
A Roadmap for Genome-Based Phage Taxonomy



{kind=link}
{kind=link}
Abstract
1. An Ongoing Revolution in Phage Taxonomy
2. The Next Steps for Tailed Phage Taxonomy
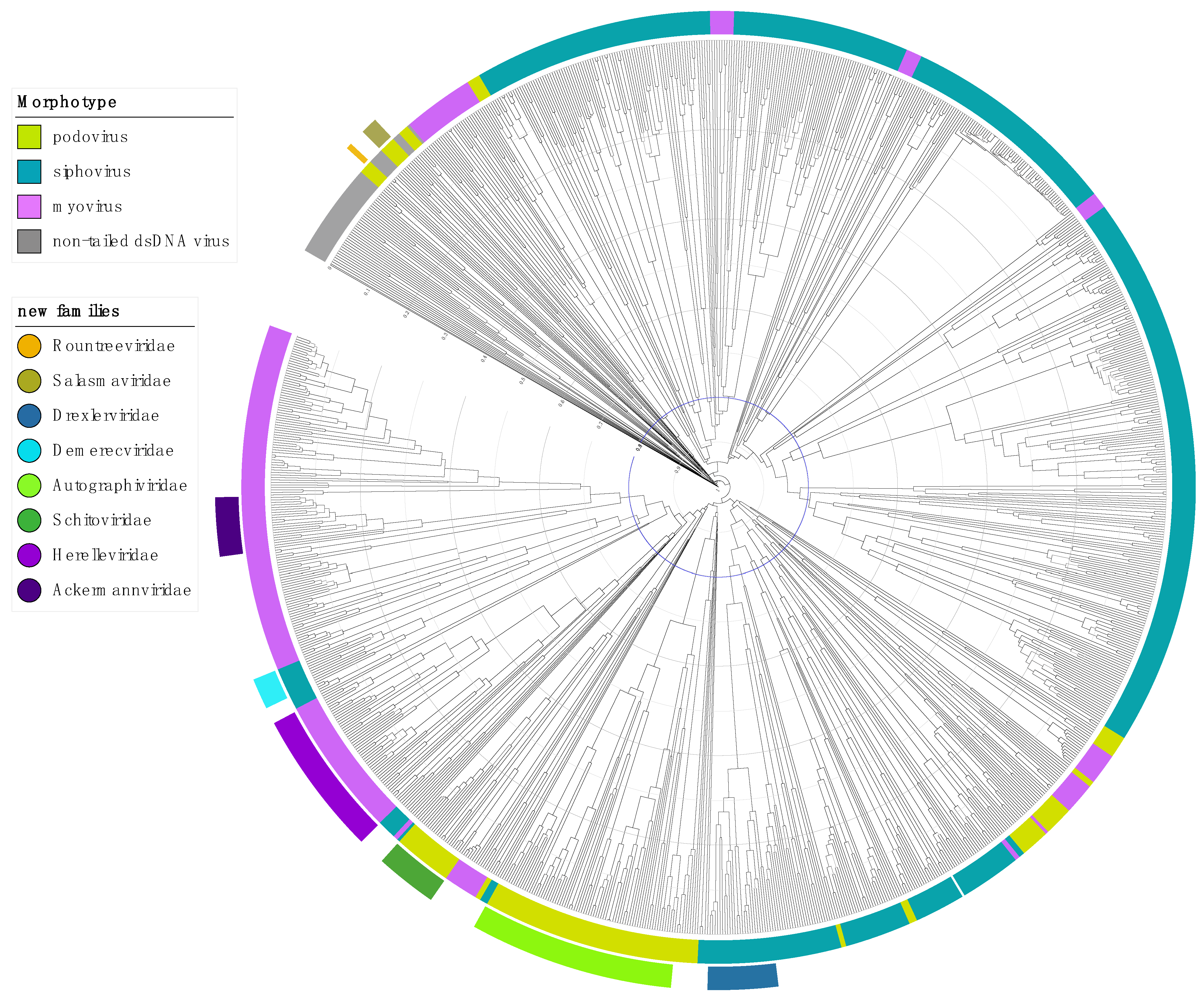
2.1. Step 1: Abolish the Order Caudovirales
2.2. Step 2: Abolish the Families Myoviridae, Podoviridae and Siphoviridae
2.3. Step 3: Elevating Existing Subfamilies to Family Rank
2.4. Step 4: Addressing the Unclassifieds
3. Rank-Specific Demarcation Criteria for Tailed Phages, Class Caudoviricetes
3.1. Species
3.2. Genus
3.3. Subfamily
3.4. Family
- The family is represented by a cohesive and monophyletic group in the main predicted proteome-based clustering tools (ViPTree, GRAViTy dendrogram, vConTACT2 network).
