
The Impacts of Low Diversity Sequence Data on Phylodynamic Inference during an Emerging Epidemic


Abstract
1. Introduction
2. Materials and Methods
2.1. Simulation Study
2.2. Empirical Data Analyses
3. Results
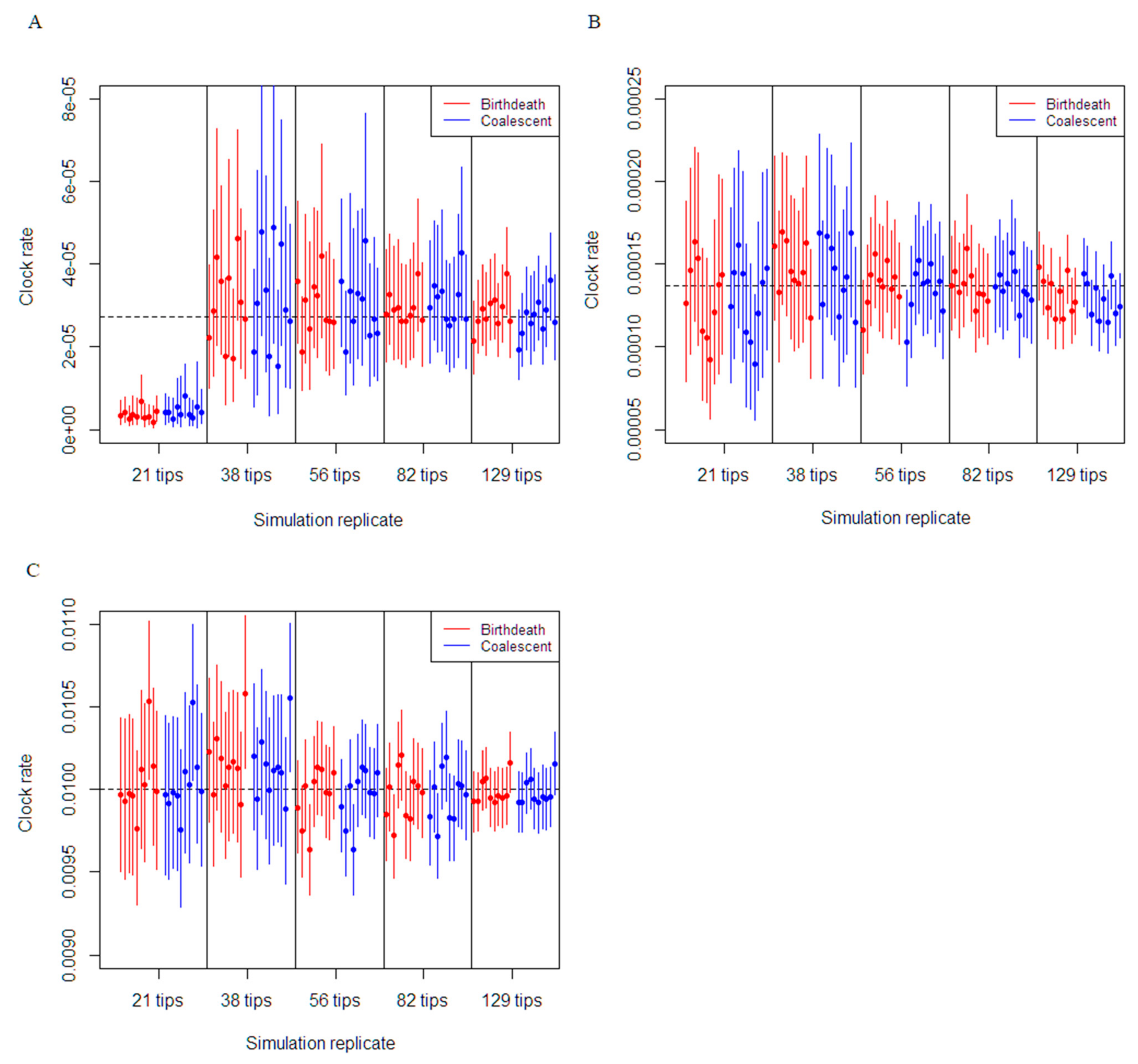
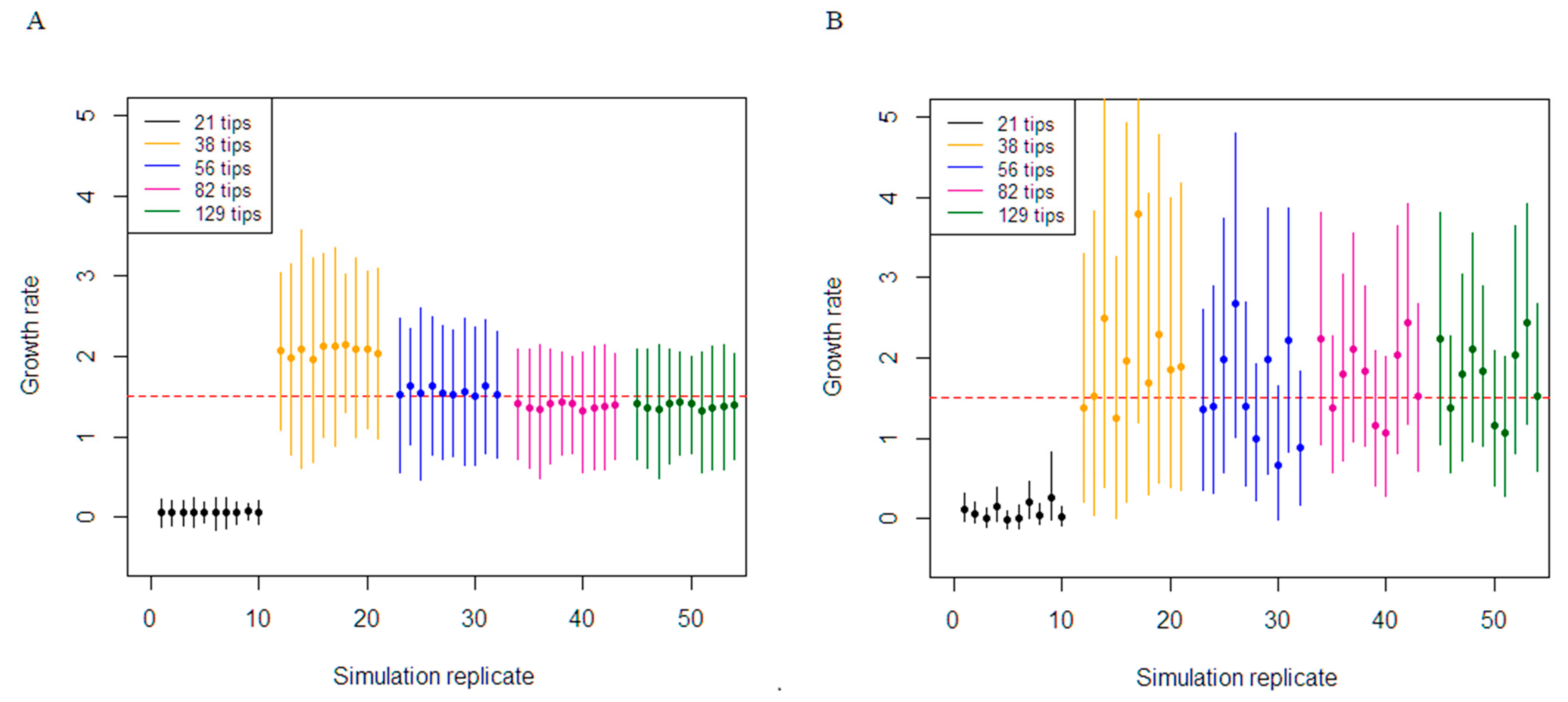
3.1. Simulation Study
3.2. Empirical Data Estimates of Molecular Clock Rate and Sampling
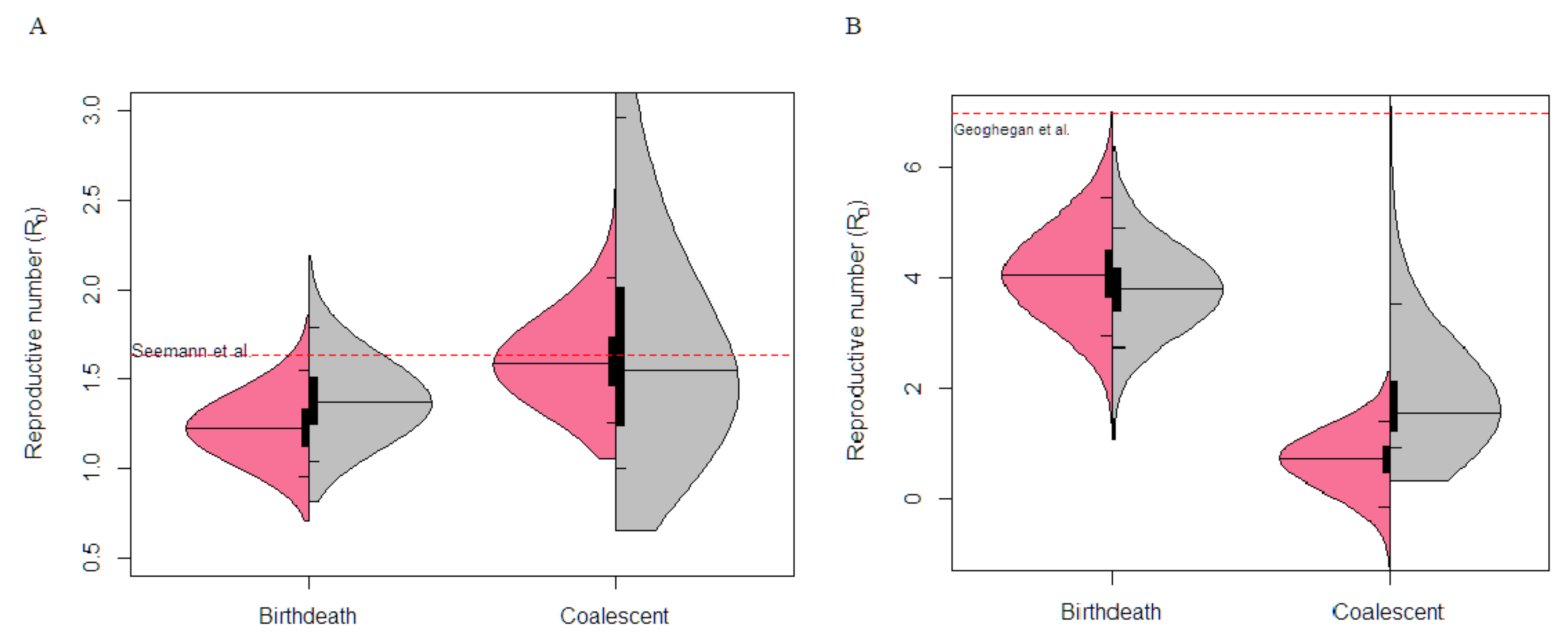
3.3. Victorian Highly Sampled Outbreak Cluster Analysis
3.4. New Zealand Exponential Cluster Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Grenfell, B.T.; Pybus, O.G.; Gog, J.R.; Wood, J.L.; Daly, J.M.; Mumford, J.A.; Holmes, E.C. Unifying the epidemiological and evolutionary dynamics of pathogens. Science 2004, 303, 327–332. [Google Scholar] [CrossRef] [PubMed]
- Pybus, O.G.; Rambaut, A. Evolutionary analysis of the dynamics of viral infectious disease. Nat. Rev. Genet. 2009, 10, 540–550. [Google Scholar] [CrossRef] [PubMed]
- Volz, E.M.; Siveroni, I. Bayesian phylodynamic inference with complex models. PLoS Comput. Biol. 2018, 14, e1006546. [Google Scholar] [CrossRef] [PubMed]
- Kuhnert, D.; Stadler, T.; Vaughan, T.G.; Drummond, A.J. Phylodynamics with migration: A computational framework to quantify population structure from genomic data. Mol. Biol. Evol. 2016, 33, 2102–2116. [Google Scholar] [CrossRef] [PubMed]
- Du Plessis, L.; Stadler, T. Getting to the root of epidemic spread with phylodynamic analysis of genomic data. Trends Microbiol. 2015, 23, 383–386. [Google Scholar] [CrossRef]
- Volz, E.M.; Koelle, K.; Bedford, T. Viral phylodynamics. PLoS Comput. Biol. 2013, 9, e1002947. [Google Scholar] [CrossRef]
