
Characterizing and Evaluating the Zoonotic Potential of Novel Viruses Discovered in Vampire Bats

 , ,
, ,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Datasets
2.2. Bioinformatic Analyses
2.3. Inclusion Criteria and Study Plan
2.4. Phylogenetic Analysis
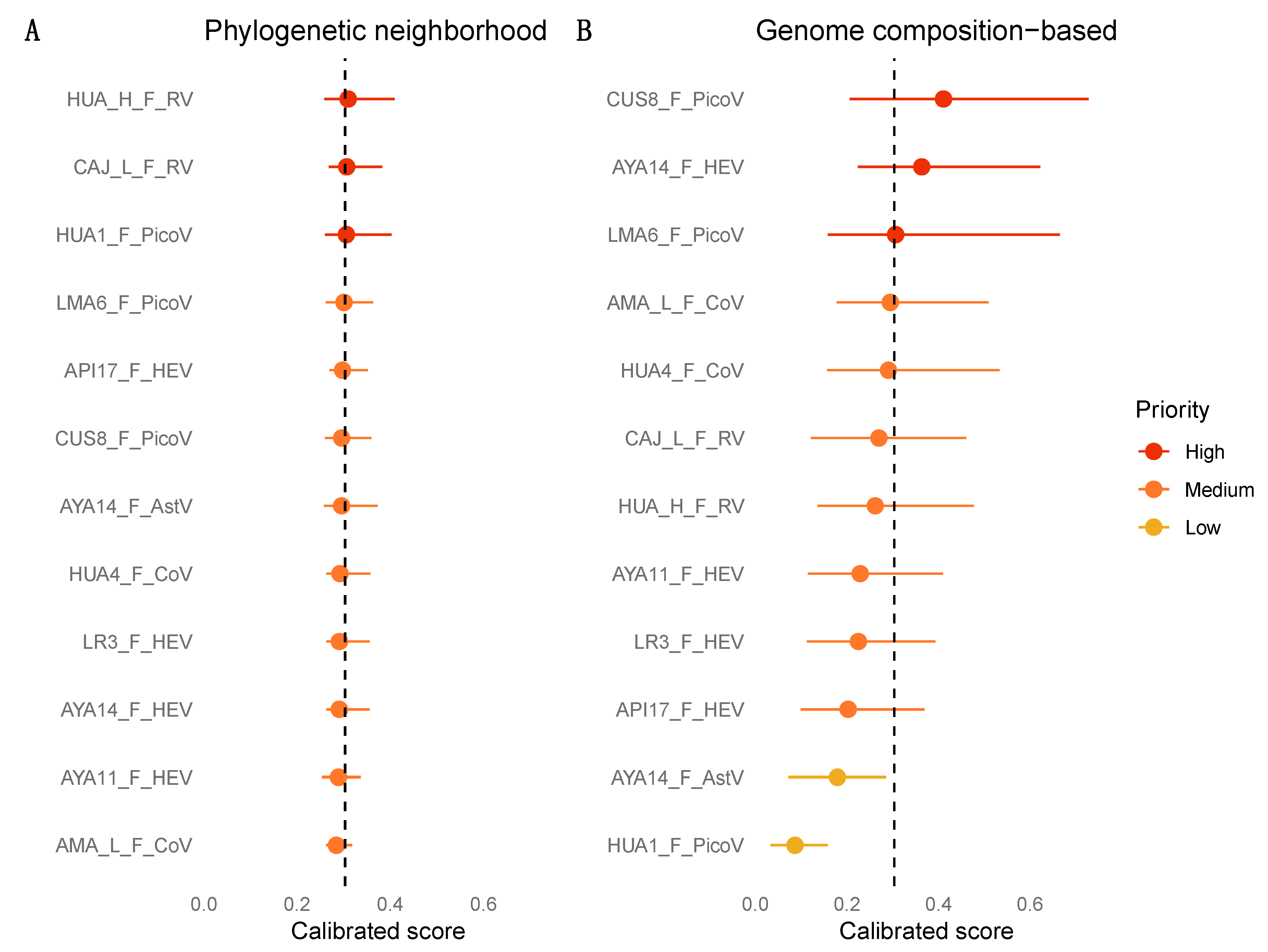
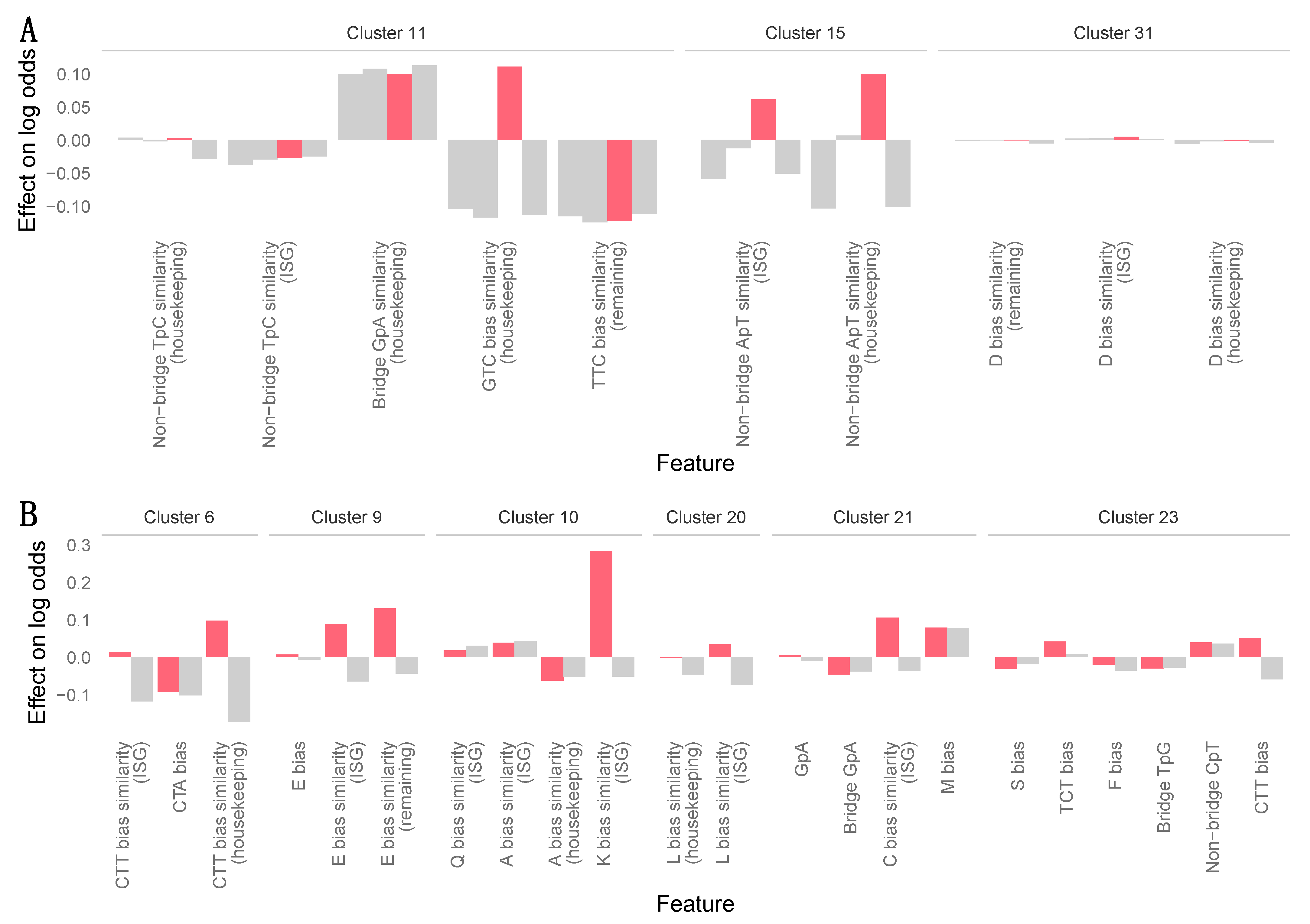
2.5. Machine Learning Analyses
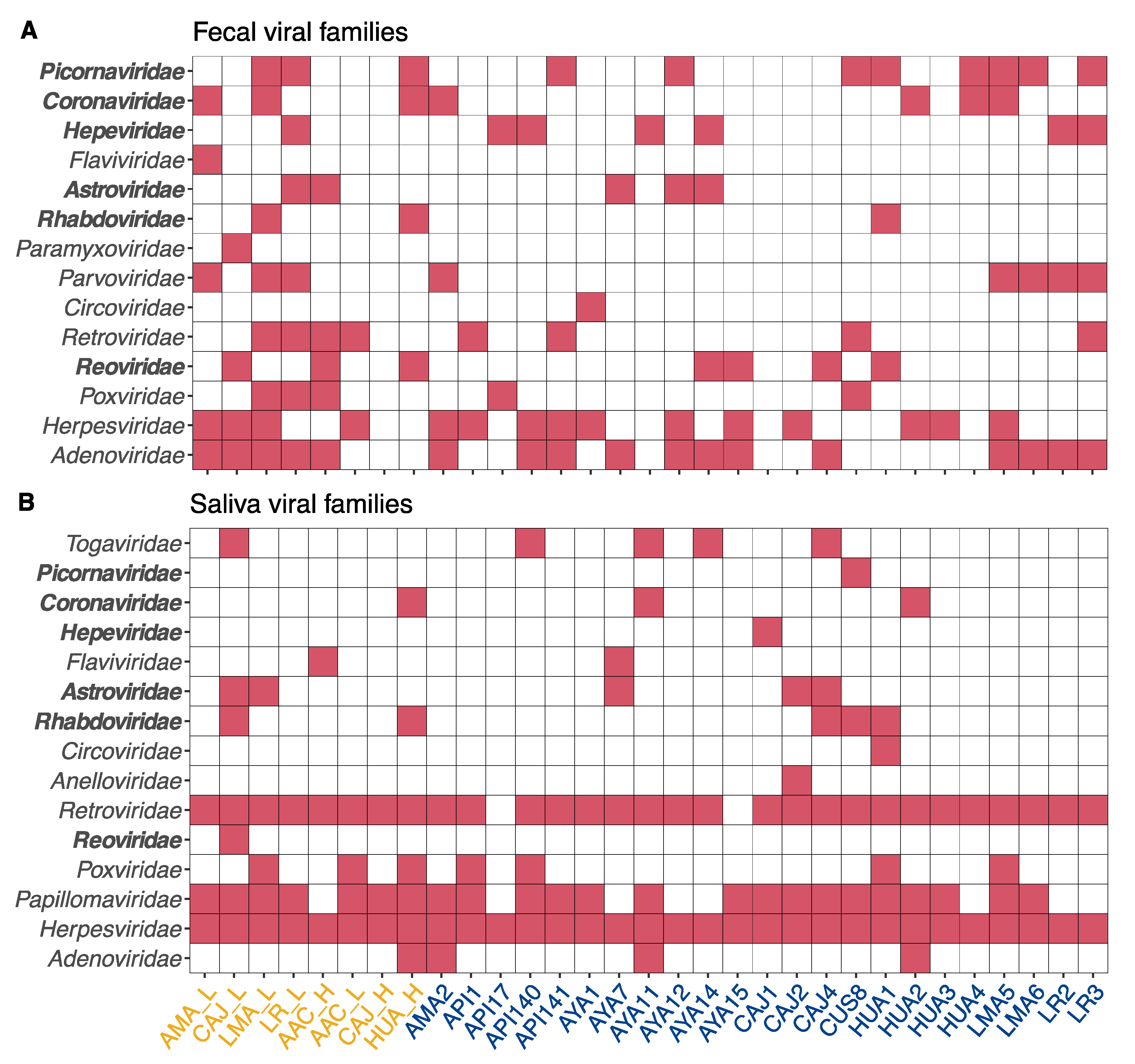
3. Results and Discussion
3.1. Rabies Virus
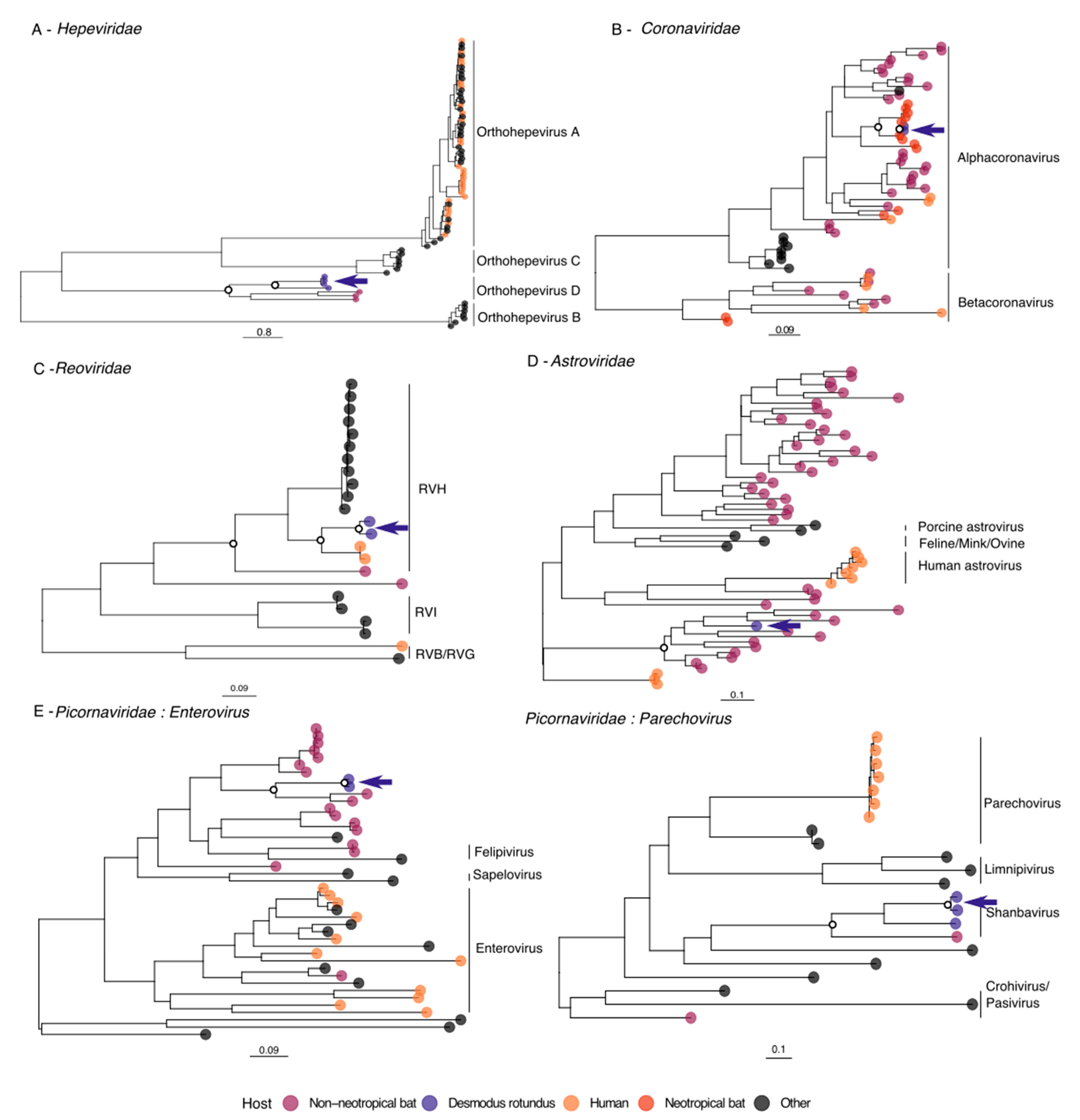
3.2. Hepeviridae
3.3. Coronaviridae
3.4. Reoviridae
3.5. Astroviridae
3.6. Picornaviridae
3.7. Zoonotic Ranking
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Wille, M.; Geoghegan, J.L.; Holmes, E.C. How accurately can we assess zoonotic risk? bioRxiv 2020. [Google Scholar] [CrossRef]
- Temmam, S.; Davoust, B.; Berenger, J.-M.; Raoult, D.; Desnues, C. Viral Metagenomics on Animals as a Tool for the Detection of Zoonoses Prior to Human Infection? Int. J. Mol Sci. 2014, 15, 10377–10397. [Google Scholar] [CrossRef]
- Firth, C.; Bhat, M.; Firth, M.A.; Williams, S.H.; Frye, M.J.; Simmonds, P.; Conte, J.M.; Ng, J.; Garcia, J.; Bhuva, N.P.; et al. Detection of Zoonotic Pathogens and Characterization of Novel Viruses Carried by Commensal Rattus norvegicus in New York City. mBio 2014, 5, e01933-14. [Google Scholar] [CrossRef]
- Williams, S.H.; Che, X.; Garcia, J.A.; Klena, J.D.; Lee, B.; Muller, D.; Ulrich, W.; Corrigan, R.M.; Nichol, S.; Jain, K.; et al. Viral Diversity of House Mice in New York City. mBio 2018, 9, e01354-17. [Google Scholar] [CrossRef]
- Coffey, L.L.; Page, B.L.; Greninger, A.L.; Herring, B.L.; Russell, R.C.; Doggett, S.L.; Haniotis, J.; Wang, C.; Deng, X.; Delwart, E.L. Enhanced arbovirus surveillance with deep sequencing: Identification of novel rhabdoviruses and bunyaviruses in Australian mosquitoes. Virology 2014, 448, 146–158. [Google Scholar] [CrossRef] [PubMed]
- Bouquet, J.; Melgar, M.; Swei, A.; Delwart, E.; Lane, R.S.; Chiu, C.Y. Metagenomic-based Surveillance of Pacific Coast tick Dermacentor occidentalis Identifies Two Novel Bunyaviruses and an Emerging Human Ricksettsial Pathogen. Sci. Rep. 2017, 7, 12234. [Google Scholar] [CrossRef]
- Marsh, G.A.; de Jong, C.; Barr, J.A.; Tachedjian, M.; Smith, C.; Middleton, D.; Yu, M.; Todd, S.; Foord, A.J.; Haring, V.; et al. Cedar Virus: A Novel Henipavirus Isolated from Australian Bats. PLoS Pathog. 2012, 8, e1002836. [Google Scholar] [CrossRef]
