
Phage Display Technology as a Powerful Platform for Antibody Drug Discovery

Abstract
1. Introduction
2. Development of Antibody Drugs Using Phage Display Technology
2.1. Development of Human Monoclonal Antibodies Using Phage Display Technology
2.2. In Vitro Affinity Maturation of Monoclonal Antibodies Using Phage Display Technology
3. Target Discovery Using Phage Display Technology
3.1. Search for Antigens that React with Autoantibodies Using Phage Display Technology
3.2. High-Throughput Validation of Therapeutic Target Candidates Using Phage Display Technology
4. Conclusions and Prospects
Author Contributions
Funding
Conflicts of Interest
References
- Smith, G.P. Filamentous fusion phage: Novel expression vectors that display cloned antigens on the virion surface. Science 1985, 228, 1315–1317. [Google Scholar] [CrossRef] [PubMed]
- Widersten, M.; Mannervik, B. Glutathione transferases with novel active sites isolated by phage display from a library of random mutants. J. Mol. Biol. 1995, 250, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Jiang, B.; Liu, W.; Qu, H.; Meng, L.; Song, S.; Ouyang, T.; Shou, C. A novel peptide isolated from a phage display peptide library with trastuzumab can mimic antigen epitope of HER-2. J. Biol. Chem. 2005, 280, 4656–4662. [Google Scholar] [CrossRef] [PubMed]
- Mukai, Y.; Sugita, T.; Yamato, T.; Yamanada, N.; Shibata, H.; Imai, S.; Abe, Y.; Nagano, K.; Nomura, T.; Tsutsumi, Y.; et al. Creation of novel Protein Transduction Domain (PTD) mutants by a phage display-based high-throughput screening system. Biol. Pharm. Bull. 2006, 29, 1570–1574. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Shibata, H.; Yoshioka, Y.; Ohkawa, A.; Minowa, K.; Mukai, Y.; Abe, Y.; Taniai, M.; Nomura, T.; Kayamuro, H.; Nabeshi, H.; et al. Creation and X-ray structure analysis of the tumor necrosis factor receptor-1-selective mutant of a tumor necrosis factor-alpha antagonist. J. Biol. Chem. 2008, 283, 998–1007. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Segers, F.; Sliedregt-Bol, K.; Bot, I.; Woltman, A.M.; Boross, P.; Verbeek, S.; Overkleeft, H.; van der Marel, G.A.; van Kooten, C.; et al. Identification of a novel CD40 ligand for targeted imaging of inflammatory plaques by phage display. FASEB J. 2013, 27, 4136–4146. [Google Scholar] [CrossRef]
- Altmann, A.; Sauter, M.; Roesch, S.; Mier, W.; Warta, R.; Debus, J.; Dyckhoff, G.; Herold-Mende, C.; Haberkorn, U. Identification of a Novel ITGalphavbeta6-Binding Peptide Using Protein Separation and Phage Display. Clin. Cancer Res. 2017, 23, 4170–4180. [Google Scholar] [CrossRef]
- Yan, X.; Xu, Z. Ribosome-display technology: Applications for directed evolution of functional proteins. Drug Discov. Today 2006, 11, 911–916. [Google Scholar] [CrossRef]
- Kunamneni, A.; Ogaugwu, C.; Bradfute, S.; Durvasula, R. Ribosome Display Technology: Applications in Disease Diagnosis and Control. Antibodies 2020, 9, 28. [Google Scholar] [CrossRef]
- Feldhaus, M.J.; Siegel, R.W. Yeast display of antibody fragments: A discovery and characterization platform. J. Immunol. Methods 2004, 290, 69–80. [Google Scholar] [CrossRef]
- Traxlmayr, M.W.; Obinger, C. Directed evolution of proteins for increased stability and expression using yeast display. Arch. Biochem. Biophys. 2012, 526, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Kristensen, P.; Ravn, P.; Jensen, K.B.; Jensen, K. Applying phage display technology in aging research. Biogerontology 2000, 1, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Gao, S.; Lv, J.; Lin, Y.; Zhou, L.; Han, L. Phage Display Technology and its Applications in Cancer Immunotherapy. Anticancer Agents Med. Chem. 2019, 19, 229–235. [Google Scholar] [CrossRef] [PubMed]
- Marks, J.D.; Griffiths, A.D.; Malmqvist, M.; Clackson, T.P.; Bye, J.M.; Winter, G. By-passing immunization: Building high affinity human antibodies by chain shuffling. Biotechnology 1992, 10, 779–783. [Google Scholar] [CrossRef]
- Cordeiro, M.F. Technology evaluation: Lerdelimumab, Cambridge Antibody Technology. Curr. Opin. Mol. Ther. 2003, 5, 199–203. [Google Scholar]
- Garnock-Jones, K.P. Necitumumab: First Global Approval. Drugs 2016, 76, 283–289. [Google Scholar] [CrossRef]
- Chowdhury, P.S.; Viner, J.L.; Beers, R.; Pastan, I. Isolation of a high-affinity stable single-chain Fv specific for mesothelin from DNA-immunized mice by phage display and construction of a recombinant immunotoxin with anti-tumor activity. Proc. Natl. Acad. Sci. USA 1998, 95, 669–674. [Google Scholar] [CrossRef]