- Duchene, S.; Bouckaert, R.; Duchene, D.A.; Stadler, T.; Drummond, A.J. Phylodynamic model adequacy using posterior predictive simulations. Syst. Biol. 2019, 68, 358–364. [Google Scholar] [CrossRef]
- Seemann, T.; Lane, C.R.; Sherry, N.L.; Duchene, S.; Goncalves da Silva, A.; Caly, L.; Sait, M.; Ballard, S.A.; Horan, K.; Schultz, M.B.; et al. Tracking the COVID-19 pandemic in Australia using genomics. Nat. Commun. 2020, 11, 4376. [Google Scholar] [CrossRef]
- Volz, E.M.; Frost, S.D. Sampling through time and phylodynamic inference with coalescent and birth-death models. J. R. Soc. Interface 2014, 11, 20140945. [Google Scholar] [CrossRef]
- Stadler, T.; Vaughan, T.G.; Gavryushkin, A.; Guindon, S.; Kuhnert, D.; Leventhal, G.E.; Drummond, A.J. How well can the exponential-growth coalescent approximate constant-rate birth-death population dynamics? Proc. Biol. Sci. 2015, 282, 20150420. [Google Scholar] [CrossRef]
- Stadler, T. Sampling-through-time in birth-death trees. J. Theor. Biol. 2010, 267, 396–404. [Google Scholar] [CrossRef] [PubMed]
- Stadler, T.; Kuhnert, D.; Bonhoeffer, S.; Drummond, A.J. Birth-death skyline plot reveals temporal changes of epidemic spread in HIV and hepatitis C virus (HCV). Proc. Natl. Acad. Sci. USA 2013, 110, 228–233. [Google Scholar] [CrossRef] [PubMed]
- Duchene, S.; Featherstone, L.; Haritopoulo-Sinanidou, M.; Rambaut, A.; Lemey, P.; Baele, G. Temporal signal and the phylodynamic threshold of SARS-CoV-2. Virus Evol. 2020. [Google Scholar] [CrossRef]
- Boskova, V.; Stadler, T.; Magnus, C. The influence of phylodynamic model specifications on parameter estimates of the Zika virus epidemic. Virus Evol. 2018, 4, vex044. [Google Scholar] [CrossRef]
- Vaughan, T.G.; Drummond, A.J. A stochastic simulator of birth-death master equations with application to phylodynamics. Mol. Biol. Evol. 2013, 30, 1480–1493. [Google Scholar] [CrossRef]