- Cantoni, D.; Hamlet, A.; Michaelis, M.; Wass, M.N.; Rossman, J.S. Risks Posed by Reston, the Forgotten Ebolavirus. mSphere 2016, 1, S65-10. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Lin, X.-D.; Tian, J.-H.; Chen, L.-J.; Chen, X.; Li, C.-X.; Qin, X.-C.; Li, J.; Cao, J.-P.; Eden, J.-S.; et al. Redefining the invertebrate RNA virosphere. Nature 2016, 540, 539–543. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Lin, X.-D.; Chen, X.; Tian, J.-H.; Chen, L.-J.; Li, K.; Wang, W.; Eden, J.-S.; Shen, J.-J.; Liu, L.; et al. The evolutionary history of vertebrate RNA viruses. Nature 2018, 556, 197–202. [Google Scholar] [CrossRef]
- Zhang, Y.-Z.; Shi, M.; Holmes, E.C. Using Metagenomics to Characterize an Expanding Virosphere. Cell 2018, 172, 1168–1172. [Google Scholar] [CrossRef]
- Holmes, E.C.; Rambaut, A.; Andersen, K.G. Pandemics: Spend on surveillance, not prediction. Nature 2018, 558, 180–182. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Cai, Z.; Tan, Z.; Lu, C.; Jiang, T.; Zhang, G.; Peng, Y. Rapid identification of human-infecting viruses. Transbound. Emerg. Dis. 2019, 66, 2517–2522. [Google Scholar] [CrossRef] [PubMed]
- Bartoszewicz, J.M.; Seidel, A.; Renard, B.Y. Interpretable detection of novel human viruses from genome sequencing data. bioRxiv 2020, 4. [Google Scholar] [CrossRef]
- Mollentze, N.; Babayan, S.; Streicker, D. Identifying and prioritizing potential human-infecting viruses from their genome sequences. bioRxiv 2020. [Google Scholar] [CrossRef]
- Delpietro, H.A.; Marchevsky, N.; Simonetti, E. Relative population densities and predation of the common vampire bat (Desmodus rotundus) in natural and cattle-raising areas in north-east Argentina. Prev. Vet. Med. 1992, 14, 13–20. [Google Scholar] [CrossRef]
- Voigt, C.C.; Kelm, D.H. Host Preference of The Common Vampire Bat (Desmodus Rotundus; Chiroptera) Assessed By Stable Isotopes. J. Mammal. 2006, 87, 1–6. [Google Scholar] [CrossRef]
- Streicker, D.G.; Allgeier, J.E. Foraging choices of vampire bats in diverse landscapes: Potential implications for land-use change and disease transmission. J. Appl. Ecol. 2016, 53, 1280–1288. [Google Scholar] [CrossRef]
- Schneider, M.C.; Aron, J.; Santos-Burgoa, C.; Uieda, W.; Ruiz-Velazco, S. Common vampire bat attacks on humans in a village of the Amazon region of Brazil. Cad. Saude. Publica 2001, 17, 1531–1536. [Google Scholar] [CrossRef]
