1 and Other Retroviruses by TRIM5. Nat. Rev. Microbiol. 2019, 17, 546–556. [Google Scholar] [CrossRef] [PubMed]
- Perron, M.J.; Stremlau, M.; Song, B.; Ulm, W.; Mulligan, R.C.; Sodroski, J. TRIM5 Mediates the Postentry Block to N-Tropic Murine Leukemia Viruses in Human Cells. Proc. Natl. Acad. Sci. USA 2004, 101, 11827–11832. [Google Scholar] [CrossRef]
- Yap, M.W.; Nisole, S.; Lynch, C.; Stoye, J.P. Trim5 Protein Restricts Both HIV-1 and Murine Leukemia Virus. Proc. Natl. Acad. Sci. USA 2004, 101, 10786–10791. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Griffero, F.; Kar, A.; Lee, M.; Stremlau, M.; Poeschla, E.; Sodroski, J. Comparative Requirements for the Restriction of Retrovirus Infection by TRIM5α and TRIMCyp. Virology 2007, 369, 400–410. [Google Scholar] [CrossRef]
- Stremlau, M.; Perron, M.; Lee, M.; Li, Y.; Song, B.; Javanbakht, H.; Diaz-Griffero, F.; Anderson, D.J.; Sundquist, W.I.; Sodroski, J. Specific Recognition and Accelerated Uncoating of Retroviral Capsids by the TRIM5 Restriction Factor. Proc. Natl. Acad. Sci. USA 2006, 103, 5514–5519. [Google Scholar] [CrossRef]
- Yu, A.; Skorupka, K.A.; Pak, A.J.; Ganser-Pornillos, B.K.; Pornillos, O.; Voth, G.A. TRIM5α Self-Assembly and Compartmentalization of the HIV-1 Viral Capsid. Nat. Commun. 2020, 11, 1307. [Google Scholar] [CrossRef]
- Roa, A.; Hayashi, F.; Yang, Y.; Lienlaf, M.; Zhou, J.; Shi, J.; Watanabe, S.; Kigawa, T.; Yokoyama, S.; Aiken, C.; et al. RING Domain Mutations Uncouple TRIM5 Restriction of HIV-1 from Inhibition of Reverse Transcription and Acceleration of Uncoating. J. Virol. 2012, 86, 1717–1727. [Google Scholar] [CrossRef] [PubMed]
- Yudina, Z.; Roa, A.; Johnson, R.; Biris, N.; de Souza Aranha Vieira, D.A.; Tsiperson, V.; Reszka, N.; Taylor, A.B.; Hart, P.J.; Demeler, B.; et al. RING Dimerization Links Higher-Order Assembly of TRIM5α to Synthesis of K63-Linked Polyubiquitin. Cell Rep. 2015, 12, 788–797. [Google Scholar] [CrossRef]
- Yamauchi, K.; Wada, K.; Tanji, K.; Tanaka, M.; Kamitani, T. Ubiquitination of E3 Ubiquitin Ligase TRIM5α and Its Potential Role: Ubiquitination of TRIM5α and Its Role. FEBS J. 2008, 275, 1540–1555. [Google Scholar] [CrossRef]
- Danielson, C.M.; Cianci, G.C.; Hope, T.J. Recruitment and Dynamics of Proteasome Association with RhTRIM5α Cytoplasmic Complexes During HIV-1 Infection: Proteasomes Associate with RhTRIM5α and HIV-1. Traffic 2012, 13, 1206–1217. [Google Scholar] [CrossRef]
- Campbell, E.M.; Weingart, J.; Sette, P.; Opp, S.; Sastri, J.; O’Connor, S.K.; Talley, S.; Diaz-Griffero, F.; Hirsch, V.; Bouamr, F. TRIM5α-Mediated Ubiquitin Chain Conjugation Is Required for Inhibition of HIV-1 Reverse Transcription and Capsid Destabilization. J. Virol. 2016, 90, 1849–1857. [Google Scholar] [CrossRef]
- Forshey, B.M.; von Schwedler, U.; Sundquist, W.I.; Aiken, C. Formation of a Human Immunodeficiency Virus Type 1 Core of Optimal Stability Is Crucial for Viral Replication. JVI 2002, 76, 5667–5677. [Google Scholar] [CrossRef]