- Silence, K.; Dreier, T.; Moshir, M.; Ulrichts, P.; Gabriels, S.M.; Saunders, M.; Wajant, H.; Brouckaert, P.; Huyghe, L.; Van Hauwermeiren, T.; et al. ARGX-110, a highly potent antibody targeting CD70, eliminates tumors via both enhanced ADCC and immune checkpoint blockade. MAbs 2014, 6, 523–532. [Google Scholar] [CrossRef]
- Rothe, C.; Urlinger, S.; Lohning, C.; Prassler, J.; Stark, Y.; Jager, U.; Hubner, B.; Bardroff, M.; Pradel, I.; Boss, M.; et al. The human combinatorial antibody library HuCAL GOLD combines diversification of all six CDRs according to the natural immune system with a novel display method for efficient selection of high-affinity antibodies. J. Mol. Biol. 2008, 376, 1182–1200. [Google Scholar] [CrossRef]
- Prassler, J.; Thiel, S.; Pracht, C.; Polzer, A.; Peters, S.; Bauer, M.; Norenberg, S.; Stark, Y.; Kolln, J.; Popp, A.; et al. HuCAL PLATINUM, a synthetic Fab library optimized for sequence diversity and superior performance in mammalian expression systems. J. Mol. Biol. 2011, 413, 261–278. [Google Scholar] [CrossRef]
- Kohler, G.; Milstein, C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 1975, 256, 495–497. [Google Scholar] [CrossRef] [PubMed]
- Raso, V. Antibodies in diagnosis and therapy. The magic bullet--nearing the century mark. Semin. Cancer Biol. 1990, 1, 227–242. [Google Scholar] [PubMed]
- Peterson, N.C. Recombinant antibodies: Alternative strategies for developing and manipulating murine-derived monoclonal antibodies. Lab. Anim. Sci. 1996, 46, 8–14. [Google Scholar] [PubMed]
- Sandhu, J.S. Protein engineering of antibodies. Crit. Rev. Biotechnol. 1992, 12, 437–462. [Google Scholar] [CrossRef] [PubMed]
- Maynard, J.; Georgiou, G. Antibody engineering. Annu. Rev. Biomed. Eng. 2000, 2, 339–376. [Google Scholar] [CrossRef] [PubMed]
- Frenzel, A.; Schirrmann, T.; Hust, M. Phage display-derived human antibodies in clinical development and therapy. MAbs 2016, 8, 1177–1194. [Google Scholar] [CrossRef]
- Green, L.L. Transgenic mouse strains as platforms for the successful discovery and development of human therapeutic monoclonal antibodies. Curr. Drug Discov. Technol. 2014, 11, 74–84. [Google Scholar] [CrossRef]
- Vaughan, T.J.; Williams, A.J.; Pritchard, K.; Osbourn, J.K.; Pope, A.R.; Earnshaw, J.C.; McCafferty, J.; Hodits, R.A.; Wilton, J.; Johnson, K.S. Human antibodies with sub-nanomolar affinities isolated from a large non-immunized phage display library. Nat. Biotechnol. 1996, 14, 309–314. [Google Scholar] [CrossRef]
- De Haard, H.J.; van Neer, N.; Reurs, A.; Hufton, S.E.; Roovers, R.C.; Henderikx, P.; de Bruine, A.P.; Arends, J.W.; Hoogenboom, H.R. A large non-immunized human Fab fragment phage library that permits rapid isolation and kinetic analysis of high affinity antibodies. J. Biol. Chem. 1999, 274, 18218–18230. [Google Scholar] [CrossRef]
- Baker, K.P.; Edwards, B.M.; Main, S.H.; Choi, G.H.; Wager, R.E.; Halpern, W.G.; Lappin, P.B.; Riccobene, T.; Abramian, D.; Sekut, L.; et al. Generation and characterization of LymphoStat-B, a human monoclonal antibody that antagonizes the bioactivities of B lymphocyte stimulator. Arthritis Rheum 2003, 48, 3253–3265. [Google Scholar] [CrossRef]
- Kim, E.S. Tivozanib: First Global Approval. Drugs 2017, 77, 1917–1923. [Google Scholar] [CrossRef] [PubMed]
- Almagro, J.C.; Fransson, J. Humanization of antibodies. Front. BioSci. 2008, 13, 1619–1633. [Google Scholar] [PubMed]
- Kim, S.J.; Hong, H.J. Humanization by guided selections. Methods Mol. Biol. 2012, 907, 247–257. [Google Scholar] [PubMed]
- Wang, Z.; Wang, Y.; Li, Z.; Li, J.; Dong, Z. Humanization of a mouse monoclonal antibody neutralizing TNF-alpha by guided selection. J. Immunol. Methods 2000, 241, 171–184. [Google Scholar] [CrossRef]
- Chen, Y.; Wiesmann, C.; Fuh, G.; Li, B.; Christinger, H.W.; McKay, P.; de Vos, A.M.; Lowman, H.B. Selection and analysis of an optimized anti-VEGF antibody: Crystal structure of an affinity-matured Fab in complex with antigen. J. Mol. Biol. 1999, 293, 865–881. [Google Scholar] [CrossRef] [PubMed]
- Mazumdar, S. Raxibacumab. MAbs 2009, 1, 531–538. [Google Scholar] [CrossRef]
- Lu, D.; Jimenez, X.; Zhang, H.; Bohlen, P.; Witte, L.; Zhu, Z. Selection of high affinity human neutralizing antibodies to VEGFR2 from a large antibody phage display library for antiangiogenesis therapy. Int. J. Cancer 2002, 97, 393–399. [Google Scholar] [CrossRef]