- Gonçalves, M.A.; Sa-Neto, R.J.; Brazil, T.K. Outbreak of aggressions and transmission of rabies in human beings by vampire bats in northeastern Brazil. Rev. Soc. Bras. Med. Trop. 2002, 35, 461–464. [Google Scholar] [CrossRef] [PubMed]
- Schneider, M.C.; Romijn, P.C.; Uieda, W.; Tamayo, H.; da Silva, D.F.; Belotto, A.; da Silva, J.B.; Leanes, L.F. Rabies transmitted by vampire bats to humans: An emerging zoonotic disease in Latin America? Rev. Panam. Salud Publica 2009, 25, 260–269. [Google Scholar] [CrossRef] [PubMed]
- Bobrowiec, P.E.D.; Lemes, M.R.; Gribel, R. Prey preference of the common vampire bat (Desmodus rotundus, Chiroptera) using molecular analysis. J. Mammal. 2015, 96, 54–63. [Google Scholar]
- Gilbert, A.T.; Petersen, B.W.; Recuenco, S.; Niezgoda, M.; Gomez, J.; Laguna-Torres, V.A.; Rupprecht, C. Evidence of rabies virus exposure among humans in the Peruvian Amazon. Am. J. Trop. Med. Hyg. 2012, 87, 206–215. [Google Scholar] [CrossRef] [PubMed]
- Benavides, J.A.; Valderrama, W.; Streicker, D.G. Spatial expansions and travelling waves of rabies in vampire bats. Proc. Royal Soc. B 2016, 283, 20160328. [Google Scholar] [CrossRef]
- Brandão, P.E.; Scheffer, K.; Villarreal, L.Y.; Achkar, S.; de Novaes Oliveira, R.; de Oliveira Fahl, W.; Castilho, J.G.; Kotait, I.; Richtzenhain, L.J. A Coronavirus Detected in the Vampire Bat Desmodus rotundus. Braz. J. Infect. Dis. 2008, 12, 466–468. [Google Scholar] [CrossRef]
- Drexler, J.F.; Corman, V.M.; Müller, M.A.; Maganga, G.D.; Vallo, P.; Binger, T.; Gloza-Rausch, F.; Rasche, A.; Yordanov, S.; Seebens, A.; et al. Bats host major mammalian paramyxoviruses. Nat. Commun. 2012, 3, 796. [Google Scholar] [CrossRef] [PubMed]
- Fagrouch, Z.; Sarwari, R.; Lavergne, A.; Delaval, M.; de Thoisy, B.; Lacoste, V.; Verschoor, E.J. Novel polyomaviruses in South American bats and their relationship to other members of the family Polyomaviridae. J. Gen. Virol. 2012, 93, 2652–2657. [Google Scholar] [CrossRef]
- de Lima, F.E.S.; Cibulski, S.P.; Elesbao, F.; Carnieli Junior, P.; de Batista, H.B.C.R.; Roehe, P.M.; Franco, A.C. First detection of adenovirus in the vampire bat (Desmodus rotundus) in Brazil. Virus Genes 2013, 47, 378–381. [Google Scholar] [CrossRef]
- Wray, A.K.; Olival, K.J.; Moran, D.; Lopez, M.R.; Alvarez, D.; Navarrete-Macias, I.; Liang, E.; Simmons, N.B.; Lipkin, W.I.; Daszak, P.; et al. Viral Diversity, Prey Preference, and Bartonella Prevalence in Desmodus rotundus in Guatemala. EcoHealth 2016, 13, 761–774. [Google Scholar] [CrossRef] [PubMed]