- Liu, Y.; Gayle, A.A.; Wilder-Smith, A.; Rocklov, J. The reproductive number of COVID-19 is higher compared to SARS coronavirus. J. Travel Med. 2020, 27. [Google Scholar] [CrossRef] [PubMed]
- Benvenuto, D.; Giovanetti, M.; Salemi, M.; Prosperi, M.; De Flora, C.; Junior Alcantara, L.C.; Angeletti, S.; Ciccozzi, M. The global spread of 2019-nCoV: A molecular evolutionary analysis. Pathog. Glob. Health 2020, 114, 64–67. [Google Scholar] [CrossRef]
- Byrne, A.W.; McEvoy, D.; Collins, A.B.; Hunt, K.; Casey, M.; Barber, A.; Butler, F.; Griffin, J.; Lane, E.A.; McAloon, C.; et al. Inferred duration of infectious period of SARS-CoV-2: Rapid scoping review and analysis of available evidence for asymptomatic and symptomatic COVID-19 cases. BMJ Open 2020, 10, e039856. [Google Scholar] [CrossRef] [PubMed]
- Dearlove, B.; Lewitus, E.; Bai, H.; Li, Y.; Reeves, D.B.; Joyce, M.G.; Scott, P.T.; Amare, M.F.; Vasan, S.; Michael, N.L.; et al. A SARS-CoV-2 vaccine candidate would likely match all currently circulating variants. Proc. Natl. Acad. Sci. USA 2020, 117, 23652–23662. [Google Scholar] [CrossRef] [PubMed]
- Ho, S.Y.; Duchene, S.; Duchene, D. Simulating and detecting autocorrelation of molecular evolutionary rates among lineages. Mol. Ecol. Resour. 2015, 15, 688–696. [Google Scholar] [CrossRef] [PubMed]
- Paradis, E.; Schliep, K. ape 5.0: An environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics 2019, 35, 526–528. [Google Scholar] [CrossRef] [PubMed]