- Lu, D.; Shen, J.; Vil, M.D.; Zhang, H.; Jimenez, X.; Bohlen, P.; Witte, L.; Zhu, Z. Tailoring in vitro selection for a picomolar affinity human antibody directed against vascular endothelial growth factor receptor 2 for enhanced neutralizing activity. J. Biol. Chem. 2003, 278, 43496–43507. [Google Scholar] [CrossRef]
- Li, S.; Kussie, P.; Ferguson, K.M. Structural basis for EGF receptor inhibition by the therapeutic antibody IMC-11F8. Structure 2008, 16, 216–227. [Google Scholar] [CrossRef]
- Liu, L.; Lu, J.; Allan, B.W.; Tang, Y.; Tetreault, J.; Chow, C.K.; Barmettler, B.; Nelson, J.; Bina, H.; Huang, L.; et al. Generation and characterization of ixekizumab, a humanized monoclonal antibody that neutralizes interleukin-17A. J. Inflamm. Res. 2016, 9, 39–50. [Google Scholar] [CrossRef]
- Markham, A. Ixekizumab: First Global Approval. Drugs 2016, 76, 901–905. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.S.; Soria, J.C.; Kowanetz, M.; Fine, G.D.; Hamid, O.; Gordon, M.S.; Sosman, J.A.; McDermott, D.F.; Powderly, J.D.; Gettinger, S.N.; et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 2014, 515, 563–567. [Google Scholar] [CrossRef] [PubMed]
- Markham, A. Atezolizumab: First Global Approval. Drugs 2016, 76, 1227–1232. [Google Scholar] [CrossRef]
- Kim, E.S. Avelumab: First Global Approval. Drugs 2017, 77, 929–937. [Google Scholar] [CrossRef] [PubMed]
- Markham, A. Guselkumab: First Global Approval. Drugs 2017, 77, 1487–1492. [Google Scholar] [CrossRef]
- Boehncke, W.H.; Brembilla, N.C.; Nissen, M.J. Guselkumab: The first selective IL-23 inhibitor for active psoriatic arthritis in adults. Expert Rev. Clin. Immunol. 2020, 1–9. [Google Scholar] [CrossRef]
- Ulrichts, H.; Silence, K.; Schoolmeester, A.; de Jaegere, P.; Rossenu, S.; Roodt, J.; Priem, S.; Lauwereys, M.; Casteels, P.; Van Bockstaele, F.; et al. Antithrombotic drug candidate ALX-0081 shows superior preclinical efficacy and safety compared with currently marketed antiplatelet drugs. Blood 2011, 118, 757–765. [Google Scholar] [CrossRef]
- Peyvandi, F.; Scully, M.; Kremer Hovinga, J.A.; Cataland, S.; Knobl, P.; Wu, H.; Artoni, A.; Westwood, J.P.; Mansouri Taleghani, M.; Jilma, B.; et al. Caplacizumab for Acquired Thrombotic Thrombocytopenic Purpura. N. Engl. J. Med. 2016, 374, 511–522. [Google Scholar] [CrossRef]
- Duggan, S. Caplacizumab: First Global Approval. Drugs 2018, 78, 1639–1642. [Google Scholar] [CrossRef]
- Al-Salama, Z.T. Emapalumab: First Global Approval. Drugs 2019, 79, 99–103. [Google Scholar] [CrossRef]
- Locatelli, F.; Jordan, M.B.; Allen, C.; Cesaro, S.; Rizzari, C.; Rao, A.; Degar, B.; Garrington, T.P.; Sevilla, J.; Putti, M.C.; et al. Emapalumab in Children with Primary Hemophagocytic Lymphohistiocytosis. N. Engl. J. Med. 2020, 382, 1811–1822. [Google Scholar] [CrossRef] [PubMed]
- Ho, M.; Nagata, S.; Pastan, I. Isolation of anti-CD22 Fv with high affinity by Fv display on human cells. Proc. Natl. Acad. Sci. USA 2006, 103, 9637–9642. [Google Scholar] [CrossRef] [PubMed]
- Kreitman, R.J.; Pastan, I. Antibody fusion proteins: Anti-CD22 recombinant immunotoxin moxetumomab pasudotox. Clin. Cancer Res. 2011, 17, 6398–6405. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, S. Moxetumomab Pasudotox: First Global Approval. Drugs 2018, 78, 1763–1767. [Google Scholar] [CrossRef]
- Kenniston, J.A.; Faucette, R.R.; Martik, D.; Comeau, S.R.; Lindberg, A.P.; Kopacz, K.J.; Conley, G.P.; Chen, J.; Viswanathan, M.; Kastrapeli, N.; et al. Inhibition of plasma kallikrein by a highly specific active site blocking antibody. J. Biol. Chem. 2014, 289, 23596–23608. [Google Scholar] [CrossRef]
- Syed, Y.Y. Lanadelumab: First Global Approval. Drugs 2018, 78, 1633–1637. [Google Scholar] [CrossRef]
- Melsheimer, R.; Geldhof, A.; Apaolaza, I.; Schaible, T. Remicade((R)) (infliximab): 20 years of contributions to science and medicine. Biologics 2019, 13, 139–178. [Google Scholar]
- Emancipator, K. Keytruda and PD-L1: A Real-World Example of Co-development of a Drug with a Predictive Biomarker. AAPS J. 2020, 23, 5. [Google Scholar] [CrossRef]
- DeMarini, D.M. The mutagenesis moonshot: The propitious beginnings of the environmental mutagenesis and genomics society. Environ. Mol. Mutagen. 2020, 61, 8–24. [Google Scholar] [CrossRef]
- Stemmer, W.P. Rapid evolution of a protein in vitro by DNA shuffling. Nature 1994, 370, 389–391. [Google Scholar] [CrossRef]
- Lantto, J.; Jirholt, P.; Barrios, Y.; Ohlin, M. Chain shuffling to modify properties of recombinant immunoglobulins. Methods Mol. Biol. 2002, 178, 303–316. [Google Scholar] [PubMed]