- Escalera-Zamudio, M.; Taboada, B.; Rojas-Anaya, E.; Löber, U.; Loza-Rubio, E.; Arias, C.F.; Greenwood, A.D. Viral Communities Among Sympatric Vampire Bats and Cattle. EcoHealth 2017, 111, 1–11. [Google Scholar] [CrossRef]
- Salmier, A.; Tirera, S.; de Thoisy, B.; Franc, A.; Darcissac, E.; Donato, D.; Bouchier, C.; Lacoste, V.; Lavergne, A. Virome analysis of two sympatric bat species (Desmodus rotundus and Molossus molossus) in French Guiana. PLoS ONE 2017, 12, e0186943-25. [Google Scholar] [CrossRef]
- Babayan, S.A.; Orton, R.J.; Streicker, D.G. Predicting reservoir hosts and arthropod vectors from evolutionary signatures in RNA virus genomes. Science 2018, 362, 577–580. [Google Scholar] [CrossRef] [PubMed]
- Bergner, L.M.; Orton, R.J.; da Silva Filipe, A.; Shaw, A.E.; Becker, D.J.; Tello, C.; Biek, R.; Streicker, D.G. Using noninvasive metagenomics to characterize viral communities from wildlife. Mol. Ecol. Resour. 2019, 19, 128–143. [Google Scholar] [CrossRef]
- Bergner, L.M.; Orton, R.J.; Benavides, J.A.; Becker, D.J.; Tello, C.; Biek, R.; Streicker, D.G. Demographic and environmental drivers of metagenomic viral diversity in vampire bats. Mol. Ecol. 2020, 29, 26–39. [Google Scholar] [CrossRef]
- Bergner, L.M.; Orton, R.J.; Streicker, D.G. Complete Genome Sequence of an Alphacoronavirus from Common Vampire Bats in Peru. Microbiol. Resour. Announc. 2020, 9, 676. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Schmieder, R.; Edwards, R. Quality control and preprocessing of metagenomic datasets. Bioinformatics 2011, 27, 863–864. [Google Scholar] [CrossRef]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, 25. [Google Scholar] [CrossRef]
- Schmieder, R.; Lim, Y.W.; Edwards, R. Identification and removal of ribosomal RNA sequences from metatranscriptomes. Bioinformatics 2011, 28, 433–435. [Google Scholar] [CrossRef] [PubMed]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef]
- Ondov, B.D.; Bergman, N.H.; Phillippy, A.M. Interactive metagenomic visualization in a Web browser. BMC Bioinform. 2011, 12, 385. [Google Scholar] [CrossRef] [PubMed]
- Rice, P.; Longden, I.; Bleasby, A. EMBOSS: The European molecular biology open software suite. Trends Genet. 2000, 16, 276–277. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2019; Available online: http://www.R-project.org/ (accessed on 25 January 2021).