- Members of the family share a significant number of orthologous genes (the number will depend on the genome sizes and number of coding sequences of members of the family), see genus section for methods.
- If a family-level cluster shares orthologues with another family-level cluster, the family cluster needs to be monophyletic in a phylogenetic analysis of the shared orthologue(s).
3.5. Order
4. Perspectives for Non-Tailed Phages
5. Concluding Statement
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ackermann, H.-W.; DuBow, M.S. Viruses of Prokaryotes; CRC Press: Boca Raton, FL, USA, 1987. [Google Scholar]
- Ackermann, H.-W. Frequency of morphological phage descriptions in the year 2000. Arch. Virol. 2001, 146, 843–857. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, H.-W. Classification of bacteriophages. In The Bacteriophages; Calendar, R., Ed.; Oxford University Press: New York, NY, USA, 2006; pp. 8–17. [Google Scholar]
- Bradley, D.E. Ultrastructure of bacteriophage and bacteriocins. Bacteriol. Rev. 1967, 31, 230–314. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, H.-W.; Eisenstark, A. The Present State of Phage Taxonomy. Intervirology 1974, 3, 201–219. [Google Scholar] [CrossRef]
- Ackermann, H.-W. Bacteriophage classification. In Bacteriophages: Biology and Applications; Kutter, E.M., Sulakvelidze, A., Eds.; CRC Press: Boca Raton, FL, USA, 2005. [Google Scholar]
- Lavigne, R.; Seto, D.; Mahadevan, P.; Ackermann, H.-W.; Kropinski, A.M. Unifying classical and molecular taxonomic classification: Analysis of the Podoviridae using BLASTP-based tools. Res. Microbiol. 2008, 159, 406–414. [Google Scholar] [CrossRef]
- Lavigne, R.; Darius, P.; Summer, E.J.; Seto, D.; Mahadevan, P.; Nilsson, A.S.; Ackermann, H.W.; Kropinski, A.M. Classification of Myoviridae bacteriophages using protein sequence similarity. BMC Microbiol. 2009, 9, 224. [Google Scholar] [CrossRef] [PubMed]
- Krupovic, M.; Dutilh, B.E.; Adriaenssens, E.M.; Wittmann, J.; Vogensen, F.K.; Sullivan, M.B.; Rumnieks, J.; Prangishvili, D.; Lavigne, R.; Kropinski, A.M.; et al. Taxonomy of prokaryotic viruses: Update from the ICTV bacterial and archaeal viruses subcommittee. Arch. Virol. 2016, 161, 1095–1099. [Google Scholar] [CrossRef]
- Rohwer, F.; Edwards, R. The Phage Proteomic Tree: A genome-based taxonomy for phage. J. Bacteriol. 2002, 184, 4529–4535. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, Y.; Yoshida, T.; Kuronishi, M.; Uehara, H.; Ogata, H.; Goto, S. ViPTree: The viral proteomic tree server. Bioinformatics 2017, 33, 2379–2380. [Google Scholar] [CrossRef]
- Lima-Mendez, G.; Van Helden, J.; Toussaint, A.; Leplae, R. Reticulate representation of evolutionary and functional relationships between phage genomes. Mol. Biol. Evol. 2008, 25, 762–777. [Google Scholar] [CrossRef]
- Iranzo, J.; Krupovic, M.; Koonin, E.V. The double-stranded DNA virosphere as a modular hierarchical network of gene sharing. MBio 2016, 7, e00978-16. [Google Scholar] [CrossRef]