- Labrou, N.E. Random mutagenesis methods for in vitro directed enzyme evolution. Curr. Protein Pept. Sci. 2010, 11, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Boder, E.T.; Midelfort, K.S.; Wittrup, K.D. Directed evolution of antibody fragments with monovalent femtomolar antigen-binding affinity. Proc. Natl. Acad. Sci. USA 2000, 97, 10701–10705. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, T.; Mizohata, E.; Nagatoishi, S.; Watanabe, T.; Nakakido, M.; Iwanari, H.; Mochizuki, Y.; Nakayama, T.; Kado, Y.; Yokota, Y.; et al. Affinity Improvement of a Cancer-Targeted Antibody through Alanine-Induced Adjustment of Antigen-Antibody Interface. Structure 2019, 27, 519–527.e515. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, P.S.; Pastan, I. Improving antibody affinity by mimicking somatic hypermutation in vitro. Nat. Biotechnol. 1999, 17, 568–572. [Google Scholar] [CrossRef]
- McConnell, A.D.; Do, M.; Neben, T.Y.; Spasojevic, V.; MacLaren, J.; Chen, A.P.; Altobell, L., 3rd; Macomber, J.L.; Berkebile, A.D.; Horlick, R.A.; et al. High affinity humanized antibodies without making hybridomas; immunization paired with mammalian cell display and in vitro somatic hypermutation. PLoS ONE 2012, 7, e49458. [Google Scholar] [CrossRef]
- Kawamura, M.; Shibata, H.; Kamada, H.; Okamoto, T.; Mukai, Y.; Sugita, T.; Abe, Y.; Imai, S.; Nomura, T.; Nagano, K.; et al. A novel method for construction of gene fragment library to searching epitopes. Biochem. Biophys. Res. Commun. 2006, 346, 198–204. [Google Scholar] [CrossRef]
- Kamada, H.; Okamoto, T.; Kawamura, M.; Shibata, H.; Abe, Y.; Ohkawa, A.; Nomura, T.; Sato, M.; Mukai, Y.; Sugita, T.; et al. Creation of novel cell-penetrating peptides for intracellular drug delivery using systematic phage display technology originated from Tat transduction domain. Biol. Pharm. Bull. 2007, 30, 218–223. [Google Scholar] [CrossRef]
- Glennie, M.J.; Johnson, P.W. Clinical trials of antibody therapy. Immunol. Today 2000, 21, 403–410. [Google Scholar] [CrossRef]
- Zhao, P.; Zhang, Y.; Li, W.; Jeanty, C.; Xiang, G.; Dong, Y. Recent advances of antibody drug conjugates for clinical applications. Acta Pharm. Sin. B 2020, 10, 1589–1600. [Google Scholar] [CrossRef]
- Wolchok, J.D.; Kluger, H.; Callahan, M.K.; Postow, M.A.; Rizvi, N.A.; Lesokhin, A.M.; Segal, N.H.; Ariyan, C.E.; Gordon, R.A.; Reed, K.; et al. Nivolumab plus ipilimumab in advanced melanoma. N. Engl. J. Med. 2013, 369, 122–133. [Google Scholar] [CrossRef] [PubMed]
- Deeks, E.D. Nivolumab: A review of its use in patients with malignant melanoma. Drugs 2014, 74, 1233–1239. [Google Scholar] [CrossRef] [PubMed]
- Bagcchi, S. Pembrolizumab for treatment of refractory melanoma. Lancet Oncol. 2014, 15, e419. [Google Scholar] [CrossRef]
- Poole, R.M. Pembrolizumab: First global approval. Drugs 2014, 74, 1973–1981. [Google Scholar] [CrossRef]
- Minami, H.; Doi, T.; Toyoda, M.; Imamura, Y.; Kiyota, N.; Mitsuma, A.; Shimokata, T.; Naito, Y.; Matsubara, N.; Tajima, T.; et al. Phase I study of the anti-PD-1 antibody spartalizumab (PDR001) in Japanese patients with advanced malignancies. Cancer Sci. 2020. [Google Scholar] [CrossRef]
- Naing, A.; Gainor, J.F.; Gelderblom, H.; Forde, P.M.; Butler, M.O.; Lin, C.C.; Sharma, S.; Ochoa de Olza, M.; Varga, A.; Taylor, M.; et al. A first-in-human phase 1 dose escalation study of spartalizumab (PDR001), an anti-PD-1 antibody, in patients with advanced solid tumors. J. Immunother. Cancer 2020, 8, e000530. [Google Scholar] [CrossRef]
- Migden, M.R.; Rischin, D.; Schmults, C.D.; Guminski, A.; Hauschild, A.; Lewis, K.D.; Chung, C.H.; Hernandez-Aya, L.; Lim, A.M.; Chang, A.L.S.; et al. PD-1 Blockade with Cemiplimab in Advanced Cutaneous Squamous-Cell Carcinoma. N. Engl. J. Med. 2018, 379, 341–351. [Google Scholar] [CrossRef]
- Sidaway, P. Cemiplimab effective in cutaneous SCC. Nat. Rev. Clin. Oncol. 2018, 15, 472. [Google Scholar] [CrossRef]
- Kaufman, H.L.; Russell, J.; Hamid, O.; Bhatia, S.; Terheyden, P.; D’Angelo, S.P.; Shih, K.C.; Lebbe, C.; Linette, G.P.; Milella, M.; et al. Avelumab in patients with chemotherapy-refractory metastatic Merkel cell carcinoma: A multicentre, single-group, open-label, phase 2 trial. Lancet Oncol. 2016, 17, 1374–1385. [Google Scholar] [CrossRef]
- Sidaway, P. Skin cancer: Avelumab effective against Merkel-cell carcinoma. Nat. Rev. Clin. Oncol. 2016, 13, 652. [Google Scholar]
- Rosenberg, J.E.; Hoffman-Censits, J.; Powles, T.; van der Heijden, M.S.; Balar, A.V.; Necchi, A.; Dawson, N.; O’Donnell, P.H.; Balmanoukian, A.; Loriot, Y.; et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: A single-arm, multicentre, phase 2 trial. Lancet 2016, 387, 1909–1920. [Google Scholar] [CrossRef]