- Katoh, K.; Misawa, K.; Kuma, K.-I.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [PubMed]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nat Methods 2012, 9, 772. [Google Scholar] [CrossRef] [PubMed]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. ProtTest 3: Fast selection of best-fit models of protein evolution. Bioinformatics 2011, 27, 1164–1165. [Google Scholar] [CrossRef]
- Stamatakis, A.; Hoover, P.; Rougemont, J. A Rapid Bootstrap Algorithm for the RAxML Web Servers. Syst. Biol. 2008, 57, 758–771. [Google Scholar] [CrossRef]
- Paradis, E.; Schliep, K. ape 5.0: An environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics 2019, 35, 526–528. [Google Scholar] [CrossRef]
- Revell, L.J. phytools: An R package for phylogenetic comparative biology (and other things). Methods Ecol. Evol. 2011, 3, 217–223. [Google Scholar] [CrossRef]
- Schliep, K.P. phangorn: Phylogenetic analysis in R. Bioinformatics 2010, 27, 592–593. [Google Scholar] [CrossRef]
- Yu, G.; Smith, D.K.; Zhu, H.; Guan, Y.; Lam, T.T.-Y. ggtree: An rpackage for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol. Evol. 2016, 8, 28–36. [Google Scholar] [CrossRef]
- Jenkins, G.M.; Holmes, E.C. The extent of codon usage bias in human RNA viruses and its evolutionary origin. Virus Res. 2003, 92, 1–7. [Google Scholar] [CrossRef]
- Shackelton, L.A.; Parrish, C.R.; Holmes, E.C. Evolutionary Basis of Codon Usage and Nucleotide Composition Bias in Vertebrate DNA Viruses. J. Mol. Evol. 2006, 62, 551–563. [Google Scholar] [CrossRef] [PubMed]
- Greenbaum, B.D.; Levine, A.J.; Bhanot, G.; Rabadan, R. Patterns of Evolution and Host Gene Mimicry in Influenza and Other RNA Viruses. PLoS Pathog. 2008, 4, e1000079. [Google Scholar] [CrossRef]
- Wong, E.H.; Smith, D.K.; Rabadan, R.; Peiris, M.; Poon, L.L. Codon usage bias and the evolution of influenza A viruses. Codon Usage Biases of Influenza Virus. BMC Evol. Biol. 2010, 10, 1–14. [Google Scholar] [CrossRef]
- Lundberg, S.M.; Lee, S.-I. A Unified Approach to Interpreting Model Predictions. arXiv 2017, arXiv:1705.07874, 4765–4774. [Google Scholar]
- Streicker, D.G.; Winternitz, J.C.; Satterfield, D.A.; Condori-Condori, R.E.; Broos, A.; Tello, C.; Recuenco, S.; Velasco-Villa, A.S.; Altizer, S.; Valderrama, W. Host-pathogen evolutionary signatures reveal dynamics and future invasions of vampire bat rabies. Proc. Natl. Acad. Sci. USA 2016, 113, 10926–10931. [Google Scholar] [CrossRef] [PubMed]
- Clark, K.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Sayers, E.W. GenBank. Nucleic Acids Res. 2016, 44, D67–D72. [Google Scholar] [CrossRef]
- Streicker, D.G.; Turmelle, A.S.; Vonhof, M.J.; Kuzmin, I.V.; McCracken, G.F.; Rupprecht, C.E. Host Phylogeny Constrains Cross-Species Emergence and Establishment of Rabies Virus in Bats. Science 2010, 329, 676–679. [Google Scholar] [CrossRef]
- Streicker, D.G.; Recuenco, S.; Valderrama, W.; Gomez Benavides, J.; Vargas, I.; Pacheco, V.; Condori Condori, R.E.; Montgomery, J.; Rupprecht, C.E.; Rohani, P.; et al. Ecological and anthropogenic drivers of rabies exposure in vampire bats: Implications for transmission and control. Proc. Royal Soc. B 2012, 279, 3384–3392. [Google Scholar] [CrossRef] [PubMed]
- Blackwood, J.C.; Streicker, D.G.; Altizer, S.; Rohani, P. Resolving the roles of immunity, pathogenesis, and immigration for rabies persistence in vampire bats. Proc. Natl. Acad. Sci. USA 2013, 110, 20837–20842. [Google Scholar] [CrossRef] [PubMed]
- Streicker, D.G.; Fallas González, S.L.; Luconi, G.; Barrientos, R.G.; Leon, B. Phylodynamics reveals extinction–recolonization dynamics underpin apparently endemic vampire bat rabies in Costa Rica. Proc. Royal Soc. B 2019, 286, 20191527. [Google Scholar] [CrossRef]
- Allendorf, S.D.; Cortez, A.; Heinemann, M.B.; Harary, C.M.A.; Antunes, J.M.A.P.; Peres, M.G.; Vicente, A.F.; Sodré, M.M.; da Rosa, A.R.; Megid, J. Rabies virus distribution in tissues and molecular characterization of strains from naturally infected non-hematophagous bats. Virus Res. 2012, 165, 119–125. [Google Scholar] [CrossRef]
- Begeman, L.; Kooi, E.A.; Weezep, E.; Bildt, M.W.G.; Reusken, C.B.E.M.; Lina, P.H.C.; Koopmans, M.P.G.; Brand, J.M.A.; Kuiken, T. Faeces as a novel material to estimate lyssavirus prevalence in bat populations. Zoonoses Public Health 2019, 67, 198–202. [Google Scholar] [CrossRef] [PubMed]
- Perez-Gracia, M.; Suay, B.; Luisa Mateos-Lindemann, M. Hepatitis E: An emerging disease. Infect. Genet. Evol. 2014, 22, 40–59. [Google Scholar] [CrossRef]
- Purdy, M.A.; Harrison, T.J.; Jameel, S.; Meng, X.J.; Okamoto, H.; Van der Poel, W.H.M.; Smith, D.B. ICTV Report Consortium ICTV Virus Taxonomy Profile: Hepeviridae. J. Gen. Virol. 2017, 98, 2645–2646. [Google Scholar] [CrossRef]