- Imam, S.; Kömürlü, S.; Mattick, J.; Selyutina, A.; Talley, S.; Eddins, A.; Diaz-Griffero, F.; Campbell, E.M. K63-Linked Ubiquitin Is Required for Restriction of HIV-1 Reverse Transcription and Capsid Destabilization by Rhesus TRIM5α. J. Virol. 2019, 93, e00558-19. [Google Scholar] [CrossRef] [PubMed]
- Nepveu-Traversy, M.-É.; Demogines, A.; Fricke, T.; Plourde, M.B.; Riopel, K.; Veillette, M.; Diaz-Griffero, F.; Sawyer, S.L.; Berthoux, L. A Putative SUMO Interacting Motif in the B30.2/SPRY Domain of Rhesus Macaque TRIM5α Important for NF-ΚB/AP-1 Signaling and HIV-1 Restriction. Heliyon 2016, 2, e00056. [Google Scholar] [CrossRef] [PubMed]
- Nepveu-Traversy, M.-É.; Berthoux, L. The Conserved Sumoylation Consensus Site in TRIM5α Modulates Its Immune Activation Functions. Virus Res. 2014, 184, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.S.; Dudley, J.P. APOBECs and Virus Restriction. Virology 2015, 479–480, 131–145. [Google Scholar] [CrossRef] [PubMed]
- Stopak, K.; de Noronha, C.; Yonemoto, W.; Greene, W.C. HIV-1 Vif Blocks the Antiviral Activity of APOBEC3G by Impairing Both Its Translation and Intracellular Stability. Mol. Cell 2003, 12, 591–601. [Google Scholar] [CrossRef]
- Harris, R.S.; Bishop, K.N.; Sheehy, A.M.; Craig, H.M.; Petersen-Mahrt, S.K.; Watt, I.N.; Neuberger, M.S.; Malim, M.H. DNA Deamination Mediates Innate Immunity to Retroviral Infection. Cell 2003, 113, 803–809. [Google Scholar] [CrossRef]
- Seissler, T.; Marquet, R.; Paillart, J.-C. Hijacking of the Ubiquitin/Proteasome Pathway by the HIV Auxiliary Proteins. Viruses 2017, 9, 322. [Google Scholar] [CrossRef]
- Zhang, W.; Du, J.; Evans, S.L.; Yu, Y.; Yu, X.-F. T-Cell Differentiation Factor CBF-β Regulates HIV-1 Vif-Mediated Evasion of Host Restriction. Nature 2012, 481, 376–379. [Google Scholar] [CrossRef]
- Jäger, S.; Kim, D.Y.; Hultquist, J.F.; Shindo, K.; LaRue, R.S.; Kwon, E.; Li, M.; Anderson, B.D.; Yen, L.; Stanley, D.; et al. Vif Hijacks CBF-β to Degrade APOBEC3G and Promote HIV-1 Infection. Nature 2012, 481, 371–375. [Google Scholar] [CrossRef]
- Anderson, B.D.; Harris, R.S. Transcriptional Regulation of APOBEC3 Antiviral Immunity through the CBF-β/RUNX Axis. Sci. Adv. 2015, 1, e1500296. [Google Scholar] [CrossRef]
- Turner, T.; Shao, Q.; Wang, W.; Wang, Y.; Wang, C.; Kinlock, B.; Liu, B. Differential Contributions of Ubiquitin-Modified APOBEC3G Lysine Residues to HIV-1 Vif-Induced Degradation. J. Mol. Biol. 2016, 428, 3529–3539. [Google Scholar] [CrossRef] [PubMed]
- Albin, J.S.; Anderson, J.S.; Johnson, J.R.; Harjes, E.; Matsuo, H.; Krogan, N.J.; Harris, R.S. Dispersed Sites of HIV Vif-Dependent Polyubiquitination in the DNA Deaminase APOBEC3F. J. Mol. Biol. 2013, 425, 1172–1182. [Google Scholar] [CrossRef]
- Iwatani, Y.; Chan, D.S.B.; Liu, L.; Yoshii, H.; Shibata, J.; Yamamoto, N.; Levin, J.G.; Gronenborn, A.M.; Sugiura, W. HIV-1 Vif-Mediated Ubiquitination/Degradation of APOBEC3G Involves Four Critical Lysine Residues in Its C-Terminal Domain. Proc. Natl. Acad. Sci. USA 2009, 106, 19539–19544. [Google Scholar] [CrossRef] [PubMed]