- Sidaway, P. Urological cancer: Atezolizumab effective against advanced disease. Nat. Rev. Clin. Oncol. 2016, 13, 266. [Google Scholar] [CrossRef] [PubMed]
- Antonia, S.; Goldberg, S.B.; Balmanoukian, A.; Chaft, J.E.; Sanborn, R.E.; Gupta, A.; Narwal, R.; Steele, K.; Gu, Y.; Karakunnel, J.J.; et al. Safety and antitumour activity of durvalumab plus tremelimumab in non-small cell lung cancer: A multicentre, phase 1b study. Lancet Oncol. 2016, 17, 299–308. [Google Scholar] [CrossRef]
- Brower, V. Anti-PD-L1 inhibitor durvalumab in bladder cancer. Lancet Oncol. 2016, 17, e275. [Google Scholar] [CrossRef]
- Chauhan, K.; Jandu, J.S.; Goyal, A.; Bansal, P.; Al-Dhahir, M.A. Rheumatoid Arthritis. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Weyand, C.M.; Goronzy, J.J. The immunology of rheumatoid arthritis. Nat. Immunol. 2020, 1108, 312–322. [Google Scholar]
- Justiz Vaillant, A.A.; Goyal, A.; Bansal, P.; Varacallo, M. Systemic Lupus Erythematosus. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Tsokos, G.C. Autoimmunity and organ damage in systemic lupus erythematosus. Nat. Immunol. 2020, 21, 605–614. [Google Scholar] [CrossRef]
- Li, R.; Patterson, K.R.; Bar-Or, A. Reassessing B cell contributions in multiple sclerosis. Nat. Immunol. 2018, 19, 696–707. [Google Scholar] [CrossRef]
- Tafti, D.; Ehsan, M.; Xixis, K.L. Multiple Sclerosis. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Taylor, P.C.; Feldmann, M. Anti-TNF biologic agents: Still the therapy of choice for rheumatoid arthritis. Nat. Rev. Rheumatol. 2009, 5, 578–582. [Google Scholar] [CrossRef]
- Kim, G.W.; Lee, N.R.; Pi, R.H.; Lim, Y.S.; Lee, Y.M.; Lee, J.M.; Jeong, H.S.; Chung, S.H. IL-6 inhibitors for treatment of rheumatoid arthritis: Past, present, and future. Arch. Pharm. Res. 2015, 38, 575–584. [Google Scholar] [CrossRef]
- Kasama, T.; Isozaki, T.; Takahashi, R.; Miwa, Y. Clinical effects of tocilizumab on cytokines and immunological factors in patients with rheumatoid arthritis. Int. Immunopharmacol. 2016, 35, 301–306. [Google Scholar] [CrossRef]
- Abbasi, M.; Mousavi, M.J.; Jamalzehi, S.; Alimohammadi, R.; Bezvan, M.H.; Mohammadi, H.; Aslani, S. Strategies toward rheumatoid arthritis therapy; the old and the new. J. Cell Physiol. 2019, 234, 10018–10031. [Google Scholar] [CrossRef] [PubMed]
- Kitching, A.R.; Anders, H.J.; Basu, N.; Brouwer, E.; Gordon, J.; Jayne, D.R.; Kullman, J.; Lyons, P.A.; Merkel, P.A.; Savage, C.O.S.; et al. ANCA-associated vasculitis. Nat. Rev. Dis. Primers 2020, 6, 71. [Google Scholar] [CrossRef] [PubMed]
- Kronbichler, A.; Lee, K.H.; Denicolo, S.; Choi, D.; Lee, H.; Ahn, D.; Kim, K.H.; Lee, J.H.; Kim, H.; Hwang, M.; et al. Immunopathogenesis of ANCA-Associated Vasculitis. Int. J. Mol. Sci. 2020, 21, 7319. [Google Scholar] [CrossRef]
- Swaak, A.J.; Huysen, V.; Nossent, J.C.; Smeenk, R.J. Antinuclear antibody profiles in relation to specific disease manifestations of systemic lupus erythematosus. Clin. Rheumatol. 1990, 9, 82–94. [Google Scholar] [CrossRef] [PubMed]
- Cabral, A.R.; Alarcon-Segovia, D. Autoantibodies in systemic lupus erythematosus. Curr. Opin. Rheumatol. 1997, 9, 387–392. [Google Scholar] [CrossRef]
- Cortini, A.; Bembich, S.; Marson, L.; Cocco, E.; Edomi, P. Identification of novel non-myelin biomarkers in multiple sclerosis using an improved phage-display approach. PLoS ONE 2019, 14, e0226162. [Google Scholar] [CrossRef]
- Vandormael, P.; Verschueren, P.; De Winter, L.; Somers, V. cDNA phage display for the discovery of theranostic autoantibodies in rheumatoid arthritis. Immunol. Res. 2017, 65, 307–325. [Google Scholar] [CrossRef]
- Wu, F.L.; Lai, D.Y.; Ding, H.H.; Tang, Y.J.; Xu, Z.W.; Ma, M.L.; Guo, S.J.; Wang, J.F.; Shen, N.; Zhao, X.D.; et al. Identification of Serum Biomarkers for Systemic Lupus Erythematosus Using a Library of Phage Displayed Random Peptides and Deep Sequencing. Mol. Cell Proteom. 2019, 18, 1851–1863. [Google Scholar] [CrossRef]
- Russo, N.; Wang, X.; Liu, M.; Banerjee, R.; Goto, M.; Scanlon, C.; Metwally, T.; Inglehart, R.C.; Tsodikov, A.; Duffy, S.; et al. A novel approach to biomarker discovery in head and neck cancer using an autoantibody signature. Oncogene 2013, 32, 5026–5037. [Google Scholar] [CrossRef]
- Dong, X.; Yang, M.; Sun, H.; Lu, J.; Zheng, Z.; Li, Z.; Zhong, L. Combined measurement of CA 15-3 with novel autoantibodies improves diagnostic accuracy for breast cancer. Onco Targets Ther. 2013, 6, 273–279. [Google Scholar]