- Bouckaert, R.; Vaughan, T.G.; Barido-Sottani, J.; Duchene, S.; Fourment, M.; Gavryushkina, A.; Heled, J.; Jones, G.; Kuhnert, D.; De Maio, N.; et al. BEAST 2.5: An advanced software platform for Bayesian evolutionary analysis. PLoS Comput. Biol. 2019, 15, e1006650. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior summarization in Bayesian phylogenetics using tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [PubMed]
- Shu, Y.; McCauley, J. GISAID: Global initiative on sharing all influenza data—From vision to reality. Euro Surveill. 2017, 22. [Google Scholar] [CrossRef]
- Ghafari, M.; Du Plessis, L.; Pybus, O.; Katzourakis, A. Time Dependence of SARS-CoV-2 Substitution Rates. Virol. Org. 2020. Available online: https://virological.org/t/time-dependence-of-sars-cov-2-substitution-rates/542 (accessed on 20 December 2020).
- Geoghegan, J.L.; Ren, X.; Storey, M.; Hadfield, J.; Jelley, L.; Jefferies, S.; Sherwood, J.; Paine, S.; Huang, S.; Douglas, J.; et al. Genomic epidemiology reveals transmission patterns and dynamics of SARS-CoV-2 in Aotearoa New Zealand. medRxiv 2020. [Google Scholar] [CrossRef]
- Biek, R.; Pybus, O.G.; Lloyd-Smith, J.O.; Didelot, X. Measurably evolving pathogens in the genomic era. Trends Ecol. Evol. 2015, 30, 306–313. [Google Scholar] [CrossRef]
- Baele, G.; Suchard, M.A.; Rambaut, A.; Lemey, P. Emerging concepts of data integration in pathogen phylodynamics. Syst. Biol. 2017, 66, e47–e65. [Google Scholar] [CrossRef]
- Boskova, V.; Bonhoeffer, S.; Stadler, T. Inference of epidemiological dynamics based on simulated phylogenies using birth-death and coalescent models. PLoS Comput. Biol. 2014, 10, e1003913. [Google Scholar] [CrossRef]
- Brown, J.M.; Thomson, R.C. Evaluating model performance in evolutionary biology. Annu. Rev. Ecol. Evol. Syst. 2018, 49, 95–114. [Google Scholar] [CrossRef]