- Pan, T.; Song, Z.; Wu, L.; Liu, G.; Ma, X.; Peng, Z.; Zhou, M.; Liang, L.; Liu, B.; Liu, J.; et al. USP49 Potently Stabilizes APOBEC3G Protein by Removing Ubiquitin and Inhibits HIV-1 Replication. eLife 2019, 8, e48318. [Google Scholar] [CrossRef] [PubMed]
- Chesarino, N.M.; Emerman, M. Polymorphisms in Human APOBEC3H Differentially Regulate Ubiquitination and Antiviral Activity. Viruses 2020, 12, 378. [Google Scholar] [CrossRef]
- Refsland, E.W.; Hultquist, J.F.; Luengas, E.M.; Ikeda, T.; Shaban, N.M.; Law, E.K.; Brown, W.L.; Reilly, C.; Emerman, M.; Harris, R.S. Natural Polymorphisms in Human APOBEC3H and HIV-1 Vif Combine in Primary T Lymphocytes to Affect Viral G-to-A Mutation Levels and Infectivity. PLoS Genet. 2014, 10, e1004761. [Google Scholar] [CrossRef]
- Jiang, Z.-Q.; Yao, X.-R.; Yu, H.; Lu, Y.-E.; Liu, B.-L.; Liu, F.-L.; Jin, Y.-B.; Zhuo, M.; Zheng, Y.-T.; Ling, F. Polymorphisms in the APOBEC3G Gene of Chinese Rhesus Macaques Affect Resistance to Ubiquitination and Degradation Mediated by HIV-2 Vif. Arch. Virol. 2019, 164, 1353–1360. [Google Scholar] [CrossRef]
- Cartier, C.; Hemonnot, B.; Gay, B.; Bardy, M.; Sanchiz, C.; Devaux, C.; Briant, L. Active CAMP-Dependent Protein Kinase Incorporated within Highly Purified HIV-1 Particles Is Required for Viral Infectivity and Interacts with Viral Capsid Protein. J. Biol. Chem. 2003, 278, 35211–35219. [Google Scholar] [CrossRef]
- Coggins, S.A.; Mahboubi, B.; Schinazi, R.F.; Kim, B. SAMHD1 Functions and Human Diseases. Viruses 2020, 12, 382. [Google Scholar] [CrossRef]
- Majer, C.; Schüssler, J.M.; König, R. Intertwined: SAMHD1 Cellular Functions, Restriction, and Viral Evasion Strategies. Med. Microbiol. Immunol. 2019, 208, 513–529. [Google Scholar] [CrossRef] [PubMed]
- Goldstone, D.C.; Ennis-Adeniran, V.; Hedden, J.J.; Groom, H.C.T.; Rice, G.I.; Christodoulou, E.; Walker, P.A.; Kelly, G.; Haire, L.F.; Yap, M.W.; et al. HIV-1 Restriction Factor SAMHD1 Is a Deoxynucleoside Triphosphate Triphosphohydrolase. Nature 2011, 480, 379–382. [Google Scholar] [CrossRef]
- Baldauf, H.-M.; Pan, X.; Erikson, E.; Schmidt, S.; Daddacha, W.; Burggraf, M.; Schenkova, K.; Ambiel, I.; Wabnitz, G.; Gramberg, T.; et al. SAMHD1 Restricts HIV-1 Infection in Resting CD4+ T Cells. Nat. Med. 2012, 18, 1682–1688. [Google Scholar] [CrossRef] [PubMed]
- Wittmann, S.; Behrendt, R.; Eissmann, K.; Volkmann, B.; Thomas, D.; Ebert, T.; Cribier, A.; Benkirane, M.; Hornung, V.; Bouzas, N.F.; et al. Phosphorylation of Murine SAMHD1 Regulates Its Antiretroviral Activity. Retrovirology 2015, 12, 103. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Hao, C.; DeLucia, M.; Swanson, S.; Florens, L.; Washburn, M.P.; Ahn, J.; Skowronski, J. CyclinA2-Cyclin-Dependent Kinase Regulates SAMHD1 Protein Phosphohydrolase Domain. J. Biol. Chem. 2015, 290, 13279–13292. [Google Scholar] [CrossRef]
- Schott, K.; Fuchs, N.V.; Derua, R.; Mahboubi, B.; Schnellbächer, E.; Seifried, J.; Tondera, C.; Schmitz, H.; Shepard, C.; Brandariz-Nuñez, A.; et al. Dephosphorylation of the HIV-1 Restriction Factor SAMHD1 Is Mediated by PP2A-B55α Holoenzymes during Mitotic Exit. Nat. Commun. 2018, 9, 2227. [Google Scholar] [CrossRef] [PubMed]