- Bolduc, B.; Jang, H.B.; Doulcier, G.; You, Z.-Q.; Roux, S.; Sullivan, M.B. vConTACT: An iVirus tool to classify double-stranded DNA viruses that infect Archaea and Bacteria. PeerJ 2017, 5, e3243. [Google Scholar] [CrossRef]
- Jang, H.B.; Bolduc, B.; Zablocki, O.; Kuhn, J.H.; Roux, S.; Adriaenssens, E.M.; Brister, J.R.; Kropinski, A.M.; Krupovic, M.; Lavigne, R.; et al. Taxonomic assignment of uncultivated prokaryotic virus genomes is enabled by gene-sharing networks. Nat. Biotechnol. 2019, 37, 632–639. [Google Scholar] [CrossRef] [PubMed]
- Aiewsakun, P.; Simmonds, P. The genomic underpinnings of eukaryotic virus taxonomy: Creating a sequence-based framework for family-level virus classification. Microbiome 2018, 6, 38. [Google Scholar] [CrossRef]
- Aiewsakun, P.; Adriaenssens, E.M.; Lavigne, R.; Kropinski, A.M.; Simmonds, P. Evaluation of the genomic diversity of viruses infecting bacteria, archaea and eukaryotes using a common bioinformatic platform: Steps towards a unified taxonomy. J. Gen. Virol. 2018, 99, 1331–1343. [Google Scholar] [CrossRef] [PubMed]
- Andrade-Martínez, J.S.; Moreno-Gallego, J.L.; Reyes, A. Defining a Core Genome for the Herpesvirales and Exploring their Evolutionary Relationship with the Caudovirales. Sci. Rep. 2019, 9, 11342. [Google Scholar] [CrossRef]
- Low, S.J.; Džunková, M.; Chaumeil, P.-A.; Parks, D.H.; Hugenholtz, P. Evaluation of a concatenated protein phylogeny for classification of tailed double-stranded DNA viruses belonging to the order Caudovirales. Nat. Microbiol. 2019, 4, 1306–1315. [Google Scholar] [CrossRef]
- Adriaenssens, E.M.; Wittmann, J.; Kuhn, J.H.; Turner, D.; Sullivan, M.B.; Dutilh, B.E.; Jang, H.B.; Van Zyl, L.J.; Klumpp, J.; Lobocka, M.; et al. Taxonomy of prokaryotic viruses: 2017 update from the ICTV Bacterial and Archaeal Viruses Subcommittee. Arch. Virol. 2018, 163, 1125–1129. [Google Scholar] [CrossRef] [PubMed]
- Adriaenssens, E.M.; Sullivan, M.B.; Knezevic, P.; Van Zyl, L.J.; Sarkar, B.L.; Dutilh, B.E.; Alfenas-Zerbini, P.; Łobocka, M.; Tong, Y.; Brister, J.R.; et al. Taxonomy of prokaryotic viruses: 2018–2019 update from the ICTV Bacterial and Archaeal Viruses Subcommittee. Arch. Virol. 2020, 165, 1253–1260. [Google Scholar] [CrossRef]
- Barylski, J.; Enault, F.; Dutilh, B.E.; Schuller, M.B.; Edwards, R.A.; Gillis, A.; Klumpp, J.; Knezevic, P.; Krupovic, M.; Kuhn, J.H.; et al. Analysis of Spounaviruses as a Case Study for the Overdue Reclassification of Tailed Phages. Syst. Biol. 2020, 69, 110–123. [Google Scholar] [CrossRef]
- Barylski, J.; Kropinski, A.M.; Alikhan, N.-F.; Adriaenssens, E.M. ICTV Virus Taxonomy Profile: Herelleviridae. J. Gen. Virol. 2020, 101, 3–4. [Google Scholar] [CrossRef]
- Kauffman, K.M.; Hussain, F.A.; Yang, J.; Arevalo, P.; Brown, J.M.; Chang, W.K.; VanInsberghe, D.; Elsherbini, J.; Sharma, R.S.; Cutler, M.B.; et al. A major lineage of non-tailed dsDNA viruses as unrecognized killers of marine bacteria. Nature 2018, 554, 118–122. [Google Scholar] [CrossRef]