- Wang, X.; Yu, J.; Sreekumar, A.; Varambally, S.; Shen, R.; Giacherio, D.; Mehra, R.; Montie, J.E.; Pienta, K.J.; Sanda, M.G.; et al. Autoantibody signatures in prostate cancer. N. Engl. J. Med. 2005, 353, 1224–1235. [Google Scholar] [CrossRef] [PubMed]
- Larman, H.B.; Zhao, Z.; Laserson, U.; Li, M.Z.; Ciccia, A.; Gakidis, M.A.; Church, G.M.; Kesari, S.; Leproust, E.M.; Solimini, N.L.; et al. Autoantigen discovery with a synthetic human peptidome. Nat. Biotechnol. 2011, 29, 535–541. [Google Scholar] [CrossRef] [PubMed]
- Lindpaintner, K. The impact of pharmacogenetics and pharmacogenomics on drug discovery. Nat. Rev. Drug Discov. 2002, 1, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Dopazo, J. Genomics and transcriptomics in drug discovery. Drug Discov. Today 2014, 19, 126–132. [Google Scholar] [CrossRef]
- Van Rensburg, I.C.; Loxton, A.G. Transcriptomics: The key to biomarker discovery during tuberculosis? Biomark. Med. 2015, 9, 483–495. [Google Scholar] [CrossRef]
- Petricoin, E.F.; Zoon, K.C.; Kohn, E.C.; Barrett, J.C.; Liotta, L.A. Clinical proteomics: Translating benchside promise into bedside reality. Nat. Rev. Drug Discov. 2002, 1, 683–695. [Google Scholar] [CrossRef]
- Kavallaris, M.; Marshall, G.M. Proteomics and disease: Opportunities and challenges. Med. J. Aust. 2005, 182, 575–579. [Google Scholar] [CrossRef]
- Hanash, S. Disease proteomics. Nature 2003, 422, 226–232. [Google Scholar] [CrossRef]
- Nishimura, T.; Ogiwara, A.; Fujii, K.; Kawakami, T.; Kawamura, T.; Anyouji, H.; Kato, H. Disease proteomics toward bedside reality. J. Gastroenterol. 2005, 40 (Suppl. S16), 7–13. [Google Scholar] [CrossRef]
- Yates, J.R.; Ruse, C.I.; Nakorchevsky, A. Proteomics by mass spectrometry: Approaches, advances, and applications. Annu. Rev. Biomed. Eng. 2009, 11, 49–79. [Google Scholar] [CrossRef]
- Robinson, M.R.; Miller, R.A.; Spellman, D.S. Mass Spectrometry-Based Biomarkers in Drug Development. Adv. Exp. Med. Biol. 2019, 1140, 435–449. [Google Scholar] [PubMed]
- Kaufmann, H.; Bailey, J.E.; Fussenegger, M. Use of antibodies for detection of phosphorylated proteins separated by two-dimensional gel electrophoresis. Proteomics 2001, 1, 194–199. [Google Scholar] [CrossRef]
- Chaga, G.S. Antibody arrays for determination of relative protein abundances. Methods Mol. Biol. 2008, 441, 129–151. [Google Scholar] [PubMed]
- Xu, Z.W.; Zhang, T.; Song, C.J.; Li, Q.; Zhuang, R.; Yang, K.; Yang, A.G.; Jin, B.Q. Application of sandwich ELISA for detecting tag fusion proteins in high throughput. Appl. Microbiol. Biotechnol. 2008, 81, 183–189. [Google Scholar] [CrossRef]
- Rimm, D.L.; Camp, R.L.; Charette, L.A.; Costa, J.; Olsen, D.A.; Reiss, M. Tissue microarray: A new technology for amplification of tissue resources. Cancer J. 2001, 7, 24–31. [Google Scholar]
- Au, N.H.; Gown, A.M.; Cheang, M.; Huntsman, D.; Yorida, E.; Elliott, W.M.; Flint, J.; English, J.; Gilks, C.B.; Grimes, H.L. P63 expression in lung carcinoma: A tissue microarray study of 408 cases. Appl Immunohistochem. Mol. Morphol. 2004, 12, 240–247. [Google Scholar] [CrossRef]
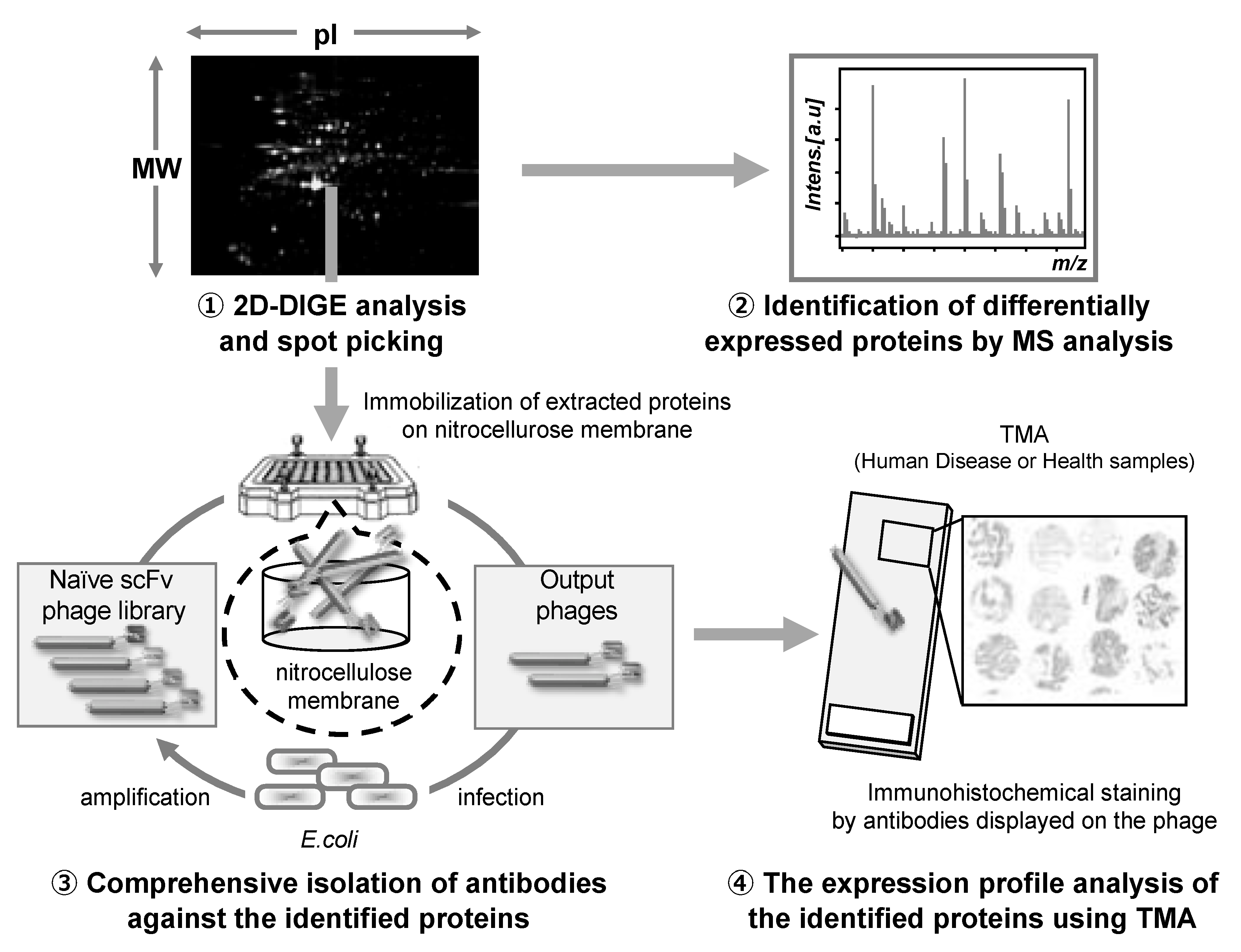
- Imai, S.; Nagano, K.; Yoshida, Y.; Okamura, T.; Yamashita, T.; Abe, Y.; Yoshikawa, T.; Yoshioka, Y.; Kamada, H.; Mukai, Y.; et al. Development of an antibody proteomics system using a phage antibody library for efficient screening of biomarker proteins. Biomaterials 2011, 32, 162–169. [Google Scholar] [CrossRef]
- Imai, S.; Mukai, Y.; Nagano, K.; Shibata, H.; Sugita, T.; Abe, Y.; Nomura, T.; Tsutsumi, Y.; Kamada, H.; Nakagawa, S.; et al. Quality enhancement of the non-immune phage scFv library to isolate effective antibodies. Biol. Pharm. Bull. 2006, 29, 1325–1330. [Google Scholar] [CrossRef]
- Nagano, K.; Imai, S.; Mukai, Y.; Nakagawa, S.; Abe, Y.; Kamada, H.; Tsunoda, S.; Tsutsumi, Y. Rapid isolation of intrabody candidates by using an optimized non-immune phage antibody library. Pharmazie 2009, 64, 238–241. [Google Scholar]
- Nagano, K.; Maeda, Y.; Kanasaki, S.; Watanabe, T.; Yamashita, T.; Inoue, M.; Higashisaka, K.; Yoshioka, Y.; Abe, Y.; Mukai, Y.; et al. Ephrin receptor A10 is a promising drug target potentially useful for breast cancers including triple negative breast cancers. J. Control. Release 2014, 189, 72–79. [Google Scholar] [CrossRef]
- Carey, L.; Winer, E.; Viale, G.; Cameron, D.; Gianni, L. Triple-negative breast cancer: Disease entity or title of convenience? Nat. Rev. Clin. Oncol. 2010, 7, 683–692. [Google Scholar] [CrossRef] [PubMed]