- Szaniawski, M.A.; Spivak, A.M.; Cox, J.E.; Catrow, J.L.; Hanley, T.; Williams, E.S.C.P.; Tremblay, M.J.; Bosque, A.; Planelles, V. SAMHD1 Phosphorylation Coordinates the Anti-HIV-1 Response by Diverse Interferons and Tyrosine Kinase Inhibition. mBio 2018, 9, e00819-18. [Google Scholar] [CrossRef]
- Tramentozzi, E.; Ferraro, P.; Hossain, M.; Stillman, B.; Bianchi, V.; Pontarin, G. The DNTP Triphosphohydrolase Activity of SAMHD1 Persists during S-Phase When the Enzyme Is Phosphorylated at T592. Cell Cycle 2018, 17, 1102–1114. [Google Scholar] [CrossRef]
- Zhang, K.; Lv, D.-W.; Li, R. Conserved Herpesvirus Protein Kinases Target SAMHD1 to Facilitate Virus Replication. Cell Rep. 2019, 28, 449–459. [Google Scholar] [CrossRef]
- Bermejo, M.; López-Huertas, M.R.; García-Pérez, J.; Climent, N.; Descours, B.; Ambrosioni, J.; Mateos, E.; Rodríguez-Mora, S.; Rus-Bercial, L.; Benkirane, M.; et al. Dasatinib Inhibits HIV-1 Replication through the Interference of SAMHD1 Phosphorylation in CD4+ T Cells. Biochem. Pharmacol. 2016, 106, 30–45. [Google Scholar] [CrossRef]
- Saiada, F.; Zhang, K.; Li, R. PIAS1 Potentiates the Anti-EBV Activity of SAMHD1 through SUMOylation. Cell Biosci. 2021, 11, 127. [Google Scholar] [CrossRef]
- Srivastava, S.; Swanson, S.K.; Manel, N.; Florens, L.; Washburn, M.P.; Skowronski, J. Lentiviral Vpx Accessory Factor Targets VprBP/DCAF1 Substrate Adaptor for Cullin 4 E3 Ubiquitin Ligase to Enable Macrophage Infection. PLoS Pathog. 2008, 4, e1000059. [Google Scholar] [CrossRef]
- Fregoso, O.I.; Ahn, J.; Wang, C.; Mehrens, J.; Skowronski, J.; Emerman, M. Evolutionary Toggling of Vpx/Vpr Specificity Results in Divergent Recognition of the Restriction Factor SAMHD1. PLoS Pathog. 2013, 9, e1003496. [Google Scholar] [CrossRef]
- Schaller, T.; Pollpeter, D.; Apolonia, L.; Goujon, C.; Malim, M.H. Nuclear Import of SAMHD1 Is Mediated by a Classical Karyopherin α/Β1 Dependent Pathway and Confers Sensitivity to VpxMAC Induced Ubiquitination and Proteasomal Degradation. Retrovirology 2014, 11, 29. [Google Scholar] [CrossRef]
- Petroski, M.D.; Deshaies, R.J. Function and Regulation of Cullin-RING Ubiquitin Ligases. Nat. Rev. Mol. Cell Biol. 2005, 6, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, H.; Norton, T.D.; Schultz, M.L.; Polsky, S.B.; Sunseri, N.; Landau, N.R. Inhibition of CUL4A Neddylation Causes a Reversible Block to SAMHD1-Mediated Restriction of HIV-1. J. Virol. 2013, 87, 11741–11750. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Li, Z.; Huan, C.; Wang, H.; Liu, Y.; Liu, X.; Su, X.; Yu, J.; Zhao, Z.; Yu, X.; Zheng, B.; et al. TRIM 21-mediated Proteasomal Degradation of SAMHD 1 Regulates Its Antiviral Activity. EMBO Rep. 2020, 21, e47528. [Google Scholar] [CrossRef] [PubMed]
- Haller, O.; Staeheli, P.; Schwemmle, M.; Kochs, G. Mx GTPases: Dynamin-like Antiviral Machines of Innate Immunity. Trends Microbiol. 2015, 23, 154–163. [Google Scholar] [CrossRef]