- Laanto, E.; Mäntynen, S.; De Colibus, L.; Marjakangas, J.; Gillum, A.; Stuart, D.I.; Ravantti, J.J.; Huiskonen, J.T.; Sundberg, L.-R. Virus found in a boreal lake links ssDNA and dsDNA viruses. Proc. Natl. Acad. Sci. USA 2017, 114, 8378–8383. [Google Scholar] [CrossRef]
- Mäntynen, S.; Laanto, E.; Sundberg, L.R.; Poranen, M.M.; Oksanen, H.M. ICTV virus taxonomy profile: Finnlakeviridae. J. Gen. Virol. 2020, 101, 894–895. [Google Scholar] [CrossRef]
- Dutilh, B.E.; Cassman, N.; McNair, K.; Sanchez, S.E.; Silva, G.G.Z.; Boling, L.; Barr, J.J.; Speth, D.R.; Seguritan, V.; Aziz, R.K.; et al. A highly abundant bacteriophage discovered in the unknown sequences of human faecal metagenomes. Nat. Commun. 2014, 5, 4498. [Google Scholar] [CrossRef]
- Guerin, E.; Shkoporov, A.; Stockdale, S.R.; Clooney, A.G.; Ryan, F.J.; Sutton, T.D.S.; Draper, L.A.; Gonzalez-Tortuero, E.; Ross, R.P.; Hill, C. Biology and Taxonomy of crAss-like Bacteriophages, the Most Abundant Virus in the Human Gut. Cell Host Microbe 2018, 24, 653–664.e6. [Google Scholar] [CrossRef] [PubMed]
- Edwards, R.A.; Vega, A.A.; Norman, H.M.; Ohaeri, M.; Levi, K.; Dinsdale, E.A.; Cinek, O.; Aziz, R.K.; McNair, K.; Barr, J.J.; et al. Global phylogeography and ancient evolution of the widespread human gut virus crAssphage. Nat. Microbiol. 2019, 4, 1727–1736. [Google Scholar] [CrossRef] [PubMed]
- Devoto, A.E.; Santini, J.M.; Olm, M.R.; Anantharaman, K.; Munk, P.; Tung, J.; Archie, E.A.; Turnbaugh, P.J.; Seed, K.D.; Blekhman, R.; et al. Megaphages infect Prevotella and variants are widespread in gut microbiomes. Nat. Microbiol. 2019, 4, 693–700. [Google Scholar] [CrossRef] [PubMed]
- Al-Shayeb, B.; Sachdeva, R.; Chen, L.-X.; Ward, F.; Munk, P.; Devoto, A.; Castelle, C.J.; Olm, M.R.; Bouma-Gregson, K.; Amano, Y.; et al. Clades of huge phages from across Earth’s ecosystems. Nature 2020, 578, 425–431. [Google Scholar] [CrossRef] [PubMed]
- Roux, S.; Krupovic, M.; Daly, R.A.; Borges, A.L.; Nayfach, S.; Schulz, F.; Sharrar, A.; Matheus Carnevali, P.B.; Cheng, J.; Ivanova, N.N.; et al. Cryptic inoviruses revealed as pervasive in bacteria and archaea across Earth’s biomes. Nat. Microbiol. 2019, 4, 1895–1906. [Google Scholar] [CrossRef]
- Krupovic, M.; Forterre, P. Microviridae goes temperate: Microvirus-related proviruses reside in the genomes of Bacteroidetes. PLoS ONE 2011, 6, e19893. [Google Scholar] [CrossRef]
- Roux, S.; Krupovic, M.; Poulet, A.; Debroas, D.; Enault, F. Evolution and diversity of the Microviridae viral family through a collection of 81 new complete genomes assembled from virome reads. PLoS ONE 2012, 7, e40418. [Google Scholar] [CrossRef]
- Quaiser, A.; Dufresne, A.; Ballaud, F.; Roux, S.; Zivanovic, Y.; Colombet, J.; Sime-Ngando, T.; Francez, A.-J. Diversity and comparative genomics of Microviridae in Sphagnum- dominated peatlands. Front. Microbiol. 2015, 6, 375. [Google Scholar] [CrossRef] [PubMed]
- Krishnamurthy, S.R.; Janowski, A.B.; Zhao, G.; Barouch, D.; Wang, D. Hyperexpansion of RNA Bacteriophage Diversity. PLoS Biol. 2016, 14, e1002409. [Google Scholar] [CrossRef]