- Podo, F.; Buydens, L.M.; Degani, H.; Hilhorst, R.; Klipp, E.; Gribbestad, I.S.; Van Huffel, S.; van Laarhoven, H.W.; Luts, J.; Monleon, D.; et al. Triple-negative breast cancer: Present challenges and new perspectives. Mol. Oncol. 2010, 4, 209–229. [Google Scholar] [CrossRef] [PubMed]
- Pal, S.K.; Childs, B.H.; Pegram, M. Triple negative breast cancer: Unmet medical needs. Breast Cancer Res. Treat. 2011, 125, 627–636. [Google Scholar] [CrossRef] [PubMed]
- Nagano, K.; Kanasaki, S.; Yamashita, T.; Maeda, Y.; Inoue, M.; Higashisaka, K.; Yoshioka, Y.; Abe, Y.; Mukai, Y.; Kamada, H.; et al. Expression of Eph receptor A10 is correlated with lymph node metastasis and stage progression in breast cancer patients. Cancer Med. 2013, 2, 972–977. [Google Scholar] [CrossRef] [PubMed]
- Taki, S.; Kamada, H.; Inoue, M.; Nagano, K.; Mukai, Y.; Higashisaka, K.; Yoshioka, Y.; Tsutsumi, Y.; Tsunoda, S. A Novel Bispecific Antibody against Human CD3 and Ephrin Receptor A10 for Breast Cancer Therapy. PLoS ONE 2015, 10, e0144712. [Google Scholar] [CrossRef] [PubMed]
- Nagano, K.; Yamashita, T.; Inoue, M.; Higashisaka, K.; Yoshioka, Y.; Abe, Y.; Mukai, Y.; Kamada, H.; Tsutsumi, Y.; Tsunoda, S. Eph receptor A10 has a potential as a target for a prostate cancer therapy. Biochem. Biophys. Res. Commun. 2014, 450, 545–549. [Google Scholar] [CrossRef] [PubMed]
- Nagano, K.; Imai, S.; Zhao, X.; Yamashita, T.; Yoshioka, Y.; Abe, Y.; Mukai, Y.; Kamada, H.; Nakagawa, S.; Tsutsumi, Y.; et al. Identification and evaluation of metastasis-related proteins, oxysterol binding protein-like 5 and calumenin, in lung tumors. Int. J. Oncol. 2015, 47, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, T.; Nagano, K.; Kanasaki, S.; Maeda, Y.; Furuya, T.; Inoue, M.; Nabeshi, H.; Yoshikawa, T.; Yoshioka, Y.; Itoh, N.; et al. Annexin A4 is a possible biomarker for cisplatin susceptibility of malignant mesothelioma cells. Biochem. Biophys. Res. Commun. 2012, 421, 140–144. [Google Scholar] [CrossRef]
- Merchant, A.M.; Zhu, Z.; Yuan, J.Q.; Goddard, A.; Adams, C.W.; Presta, L.G.; Carter, P. An efficient route to human bispecific IgG. Nat. Biotechnol. 1998, 16, 677–681. [Google Scholar] [CrossRef]
- Yu, J.; Wang, W.; Huang, H. Efficacy and safety of bispecific T-cell engager (BiTE) antibody blinatumomab for the treatment of relapsed/refractory acute lymphoblastic leukemia and non-Hodgkin’s lymphoma: A systemic review and meta-analysis. Hematology 2019, 24, 199–207. [Google Scholar] [CrossRef]
- Hosseini, S.S.; Khalili, S.; Baradaran, B.; Bidar, N.; Shahbazi, M.A.; Mosafer, J.; Hashemzaei, M.; Mokhtarzadeh, A.; Hamblin, M.R. Bispecific monoclonal antibodies for targeted immunotherapy of solid tumors: Recent advances and clinical trials. Int. J. Biol. Macromol. 2020, 167, 1030–1047. [Google Scholar] [CrossRef] [PubMed]
- McGuinness, B.T.; Walter, G.; FitzGerald, K.; Schuler, P.; Mahoney, W.; Duncan, A.R.; Hoogenboom, H.R. Phage diabody repertoires for selection of large numbers of bispecific antibody fragments. Nat. Biotechnol. 1996, 14, 1149–1154. [Google Scholar] [CrossRef] [PubMed]
- Fagete, S.; Botas-Perez, L.; Rossito-Borlat, I.; Adea, K.; Gueneau, F.; Ravn, U.; Rousseau, F.; Kosco-Vilbois, M.; Fischer, N.; Hartley, O. Dual display: Phage selection driven by co-engagement of two targets by two different antibody fragments. Protein Eng. Des. Sel. 2017, 30, 575–582. [Google Scholar] [CrossRef] [PubMed]
- Luthra, A.; Langley, D.B.; Schofield, P.; Jackson, J.; Abdelatti, M.; Rouet, R.; Nevoltris, D.; Mazigi, O.; Crossett, B.; Christie, M.; et al. Human Antibody Bispecifics through Phage Display Selection. Biochemistry 2019, 58, 1701–1704. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, D.; Ye, Z.; Gouda, M.; Yokota, K.; Tsumuraya, T.; Fujii, I. Selection of inhibitory peptides for Aurora-A kinase from a phage-displayed library of helix-loop-helix peptides. Bioorg. Med. Chem. Lett. 2010, 20, 1776–1778. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, D.; Fujii, I. Phage selection of peptide “microantibodies”. Curr. Protoc. Chem. Biol. 2013, 5, 171–194. [Google Scholar] [CrossRef]
- Nagano, K.; Tsutsumi, Y. Development of novel drug delivery systems using phage display technology for clinical application of protein drugs. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2016, 92, 156–166. [Google Scholar] [CrossRef]




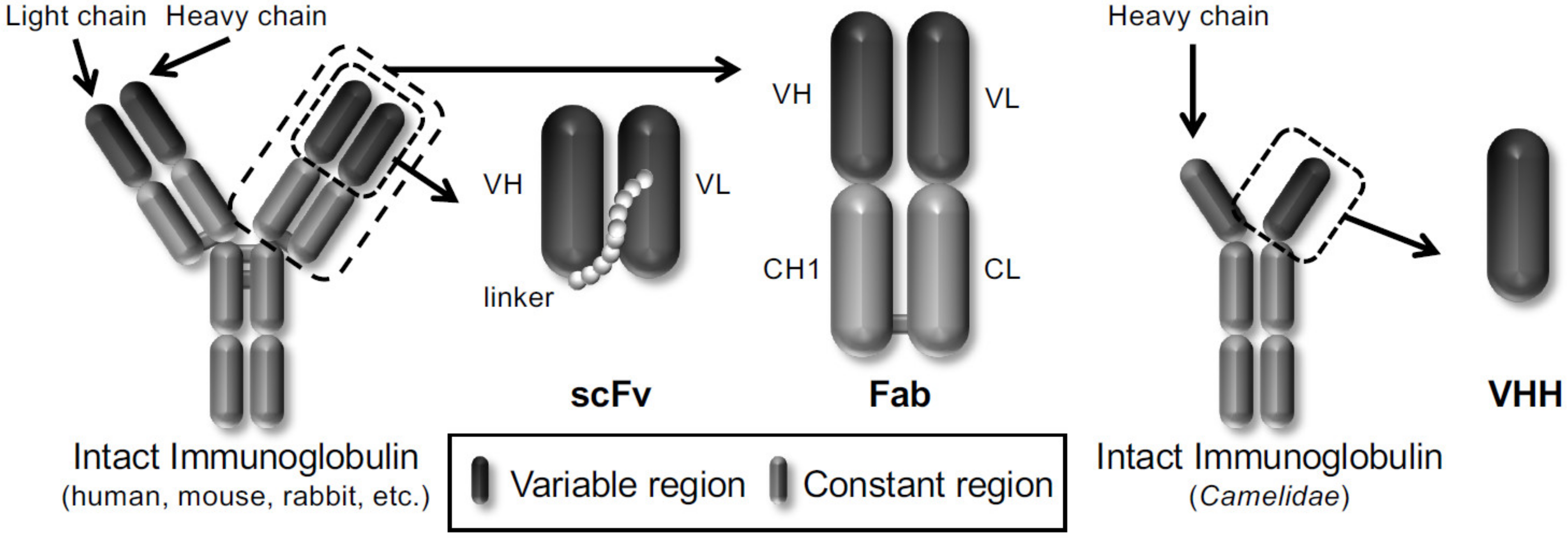{kind=link}
{kind=link}
{kind=link}
| Product Name | Nonproprietary Name | Target Antigen | First Application | Approved Year | Special Note on Phage Display Technology |