- Betancor, G.; Jimenez-Guardeño, J.M.; Lynham, S.; Antrobus, R.; Khan, H.; Sobala, A.; Dicks, M.D.J.; Malim, M.H. MX2-Mediated Innate Immunity against HIV-1 Is Regulated by Serine Phosphorylation. Nat. Microbiol. 2021, 6, 1031–1042. [Google Scholar] [CrossRef]
- Jouvenet, N.; Neil, S.J.D.; Zhadina, M.; Zang, T.; Kratovac, Z.; Lee, Y.; McNatt, M.; Hatziioannou, T.; Bieniasz, P.D. Broad-Spectrum Inhibition of Retroviral and Filoviral Particle Release by Tetherin. JVI 2009, 83, 1837–1844. [Google Scholar] [CrossRef]
- Sakuma, T.; Noda, T.; Urata, S.; Kawaoka, Y.; Yasuda, J. Inhibition of Lassa and Marburg Virus Production by Tetherin. JVI 2009, 83, 2382–2385. [Google Scholar] [CrossRef] [PubMed]
- Perez-Caballero, D.; Zang, T.; Ebrahimi, A.; McNatt, M.W.; Gregory, D.A.; Johnson, M.C.; Bieniasz, P.D. Tetherin Inhibits HIV-1 Release by Directly Tethering Virions to Cells. Cell 2009, 139, 499–511. [Google Scholar] [CrossRef]
- Venkatesh, S.; Bieniasz, P.D. Mechanism of HIV-1 Virion Entrapment by Tetherin. PLoS Pathog. 2013, 9, e1003483. [Google Scholar] [CrossRef] [PubMed]
- Iwabu, Y.; Fujita, H.; Kinomoto, M.; Kaneko, K.; Ishizaka, Y.; Tanaka, Y.; Sata, T.; Tokunaga, K. HIV-1 Accessory Protein Vpu Internalizes Cell-Surface BST-2/Tetherin through Transmembrane Interactions Leading to Lysosomes. J. Biol. Chem. 2009, 284, 35060–35072. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Pang, X.; Li, J.; Cen, S.; Jin, Q.; Guo, F. The Role of the Structural Domains of Human BST-2 in Inhibiting the Release of Xenotropic Murine Leukemia Virus-Related Virus. Biochem. Biophys. Res. Commun. 2012, 428, 17–23. [Google Scholar] [CrossRef]
- Liang, Z.; Zhang, Y.; Song, J.; Zhang, H.; Zhang, S.; Li, Y.; Tan, J.; Qiao, W. The Effect of Bovine BST2A1 on the Release and Cell-to-Cell Transmission of Retroviruses. Virol. J. 2017, 14, 173. [Google Scholar] [CrossRef]
- Andrew, A.J.; Miyagi, E.; Kao, S.; Strebel, K. The Formation of Cysteine-Linked Dimers of BST-2/Tetherin Is Important for Inhibition of HIV-1 Virus Release but Not for Sensitivity to Vpu. Retrovirology 2009, 6, 80. [Google Scholar] [CrossRef] [PubMed]
- Waheed, A.; Gitzen, A.; Swiderski, M.; Freed, E. High-Mannose But Not Complex-Type Glycosylation of Tetherin Is Required for Restriction of HIV-1 Release. Viruses 2018, 10, 26. [Google Scholar] [CrossRef]
- Fukuma, A.; Abe, M.; Morikawa, Y.; Miyazawa, T.; Yasuda, J. Cloning and Characterization of the Antiviral Activity of Feline Tetherin/BST-2. PLoS ONE 2011, 6, e18247. [Google Scholar] [CrossRef]
- Wang, W.; Wang, J.; Qu, M.; Li, X.; Zhang, J.; Zhang, H.; Wu, J.; Yu, B.; Wu, H.; Kong, W.; et al. Viral Restriction Activity of Feline BST2 Is Independent of Its N-Glycosylation and Induction of NF-ΚB Activation. PLoS ONE 2015, 10, e0138190. [Google Scholar] [CrossRef]
- Bai, B.; Wang, X.-F.; Zhang, M.; Na, L.; Zhang, X.; Zhang, H.; Yang, Z.; Wang, X. The N-Glycosylation of Equine Tetherin Affects Antiviral Activity by Regulating Its Subcellular Localization. Viruses 2020, 12, 220. [Google Scholar] [CrossRef]