- Drexler, J.F.; Seelen, A.; Corman, V.M.; Fumie Tateno, A.; Cottontail, V.; Melim Zerbinati, R.; Gloza-Rausch, F.; Klose, S.M.; Adu-Sarkodie, Y.; Oppong, S.K.; et al. Bats Worldwide Carry Hepatitis E Virus-Related Viruses That Form a Putative Novel Genus within the Family Hepeviridae. J. Virol. 2012, 86, 9134–9147. [Google Scholar] [CrossRef]
- Smith, D.B.; Simmonds, P.; Members of the International Committee on the Taxonomy of Viruses Hepeviridae Study Group; Jameel, S.; Emerson, S.U.; Harrison, T.J.; Meng, X.J.; Okamoto, H.; Van der Poel, W.H.M.; Purdy, M.A. Consensus proposals for classification of the family Hepeviridae. J. Gen. Virol. 2014, 95, 2223–2232. [Google Scholar] [CrossRef]
- Pavio, N.; Meng, X.-J.; Renou, C. Zoonotic hepatitis E: Animal reservoirs and emerging risks. Vet. Res. 2010, 41, 46. [Google Scholar] [CrossRef]
- Wang, B.; Yang, X.-L.; Li, W.; Zhu, Y.; Ge, X.-Y.; Zhang, L.-B.; Zhang, Y.-Z.; Bock, C.-T.; Shi, Z.-L. Detection and genome characterization of four novel bat hepadnaviruses and a hepevirus in China. Virol. J. 2017, 14, 40. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Murakami, S.; Yamamoto, T.; Mineshita, K.; Sakuyama, M.; Sasaki, R.; Maeda, K.; Horimoto, T. Detection of bat hepatitis E virus RNA in microbats in Japan. Virus Genes 2018, 54, 599–602. [Google Scholar] [CrossRef]
- Sridhar, S.; Yip, C.C.Y.; Wu, S.; Chew, N.F.S.; Leung, K.H.; Chan, J.F.W.; Zhao, P.S.; Chan, W.M.; Poon, R.W.S.; Tsoi, H.-W.; et al. Transmission of rat hepatitis E virus infection to humans in Hong Kong: A clinical and epidemiological analysis. Hepatology 2020, 31138-29. [Google Scholar] [CrossRef]
- Poon, L.L.M.; Chu, D.K.W.; Chan, K.H.; Wong, O.K.; Ellis, T.M.; Leung, Y.H.C.; Lau, S.K.P.; Woo, P.C.Y.; Suen, K.Y.; Yuen, K.Y.; et al. Identification of a Novel Coronavirus in Bats. J. Virol. 2005, 79, 2001–2009. [Google Scholar] [CrossRef] [PubMed]
- Drexler, J.F.; Corman, V.M.; Wegner, T.; Tateno, A.F.; Zerbinati, R.M.; Gloza-Rausch, F.; Seebens, A.; Mueller, M.A.; Drosten, C. Amplification of Emerging Viruses in a Bat Colony. Emerg. Infect. Dis. 2011, 17, 449–456. [Google Scholar] [CrossRef]
- Li, W.; Shi, Z.; Yu, M.; Ren, W.; Smith, C.; Epstein, J.H.; Wang, H.; Crameri, G.; Hu, Z.; Zhang, H.; et al. Bats are natural reservoirs of SARS-like coronaviruses. Science 2005, 310, 676–679. [Google Scholar] [CrossRef]
- Memish, Z.A.; Mishra, N.; Olival, K.J.; Fagbo, S.F.; Kapoor, V.; Epstein, J.H.; AlHakeem, R.; Durosinloun, A.; Asmari, A.M.; Islam, A.; et al. Middle East Respiratory Syndrome Coronavirus in Bats, Saudi Arabia. Emerg. Infect. Dis. 2013, 19, 1819–1823. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed]
- Carrington, C.V.F.; Foster, J.E.; Zhu, H.C.; Zhang, J.X.; Smith, G.J.D.; Thompson, N.; Auguste, A.J.; Ramkissoon, V.; Adesiyun, A.A.; Guan, Y. Detection and Phylogenetic Analysis of Group 1 Coronaviruses in South American Bats. Emerg. Infect. Dis. 2008, 14, 1890–1893. [Google Scholar] [CrossRef]
- Anthony, S.J.; Ojeda-Flores, R.; Rico-Chavez, O.; Navarrete-Macias, I.; Zambrana-Torrelio, C.M.; Rostal, M.K.; Epstein, J.H.; Tipps, T.; Liang, E.; Sanchez-Leon, M.; et al. Coronaviruses in bats from Mexico. J. Gen. Virol. 2013, 94, 1028–1038. [Google Scholar] [CrossRef]
- Corman, V.M.; Rasche, A.; Diallo, T.D.; Cottontail, V.M.; Stocker, A.; Souza, B.F.D.C.D.; Correa, J.I.; Carneiro, A.J.B.; Franke, C.R.; Nagy, M.; et al. Highly diversified coronaviruses in neotropical bats. J. Gen. Virol. 2013, 94, 1984–1994. [Google Scholar] [CrossRef] [PubMed]
- Asano, K.M.; Hora, A.S.; Scheffer, K.C.; Fahl, W.O.; Iamamoto, K.; Mori, E.; Brandão, P.E. Alphacoronavirus in urban Molossidae and Phyllostomidae bats, Brazil. Virol. J. 2016, 13, 1–5. [Google Scholar]
- Huynh, J.; Li, S.; Yount, B.; Smith, A.; Sturges, L.; Olsen, J.C.; Nagel, J.; Johnson, J.B.; Agnihothram, S.; Gates, J.E.; et al. Evidence supporting a zoonotic origin of human coronavirus strain NL63. J. Virol. 2012, 86, 12816–12825. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Fang, B.; Liu, Y.; Cai, M.; Jun, J.; Ma, J.; Bu, D.; Wang, L.; Zhou, P.; Wang, H.; et al. Newly emerged porcine enteric alphacoronavirus in southern China_ Identification, origin and evolutionary history analysis. Infect. Genet. Evol. 2018, 62, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Li, F.; Shi, Z.-L. Origin and evolution of pathogenic coronaviruses. Nature 2019, 17, 181–192. [Google Scholar] [CrossRef]