- Featherstone, L.A.; Di Giallonardo, F.; Holmes, E.C.; Vaughan, T.G.; Duchêne, S. Infectious disease phylodynamics with occurrence data. bioRxiv 2020. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Phylodynamic Model | Parameter | Value | Substitution Model | Clock Model |
|---|---|---|---|---|
| Constant rate birth–death | Effective reproductive number (R0) | Estimated. Prior; Log-normal distribution with mean = 0, sigma = 1 | HKY+Γ | Strict clock |
| Become uninfectious rate (δ) | Fixed; 36.5 years−1 | |||
| Sampling probability (p) | Estimated. Prior; Beta distribution with (α and β) = 1 | |||
| Coalescent exponential | Effective population size (Ne) | Estimated. Prior; Log-normal distribution with mean = 1, sigma = 2 | HKY+Γ | Strict clock |
| Growth rate (r) | Estimated. Prior; Laplace distribution with μ = 0, scale = 30.70 |
| Dataset (Samples) | Variable Sites (Mean) | Coverage (%) | Relative Bias | Relative Precision (95% CI Width) | |
|---|---|---|---|---|---|
| Birth–death estimation of growth rate (Truth—1.5) | 21 tips | 3154 | 100 | 0.07 | 2.66 |
| 38 tips | 4857 | 100 | 0.21 | 2.19 | |
| 56 tips | 7875 | 100 | 0.01 | 1.51 | |
| 82 tips | 9956 | 100 | −0.13 | 1.32 | |
| 129 tips | 15537 | 100 | −0.15 | 0.93 | |
| Coalescent estimation of growth rate (Truth—1.5) | 21 tips | 3154 | 100 | −0.05 | 1.47 |
| 38 tips | 4857 | 100 | 0.12 | 1.72 | |
| 56 tips | 7875 | 100 | −0.07 | 0.89 | |
| 82 tips | 9956 | 100 | −0.02 | 0.83 | |
| 129 tips | 15537 | 100 | −0.09 | 0.58 |
| Dataset | Variable Sites (Mean) | Coverage (%) | Relative Bias | Relative Precision (95% CI Width) | |
|---|---|---|---|---|---|
| Birth–death estimation of growth rate (Truth—1.5) | 21 tips | 9 | 0 | −0.96 | 0.31 |
| 38 tips | 16 | 100 | 0.39 | 2.27 | |
| 56 tips | 24 | 100 | 0.04 | 1.73 | |
| 82 tips | 34 | 100 | −0.08 | 1.44 | |