- Han, Z.; Lv, M.; Shi, Y.; Yu, J.; Niu, J.; Yu, X.-F.; Zhang, W. Mutation of Glycosylation Sites in BST-2 Leads to Its Accumulation at Intracellular CD63-Positive Vesicles without Affecting Its Antiviral Activity against Multivesicular Body-Targeted HIV-1 and Hepatitis B Virus. Viruses 2016, 8, 62. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Tan, J.; Liu, R.; Xu, D.; Li, Y.; Geng, Y.; Liang, C.; Qiao, W. Tetherin Inhibits Prototypic Foamy Virus Release. Virol. J. 2011, 8, 198. [Google Scholar] [CrossRef]
- Taylor, J.K.; Coleman, C.M.; Postel, S.; Sisk, J.M.; Bernbaum, J.G.; Venkataraman, T.; Sundberg, E.J.; Frieman, M.B. Severe Acute Respiratory Syndrome Coronavirus ORF7a Inhibits Bone Marrow Stromal Antigen 2 Virion Tethering through a Novel Mechanism of Glycosylation Interference. J. Virol. 2015, 89, 11820–11833. [Google Scholar] [CrossRef] [PubMed]
- Chu, H.; Wang, J.-J.; Qi, M.; Yoon, J.-J.; Chen, X.; Wen, X.; Hammonds, J.; Ding, L.; Spearman, P. Tetherin/BST-2 Is Essential for the Formation of the Intracellular Virus-Containing Compartment in HIV-Infected Macrophages. Cell Host Microbe 2012, 12, 360–372. [Google Scholar] [CrossRef] [PubMed]
- Mansouri, M.; Viswanathan, K.; Douglas, J.L.; Hines, J.; Gustin, J.; Moses, A.V.; Früh, K. Molecular Mechanism of BST2/Tetherin Downregulation by K5/MIR2 of Kaposi’s Sarcoma-Associated Herpesvirus. JVI 2009, 83, 9672–9681. [Google Scholar] [CrossRef] [PubMed]
- Agromayor, M.; Soler, N.; Caballe, A.; Kueck, T.; Freund, S.M.; Allen, M.D.; Bycroft, M.; Perisic, O.; Ye, Y.; McDonald, B.; et al. The UBAP1 Subunit of ESCRT-I Interacts with Ubiquitin via a SOUBA Domain. Structure 2012, 20, 414–428. [Google Scholar] [CrossRef]
- Mitchell, R.S.; Katsura, C.; Skasko, M.A.; Fitzpatrick, K.; Lau, D.; Ruiz, A.; Stephens, E.B.; Margottin-Goguet, F.; Benarous, R.; Guatelli, J.C. Vpu Antagonizes BST-2–Mediated Restriction of HIV-1 Release via β-TrCP and Endo-Lysosomal Trafficking. PLoS Pathog. 2009, 5, e1000450. [Google Scholar] [CrossRef]
- Janvier, K.; Pelchen–Matthews, A.; Renaud, J.-B.; Caillet, M.; Marsh, M.; Berlioz-Torrent, C. The ESCRT-0 Component HRS Is Required for HIV-1 Vpu-Mediated BST-2/Tetherin Down-Regulation. PLoS Pathog. 2011, 7, e1001265. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Cheng, M.; Chi, X.; Liu, X.; Zhou, J.; Lin, T.; Yang, W. High-Throughput Screening Identifies Mixed-Lineage Kinase 3 as a Key Host Regulatory Factor in Zika Virus Infection. J. Virol. 2019, 93, e00758-19. [Google Scholar] [CrossRef]
- Pfaender, S.; Mar, K.B.; Michailidis, E.; Kratzel, A.; Boys, I.N.; V’kovski, P.; Fan, W.; Kelly, J.N.; Hirt, D.; Ebert, N.; et al. LY6E Impairs Coronavirus Fusion and Confers Immune Control of Viral Disease. Nat. Microbiol. 2020, 5, 1330–1339. [Google Scholar] [CrossRef]
- Chougui, G.; Munir-Matloob, S.; Matkovic, R.; Martin, M.M.; Morel, M.; Lahouassa, H.; Leduc, M.; Ramirez, B.C.; Etienne, L.; Margottin-Goguet, F. HIV-2/SIV Viral Protein X Counteracts HUSH Repressor Complex. Nat. Microbiol. 2018, 3, 891–897. [Google Scholar] [CrossRef]