- Callanan, J.; Stockdale, S.R.; Shkoporov, A.; Draper, L.A.; Ross, R.P.; Hill, C. Expansion of known ssRNA phage genomes: From tens to over a thousand. Sci. Adv. 2020, 6, eaay5981. [Google Scholar] [CrossRef]
- Gorbalenya, A.E.; Krupovic, M.; Mushegian, A.; Kropinski, A.M.; Siddell, S.G.; Varsani, A.; Adams, M.J.; Davison, A.J.; Dutilh, B.E.; Harrach, B.; et al. The new scope of virus taxonomy: Partitioning the virosphere into 15 hierarchical ranks. Nat. Microbiol. 2020, 5, 668–674. [Google Scholar]
- Koonin, E.V.; Dolja, V.V.; Krupovic, M.; Varsani, A.; Wolf, Y.I.; Yutin, N.; Zerbini, F.M.; Kuhn, J.H. Global Organization and Proposed Megataxonomy of the Virus World. Microbiol. Mol. Biol. Rev. 2020, 84, e00061-19. [Google Scholar] [CrossRef] [PubMed]
- Wittmann, J.; Turner, D.; Millard, A.; Mahadevan, P.; Kropinski, A.; Adriaenssens, E. From Orphan Phage to a Proposed New Family–the Diversity of N4-Like Viruses. Antibiotics 2020, 9, 663. [Google Scholar] [CrossRef]
- Turner, D.; Reynolds, D.; Seto, D.; Mahadevan, P. CoreGenes3.5: A webserver for the determination of core genes from sets of viral and small bacterial genomes. BMC Res. Notes 2013, 6, 16–19. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. 2019, 47, W256–W259. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Moraru, C.; Varsani, A.; Kropinski, A.M. VIRIDIC-A Novel Tool to Calculate the Intergenomic Similarities of Prokaryote-Infecting Viruses. Viruses 2020, 12, 1268. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef] [PubMed]
- Adriaenssens, E.M.; Edwards, R.; Nash, J.H.E.; Mahadevan, P.; Seto, D.; Ackermann, H.-W.; Lavigne, R.; Kropinski, A.M. Integration of genomic and proteomic analyses in the classification of the Siphoviridae family. Virology 2015, 477, 144–154. [Google Scholar] [CrossRef] [PubMed]
- Gregory, A.C.; Solonenko, S.A.; Ignacio-Espinoza, J.C.; LaButti, K.; Copeland, A.; Sudek, S.; Maitland, A.; Chittick, L.; Dos Santos, F.; Weitz, J.S.; et al. Genomic differentiation among wild cyanophages despite widespread horizontal gene transfer. BMC Genom. 2016, 17, 930. [Google Scholar] [CrossRef]
- Roux, S.; Adriaenssens, E.M.; Dutilh, B.E.; Koonin, E.V.; Kropinski, A.M.; Krupovic, M.; Kuhn, J.H.; Lavigne, R.; Brister, J.R.; Varsani, A.; et al. Minimum Information about an Uncultivated Virus Genome (MIUViG). Nat. Biotechnol. 2019, 37, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Gregory, A.C.; Zablocki, O.; Zayed, A.A.; Howell, A.; Bolduc, B.; Sullivan, M.B. The Gut Virome Database Reveals Age-Dependent Patterns of Virome Diversity in the Human Gut. Cell Host Microbe 2020, 28, 724–740.e8. [Google Scholar] [CrossRef] [PubMed]
- Ondov, B.D.; Treangen, T.J.; Melsted, P.; Mallonee, A.B.; Bergman, N.H.; Koren, S.; Phillippy, A.M. Mash: Fast genome and metagenome distance estimation using MinHash. Genome Biol. 2016, 17, 132. [Google Scholar] [CrossRef]
- Page, A.J.; Cummins, C.A.; Hunt, M.; Wong, V.K.; Reuter, S.; Holden, M.T.G.; Fookes, M.; Falush, D.; Keane, J.A.; Parkhill, J. Roary: Rapid large-scale prokaryote pan genome analysis. Bioinformatics 2015, 31, 3691–3693. [Google Scholar] [CrossRef]