| 129 tips | 62 | 100 | −0.17 | 0.97 | |
| Coalescent estimation of growth rate (Truth—1.5) | 21 tips | 9 | 0 | −0.94 | 0.36 |
| 38 tips | 16 | 100 | 0.34 | 4.14 | |
| 56 tips | 24 | 100 | 0.04 | 2.55 | |
| 82 tips | 34 | 100 | 0.17 | 2.26 | |
| 129 tips | 62 | 100 | −0.08 | 1.42 |
| Dataset | Variable Sites (Mean) | Coverage (%) | Relative Bias | Relative Precision (95% CI Width) | |
|---|---|---|---|---|---|
| Birth–death estimation of growth rate (Truth—1.5) | 21 tips | 45 | 100 | 0.11 | 2.61 |
| 38 tips | 72 | 100 | 0.31 | 2.41 | |
| 56 tips | 127 | 100 | 0.00 | 1.53 | |
| 82 tips | 158 | 100 | −0.11 | 1.40 | |
| 129 tips | 309 | 100 | −0.17 | 0.89 | |
| Coalescent estimation of growth rate (Truth—1.5) | 21 tips | 45 | 100 | 0.18 | 2.52 |
| 38 tips | 72 | 100 | 0.26 | 2.64 | |
| 56 tips | 127 | 90 | −0.13 | 1.28 | |
| 82 tips | 158 | 100 | −0.01 | 1.23 | |
| 129 tips | 309 | 100 | −0.11 | 0.80 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lam, A.; Duchene, S. The Impacts of Low Diversity Sequence Data on Phylodynamic Inference during an Emerging Epidemic. Viruses 2021, 13, 79. https://doi.org/10.3390/v13010079
Lam A, Duchene S. The Impacts of Low Diversity Sequence Data on Phylodynamic Inference during an Emerging Epidemic. Viruses. 2021; 13(1):79. https://doi.org/10.3390/v13010079
Chicago/Turabian StyleLam, Anthony, and Sebastian Duchene. 2021. "The Impacts of Low Diversity Sequence Data on Phylodynamic Inference during an Emerging Epidemic" Viruses 13, no. 1: 79. https://doi.org/10.3390/v13010079
APA StyleLam, A., & Duchene, S. (2021). The Impacts of Low Diversity Sequence Data on Phylodynamic Inference during an Emerging Epidemic. Viruses, 13(1), 79. https://doi.org/10.3390/v13010079