- Yurkovetskiy, L.; Guney, M.H.; Kim, K.; Goh, S.L.; McCauley, S.; Dauphin, A.; Diehl, W.E.; Luban, J. Primate Immunodeficiency Virus Proteins Vpx and Vpr Counteract Transcriptional Repression of Proviruses by the HUSH Complex. Nat. Microbiol. 2018, 3, 1354–1361. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Zheng, S.; Chen, D.; Zheng, M.; Li, X.; Li, G.; Lin, H.; Chang, J.; Zeng, H.; Guo, J.-T. LY6E Restricts Entry of Human Coronaviruses, Including Currently Pandemic SARS-CoV-2. J. Virol. 2020, 94, e00562-20. [Google Scholar] [CrossRef] [PubMed]
- Hackett, B.A.; Cherry, S. Flavivirus Internalization Is Regulated by a Size-Dependent Endocytic Pathway. Proc. Natl. Acad. Sci. USA 2018, 115, 4246–4251. [Google Scholar] [CrossRef] [PubMed]
- Mar, K.B.; Rinkenberger, N.R.; Boys, I.N.; Eitson, J.L.; McDougal, M.B.; Richardson, R.B.; Schoggins, J.W. LY6E Mediates an Evolutionarily Conserved Enhancement of Virus Infection by Targeting a Late Entry Step. Nat. Commun. 2018, 9, 3603. [Google Scholar] [CrossRef]
- Yu, J.; Liang, C.; Liu, S.-L. CD4-Dependent Modulation of HIV-1 Entry by LY6E. J. Virol. 2019, 93, e01866-18. [Google Scholar] [CrossRef] [PubMed]
- Dutrieux, J.; Maarifi, G.; Portilho, D.M.; Arhel, N.J.; Chelbi-Alix, M.K.; Nisole, S. PML/TRIM19-Dependent Inhibition of Retroviral Reverse-Transcription by Daxx. PLoS Pathog. 2015, 11, e1005280. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.-Y.; Huang, Y.-S.; Jeng, J.-C.; Kuo, H.-Y.; Chang, C.-C.; Chao, T.-T.; Ho, C.-C.; Chen, Y.-C.; Lin, T.-P.; Fang, H.-I.; et al. Role of SUMO-Interacting Motif in Daxx SUMO Modification, Subnuclear Localization, and Repression of Sumoylated Transcription Factors. Mol. Cell 2006, 24, 341–354. [Google Scholar] [CrossRef] [PubMed]
- Jang, M.-S.; Ryu, S.-W.; Kim, E. Modification of Daxx by Small Ubiquitin-Related Modifier-1. Biochem. Biophys. Res. Commun. 2002, 295, 495–500. [Google Scholar] [CrossRef]
- Chougui, G.; Margottin-Goguet, F. HUSH, a Link Between Intrinsic Immunity and HIV Latency. Front. Microbiol. 2019, 10, 224. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chamontin, C.; Bossis, G.; Nisole, S.; Arhel, N.J.; Maarifi, G. Regulation of Viral Restriction by Post-Translational Modifications. Viruses 2021, 13, 2197. https://doi.org/10.3390/v13112197
Chamontin C, Bossis G, Nisole S, Arhel NJ, Maarifi G. Regulation of Viral Restriction by Post-Translational Modifications. Viruses. 2021; 13(11):2197. https://doi.org/10.3390/v13112197
Chicago/Turabian StyleChamontin, Célia, Guillaume Bossis, Sébastien Nisole, Nathalie J. Arhel, and Ghizlane Maarifi. 2021. "Regulation of Viral Restriction by Post-Translational Modifications" Viruses 13, no. 11: 2197. https://doi.org/10.3390/v13112197
APA StyleChamontin, C., Bossis, G., Nisole, S., Arhel, N. J., & Maarifi, G. (2021). Regulation of Viral Restriction by Post-Translational Modifications. Viruses, 13(11), 2197. https://doi.org/10.3390/v13112197