|---|---|---|---|---|---|
| Humira® | Adalimumab | TNFα | RA | 2002 | Humanization using guided selection method [34] |
| Lucentis® | Ranibizumab | VEGFA | nAMD | 2006 | In vitro affinity maturation [35]. |
| Benlysta® | Belimumab | BLyS | SLE | 2011 | Isolation from CAT’s library (human naïve scFv library) [30] |
| ABthrax® | Raxibacumab | Bacillus anthracis PA | Inhaled anthrax | 2012 | Isolation from CAT’s library (human naïve scFv library) [36] |
| Cyramza® | Ramucirumab | VEGFR2 | GC NSCLC | 2014 | Isolation from Dyax’s library (human naïve Fab library) [37,38] |
| Portrazza® | Necitumumab | EGFR | NSCLC | 2015 | Isolation from Dyax’s library (human naïve Fab library) [16,39] |
| Taltz® | Ixekizumab | IL-17A | Psoriasis | 2016 | Isolation from mouse immune Fab library [40,41] |
| Tecentriq® | Atezolizumab | PD-L1 | UC NSCLC | 2016 | Isolation from Genentech’s library (human naïve library) [42,43] |
| Bavencio® | Avelumab | PD-L1 | MCC | 2017 | Isolation from Dyax’s library (human naïve Fab library) [31,44] |
| Tremfya® | Guselkumab | IL-23 | Psoriasis | 2017 | Isolation from HuCAL GOLD® library (Synthetic Fab library) [45,46] |
| Cablivi® | Caplacizumab | vWF | aTTP | 2018 | Isolation from Camelidae-derived nanobody library [47,48,49] |
| Gamifant® | Emapalumab | IFNγ | HLH | 2018 | Isolation from CAT’s library (human naïve scFv library) [50,51] |
| Lumoxiti® | Moxetumomab pasudotox | CD22 | HCL | 2018 | In vitro affinity maturation [52,53,54]. |
| Takhzyro® | Lanadelumab | pKal | HAE | 2018 | Isolation from Dyax’s library (human naïve Fab library) [55,56]. |
| Advantages | Disadvantages | |
|---|---|---|
| Random mutagenesis | The introduction of logically inconceivable mutation can enhance the interaction of antigen-antibody and increase the stability of the structure of the antibody. | The three-dimensional structure of the antibody contact region may be disrupted by introducing random mutations. |
| Site-specific mutagenesis | The antibody with higher affinity could be efficiently prepared without disrupting the structure of the antibody. | The introduction of logically inconceivable mutations cannot be established. |
| Product Name | Nonproprietary Name | Drug Format | Target Antigen | Main Application | Sales Amount 1 | |
|---|---|---|---|---|---|---|
| 1 | Humira® | Adalimumab | Antibody | TNFα | RA 2 | 26.85 |
| 2 | Eliquis® | Apixaban | Organic compound | Fxa | Thrombocytopenia | 13.47 |
| 3 | Keytruda® | Pembrolizumab | Antibody | PD-1 | Cancer | 11.36 |
| 4 | Xarelto® | Rivaroxaban | Organic compound | Fxa | Thrombocytopenia | 10.38 |
| 5 | Lantus® | Insulin glargine | Peptide | Insulin receptor | Diabetes | 10.01 |
| 6 | Enbrel® | Etanercept | Fc fusion protein 3 | TNFα | RA | 9.71 |
| 7 | Stelara® | Ustekinumab | Antibody | IL-12/23 | Psoriasis | 8.79 |
| 8 | Opdivo® | Nivolumab | Antibody | PD-1 | Cancer | 8.03 |
| 9 | Januvia® | Sitagliptin | Organic compound | DPP-4 | Diabetes | 7.47 |
| 10 | NovoRapid® | Insulin aspart | Peptide | Insulin receptor | Diabetes | 7.39 |
| 11 | Trulicity® | Dulaglutide | Fc fusion protein 4 | GLP-1 receptor | Diabetes | 7.30 |
| 12 | Remicade® | Infliximab | Antibody | TNFα | RA | 6.96 |
| 13 | Avastin® | Bevacizumab | Antibody | VEGF | Cancer | 6.47 |
| 14 | Rituxan® | Rituximab | Antibody | CD20 | Cancer | 5.90 |
| 15 | Humalog® | Insulin lispro | Peptide | Insulin receptor | Diabetes | 5.83 |
| 16 | Herceptin® | Trastuzumab | Antibody | HER2 | Cancer | 5.72 |
| 17 | Imbruvica® | Ibrutinib | Organic compound | Tyrosine kinase | Cancer | 5.69 |
| 18 | Symbicort® | Budesonide | Organic Compound | - | Asthma | 5.60 |
| 19 | Revlimid® | Lenalidomide | Organic Compound | - | Cancer | 5.59 |
| 20 | Ibrance® | Palbociclib | Organic Compound | CDK4/6 | Cancer | 5.54 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nagano, K.; Tsutsumi, Y. Phage Display Technology as a Powerful Platform for Antibody Drug Discovery. Viruses 2021, 13, 178. https://doi.org/10.3390/v13020178
Nagano K, Tsutsumi Y. Phage Display Technology as a Powerful Platform for Antibody Drug Discovery. Viruses. 2021; 13(2):178. https://doi.org/10.3390/v13020178
Chicago/Turabian StyleNagano, Kazuya, and Yasuo Tsutsumi. 2021. "Phage Display Technology as a Powerful Platform for Antibody Drug Discovery" Viruses 13, no. 2: 178. https://doi.org/10.3390/v13020178
APA StyleNagano, K., & Tsutsumi, Y. (2021). Phage Display Technology as a Powerful Platform for Antibody Drug Discovery. Viruses, 13(2), 178. https://doi.org/10.3390/v13020178