- Lechner, M.; Findeiß, S.; Steiner, L.; Marz, M.; Stadler, P.F.; Prohaska, S.J. Proteinortho: Detection of (Co-)orthologs in large-scale analysis. BMC Bioinform. 2011, 12, 124. [Google Scholar] [CrossRef]
- Bayliss, S.C.; Thorpe, H.A.; Coyle, N.M.; Sheppard, S.K.; Feil, E.J. PIRATE: A fast and scalable pangenomics toolbox for clustering diverged orthologues in bacteria. Gigascience 2019, 8, 1–9. [Google Scholar] [CrossRef]
- Contreras-Moreira, B.; Vinuesa, P. GET_HOMOLOGUES, a versatile software package for scalable and robust microbial pangenome analysis. Appl. Environ. Microbiol. 2013, 79, 7696–7701. [Google Scholar] [CrossRef] [PubMed]
- Söding, J.; Biegert, A.; Lupas, A.N. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 2005, 33, W244–W2488. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Clements, J.; Eddy, S.R. HMMER web server: Interactive sequence similarity searching. Nucleic Acids Res. 2011, 39, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Punta, M.; Coggill, P.C.; Eberhardt, R.Y.; Mistry, J.; Tate, J.; Boursnell, C.; Pang, N.; Forslund, K.; Ceric, G.; Clements, J.; et al. The Pfam protein families database. Nucleic Acids Res. 2012, 40, D290–D301. [Google Scholar] [CrossRef] [PubMed]
- Hunter, S.; Jones, P.; Mitchell, A.; Apweiler, R.; Attwood, T.K.; Bateman, A.; Bernard, T.; Binns, D.; Bork, P.; Burge, S.; et al. InterPro in 2011: New developments in the family and domain prediction database. Nucleic Acids Res. 2012, 40, D306–D312. [Google Scholar] [CrossRef]
- Marchler-Bauer, A.; Anderson, J.B.; Chitsaz, F.; Derbyshire, M.K.; DeWeese-Scott, C.; Fong, J.H.; Geer, L.Y.; Geer, R.C.; Gonzales, N.R.; Gwadz, M.; et al. CDD: Specific functional annotation with the Conserved Domain Database. Nucleic Acids Res. 2009, 37, D205–D210. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; Von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Hoang, D.T.; Chernomor, O.; Von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the ultrafast bootstrap approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef]
- Dereeper, A.; Guignon, V.; Blanc, G.; Audic, S.; Buffet, S.; Chevenet, F.; Dufayard, J.-F.; Guindon, S.; Lefort, V.; Lescot, M.; et al. Phylogeny.fr: Robust phylogenetic analysis for the non-specialist. Nucleic Acids Res. 2008, 36, W465–W469. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Turner, D.; Kropinski, A.M.; Adriaenssens, E.M. A Roadmap for Genome-Based Phage Taxonomy. Viruses 2021, 13, 506. https://doi.org/10.3390/v13030506
Turner D, Kropinski AM, Adriaenssens EM. A Roadmap for Genome-Based Phage Taxonomy. Viruses. 2021; 13(3):506. https://doi.org/10.3390/v13030506
Chicago/Turabian StyleTurner, Dann, Andrew M. Kropinski, and Evelien M. Adriaenssens. 2021. "A Roadmap for Genome-Based Phage Taxonomy" Viruses 13, no. 3: 506. https://doi.org/10.3390/v13030506
APA StyleTurner, D., Kropinski, A. M., & Adriaenssens, E. M. (2021). A Roadmap for Genome-Based Phage Taxonomy. Viruses, 13(3), 506. https://doi.org/10.3390/v13030506