- Asano, K.M.; Gregori, F.; Hora, A.S.; Scheffer, K.C.; Fahl, W.O.; Iamamoto, K.; Mori, E.; Silva, F.D.F.; Taniwaki, S.A.; Brandão, P.E. Group A rotavirus in Brazilian bats: Description of novel T15 and H15 genotypes. Arch. Virol. 2016, 161, 3225–3230. [Google Scholar] [CrossRef]
- Yinda, C.K.; Zeller, M.; Conceição-Neto, N.; Maes, P.; Deboutte, W.; Beller, L.; Heylen, E.; Ghogomu, S.M.; Van Ranst, M.; Matthijnssens, J. Novel highly divergent reassortant bat rotaviruses in Cameroon, without evidence of zoonosis. Sci. Rep. 2016, 6, srep34209. [Google Scholar] [CrossRef]
- Kim, H.K.; Yoon, S.W.; Kim, D.J.; Koo, B.S.; Noh, J.Y.; Kim, J.H.; Choi, Y.G.; Na, W.; Chang, K.T.; Song, D.; et al. Detection of Severe Acute Respiratory Syndrome-Like, Middle East Respiratory Syndrome-Like Bat Coronaviruses and Group H Rotavirus in Faeces of Korean Bats. Transbound. Emerg. Dis. 2016, 63, 365–372. [Google Scholar] [CrossRef]
- Bányai, K.; Kemenesi, G.; Budinski, I.; Földes, F.; Zana, B.; Marton, S.; Varga-Kugler, R.; Oldal, M.; Kurucz, K.; Jakab, F. Candidate new rotavirus species in Schreiber’s bats, Serbia. Infect. Genet. Evol. 2017, 48, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Yinda, C.K.; Ghogomu, S.M.; Conceição-Neto, N.; Beller, L.; Deboutte, W.; Vanhulle, E.; Maes, P.; Van Ranst, M.; Matthijnssens, J. Cameroonian fruit bats harbor divergent viruses, including rotavirus H, bastroviruses, and picobirnaviruses using an alternative genetic code. Virus Evol. 2018, 4, vey008. [Google Scholar] [CrossRef]
- Matthijnssens, J.; Otto, P.H.; Ciarlet, M.; Desselberger, U.; Van Ranst, M.; Johne, R. VP6-sequence-based cutoff values as a criterion for rotavirus species demarcation. Arch. Virol. 2012, 157, 1177–1182. [Google Scholar] [CrossRef] [PubMed]
- Alam, M.M.; Kobayashi, N.; Ishino, M.; Ahmed, M.S.; Ahmed, M.U.; Paul, S.K.; Muzumdar, B.K.; Hussain, Z.; Wang, Y.H.; Naik, T.N. Genetic analysis of an ADRV-N-like novel rotavirus strain B219 detected in a sporadic case of adult diarrhea in Bangladesh. Arch. Virol. 2006, 152, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Ji, S.; Tang, Q.; Cui, X.; Yang, H.; Kan, B.; Gao, S. Molecular characterization of a novel adult diarrhoea rotavirus strain J19 isolated in China and its significance for the evolution and origin of group B rotaviruses. J. Gen. Virol. 2008, 89, 2622–2629. [Google Scholar] [CrossRef]
- Nagashima, S.; Kobayashi, N.; Ishino, M.; Alam, M.M.; Ahmed, M.U.; Paul, S.K.; Ganesh, B.; Chawla-Sarkar, M.; Krishnan, T.; Naik, T.N.; et al. Whole genomic characterization of a human rotavirus strain B219 belonging to a novel group of the genus rotavirus. J. Med. Virol. 2008, 80, 2023–2033. [Google Scholar] [CrossRef]
- Wakuda, M. Porcine rotavirus closely related to novel group of human rotaviruses. Emerg. Infect. Dis. 2011, 17, 1491–1493. [Google Scholar] [CrossRef] [PubMed]
- Esona, M.D.; Mijatovic-Rustempasic, S.; Conrardy, C.; Tong, S.; Kuzmin, I.V.; Agwanda, B.; Breiman, R.F.; Bányai, K.; Niezgoda, M.; Rupprecht, C.E.; et al. Reassortant Group A Rotavirus from Straw-colored Fruit Bat (Eidolon helvum). Emerg. Infect. Dis. 2010, 16, 1844–1852. [Google Scholar] [CrossRef]
- Xia, L.; Fan, Q.; He, B.; Xu, L.; Zhang, F.; Hu, T.; Wang, Y.; Li, N.; Qiu, W.; Zheng, Y.; et al. The complete genome sequence of a G3P[10] Chinese bat rotavirus suggests multiple bat rotavirus inter-host species transmission events. Infect. Genet. Evol. 2014, 28, 1–4. [Google Scholar] [CrossRef]
- Simsek, C.; Corman, V.M.; Everling, H.U.; Lukashev, A.N.; Rasche, A.; Maganga, G.D.; Binger, T.; Jansen, D.; Beller, L.; Deboutte, W.; et al. At least seven distinct rotavirus genotype constellations in bats with evidence of reassortment and zoonotic transmissions. bioRxiv 2020, 12. [Google Scholar] [CrossRef]
- De Benedictis, P.; Schultz-Cherry, S.; Burnham, A.; Cattoli, G. Astrovirus infections in humans and animals–Molecular biology, genetic diversity, and interspecies transmissions. Infect. Genet. Evol. 2011, 11, 1529–1544. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.C.; Chu, D.K.W.; Liu, W.; Dong, B.Q.; Zhang, S.-Y.; Zhang, J.X.; Li, L.F.; Vijaykrishna, D.; Smith, G.J.D.; Chen, H.L.; et al. Detection of diverse astroviruses from bats in China. J. Gen. Virol. 2009, 90, 883–887. [Google Scholar] [CrossRef]
- Lacroix, A.; Duong, V.; Hul, V.; San, S.; Davun, H.; Omaliss, K.; Chea, S.; Hassanin, A.; Theppangna, W.; Silithammavong, S.; et al. Diversity of bat astroviruses in Lao PDR and Cambodia. Infect. Genet. Evol. 2017, 47, 41–50. [Google Scholar] [CrossRef]
- Karlsson, E.A.; Small, C.T.; Freiden, P.; Feeroz, M.M.; Matsen, F.A.; San, S.; Hasan, M.K.; Wang, D.; Jones-Engel, L.; Schultz-Cherry, S. Non-Human Primates Harbor Diverse Mammalian and Avian Astroviruses Including Those Associated with Human Infections. PLoS Pathog. 2015, 11, e1005225. [Google Scholar] [CrossRef]
- Japhet, M.O.; Famurewa, O.; Adesina, O.A.; Opaleye, O.O.; Wang, B.; Höhne, M.; Bock, C.T.; Marques, A.M.; Niendorf, S. Viral gastroenteritis among children of 0–5 years in Nigeria-Characterization of the first Nigerian aichivirus, recombinant noroviruses and detection of a zoonotic astrovirus. J. Clin. Virol. 2019, 111, 4–11. [Google Scholar] [CrossRef] [PubMed]
- Pankovics, P.; Boros, Á.; Kiss, T.; Delwart, E.; Reuter, G. Detection of a mammalian-like astrovirus in bird, European roller (Coracias garrulus). Infect. Genet. Evol. 2015, 34, 114–121. [Google Scholar] [CrossRef]
- Letko, M.; Seifert, S.N.; Olival, K.J.; Plowright, R.K.; Munster, V.J. Bat-borne virus diversity, spillover and emergence. Nature 2020, 18, 461–471. [Google Scholar] [CrossRef]
- Olival, K.J.; Hosseini, P.R.; Zambrana-Torrelio, C.; Ross, N.; Bogich, T.L.; Daszak, P. Host and viral traits predict zoonotic spillover from mammals. Nature 2017, 546, 646–650. [Google Scholar] [CrossRef] [PubMed]
- Mollentze, N.; Streicker, D.G. Viral zoonotic risk is homogenous among taxonomic orders of mammalian and avian reservoir hosts. Proc. Natl. Acad. Sci. USA 2020, 117, 9423–9430. [Google Scholar] [CrossRef] [PubMed]
- Lau, S.K.P.; Woo, P.C.Y.; Lai, K.K.Y.; Huang, Y.; Yip, C.C.Y.; Shek, C.T.; Lee, P.; Lam, C.S.F.; Chan, K.H.; Yuen, K.Y. Complete Genome Analysis of Three Novel Picornaviruses from Diverse Bat Species. J. Virol. 2011, 85, 8819–8828. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Victoria, J.G.; Wang, C.; Jones, M.; Fellers, G.M.; Kunz, T.H.; Delwart, E. Bat Guano Virome: Predominance of Dietary Viruses from Insects and Plants plus Novel Mammalian Viruses. J. Virol. 2010, 84, 6955–6965. [Google Scholar] [CrossRef] [PubMed]
- Kemenesi, G.; Zhang, D.; Marton, S.; Dallos, B.; Gorfol, T.; Estok, P.; Boldogh, S.; Kurucz, K.; Oldal, M.; Kutas, A.; et al. Genetic characterization of a novel picornavirus detected in Miniopterus schreibersii bats. J. Gen. Virol. 2015, 96, 815–821. [Google Scholar] [CrossRef]
- Wu, Z.; Yang, L.; Ren, X.; He, G.; Zhang, J.; Yang, J.; Qian, Z.; Dong, J.; Sun, L.; Zhu, Y.; et al. Deciphering the bat virome catalog to better understand the ecological diversity of bat viruses and the bat origin of emerging infectious diseases. ISME J. 2016, 10, 609–620. [Google Scholar] [CrossRef]
- Lukashev, A.N.; Corman, V.M.; Schacht, D.; Gloza-Rausch, F.; Seebens-Hoyer, A.; Gmyl, A.P.; Drosten, C.; Drexler, J.F. Close genetic relatedness of picornaviruses from European and Asian bats. J. Gen. Virol. 2017, 98, 955–961. [Google Scholar] [CrossRef] [PubMed]
- Zell, R.; Delwart, E.; Gorbalenya, A.E.; Hovi, T.; King, A.M.Q.; Knowles, N.J.; Lindberg, A.M.; Pallansch, M.A.; Palmenberg, A.C.; Reuter, G.; et al. ICTV Report Consortium ICTV Virus Taxonomy Profile: Picornaviridae. J. Gen. Virol. 2017, 98, 2421–2422. [Google Scholar] [CrossRef]
- Drexler, J.F.; Corman, V.M.; Lukashev, A.N.; van den Brand, J.M.A.; Gmyl, A.P.; Brünink, S.; Rasche, A.; Seggewiβ, N.; Feng, H.; Leijten, L.M.; et al. The Hepatovirus Ecology Consortium Evolutionary origins of hepatitis A virus in small mammals. Proc. Natl. Acad. Sci. USA 2015, 112, 15190–15195. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Lu, L.; Liu, F.; Su, H.; Dong, J.; Sun, L.; Zhu, Y.; Ren, X.; Yang, F.; Guo, F.; et al. Distribution and characteristics of rodent picornaviruses in China. Sci. Rep. 2016, 6, 34381. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bergner, L.M.; Mollentze, N.; Orton, R.J.; Tello, C.; Broos, A.; Biek, R.; Streicker, D.G. Characterizing and Evaluating the Zoonotic Potential of Novel Viruses Discovered in Vampire Bats. Viruses 2021, 13, 252. https://doi.org/10.3390/v13020252
Bergner LM, Mollentze N, Orton RJ, Tello C, Broos A, Biek R, Streicker DG. Characterizing and Evaluating the Zoonotic Potential of Novel Viruses Discovered in Vampire Bats. Viruses. 2021; 13(2):252. https://doi.org/10.3390/v13020252
Chicago/Turabian StyleBergner, Laura M., Nardus Mollentze, Richard J. Orton, Carlos Tello, Alice Broos, Roman Biek, and Daniel G. Streicker. 2021. "Characterizing and Evaluating the Zoonotic Potential of Novel Viruses Discovered in Vampire Bats" Viruses 13, no. 2: 252. https://doi.org/10.3390/v13020252
APA StyleBergner, L. M., Mollentze, N., Orton, R. J., Tello, C., Broos, A., Biek, R., & Streicker, D. G. (2021). Characterizing and Evaluating the Zoonotic Potential of Novel Viruses Discovered in Vampire Bats. Viruses, 13(2), 252. https://doi.org/10.3390/v13020252