
Recent Issues in Varicella-Zoster Virus Latency



{kind=link}
Abstract
:1. Introduction
2. The Nature of VZV Latency
3. Establishment of Alphaherpesvirus Lytic and Latent Infection
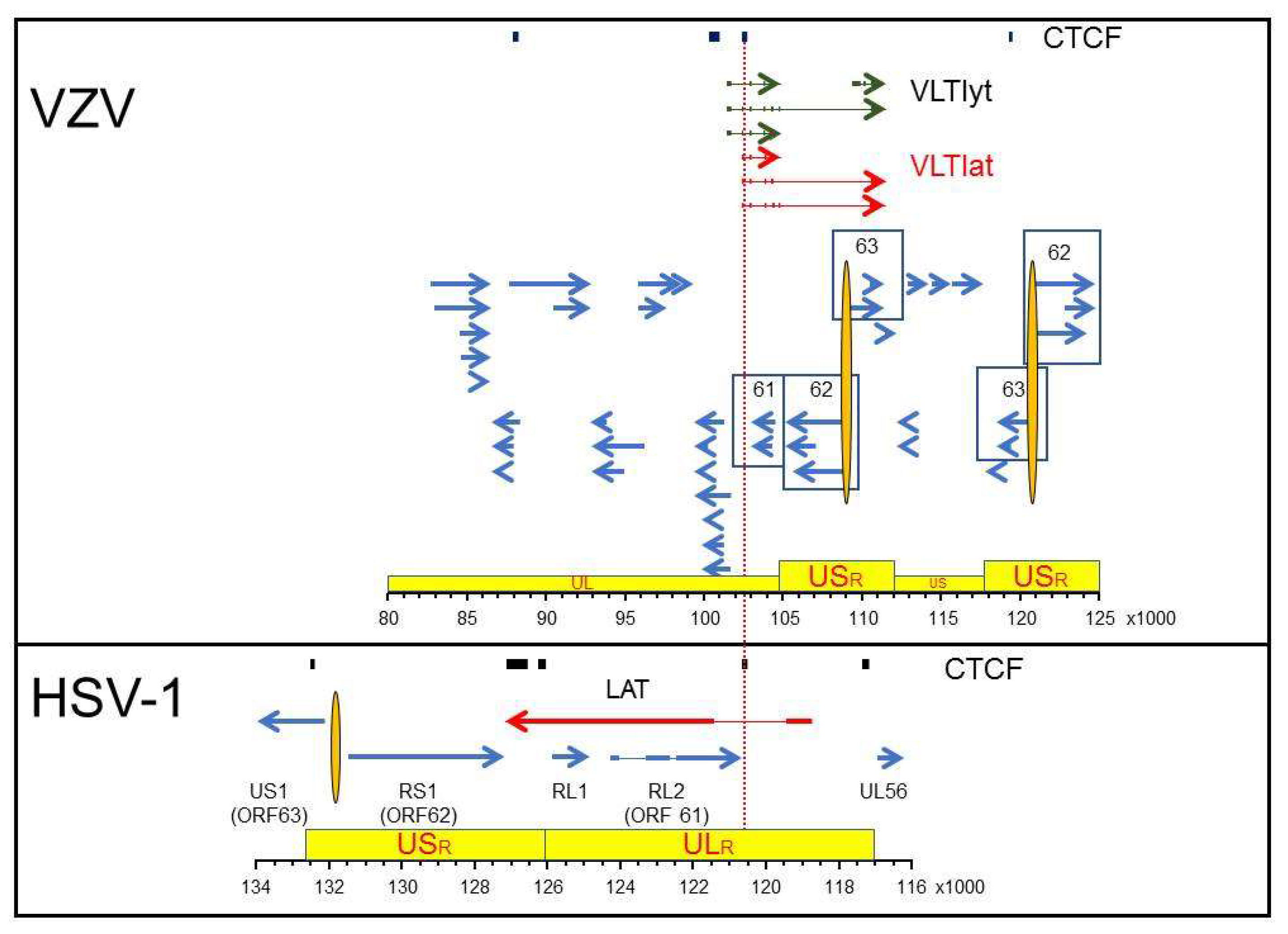
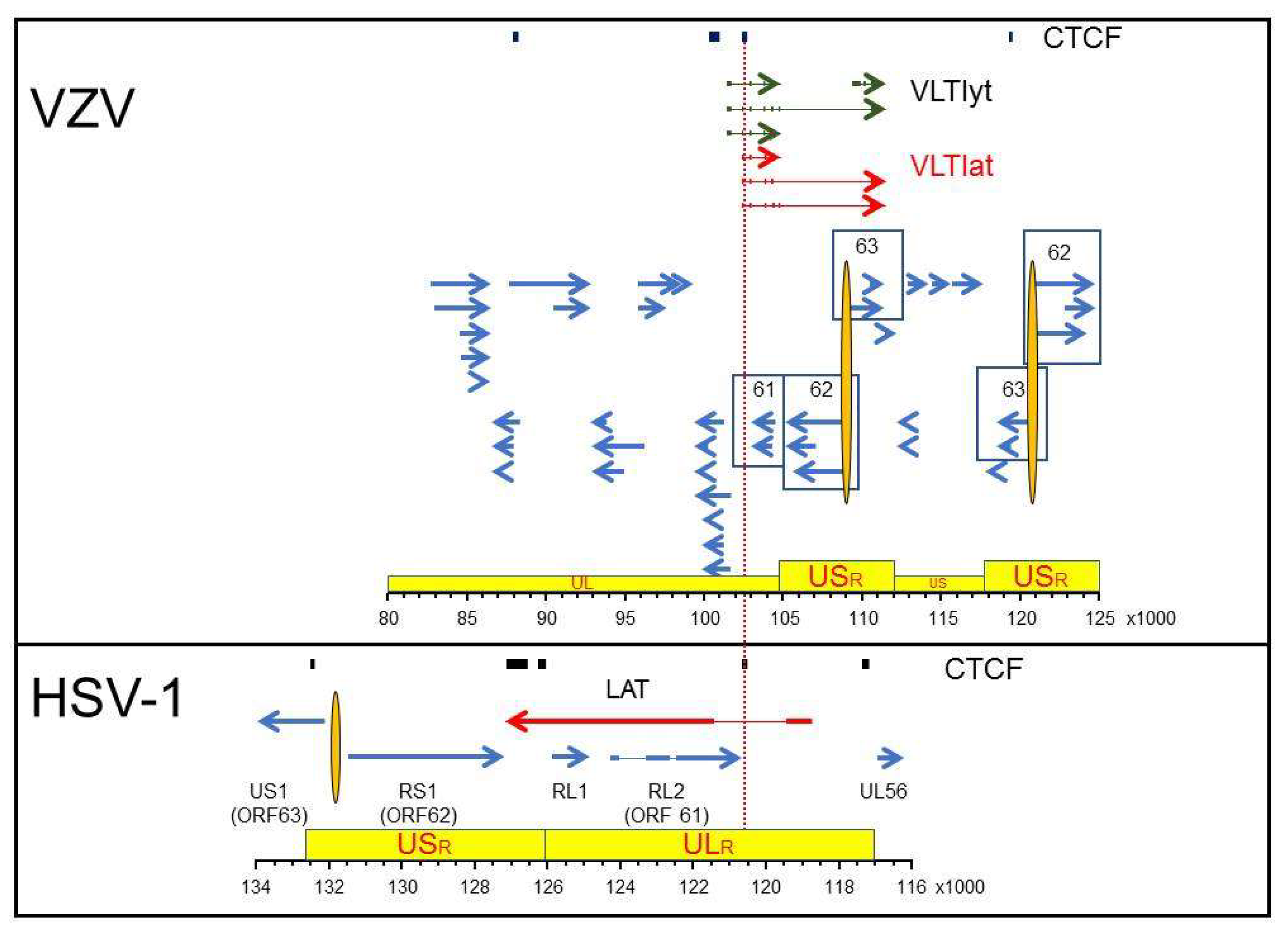
4. Alphaherpesvirus Latency Mediated through Expression of Viral Latency Transcripts
5. Epigenetic Control of Alphaherpesvirus Latency
6. Immunological Aspects of Alphaherpesvirus Latency with a Focus on VZV
6.1. Immune Protection against VZV Infection and Reactivation
6.2. Immune Histology in Ganglia
6.3. Inborn Errors of Immunity with Genetic Defects Conferring Increased Susceptibility to VZV Infection and Reactivation
7. Role of Autophagy in Reactivation of Other Alphaherpesviruses, the Example of Mollaret’s Recurrent Lymphocytic Meningitis
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kennedy, P.G.E.; Grinfeld, E.; Gow, J.W. Latent varicella-zoster virus is located predominantly in neurons in human trigeminal ganglia. Proc. Natl. Acad. Sci. USA 1998, 95, 4658–4662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gershon, A.A.; Breuer, J.; Cohen, J.I.; Cohrs, R.J.; Gershon, M.D.; Gilden, D.; Grose, C.; Hambleton, S.; Kennedy, P.G.E.; Oxman, M.N.; et al. Varicella zoster virus infection. Nat. Rev. Dis. Prim. 2015, 1, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kennedy, P.G.E.; Gershon, A.A. Clinical features of Varicella-Zoster virus infection of the nervous system. Viruses 2018, 10, 609. [Google Scholar] [CrossRef] [Green Version]
- Nagel, M.A.; Gilden, D.H. The protean neurologic manifestations of varicella-zoster virus. Clevel. Clin. J. Med. 2007, 74, 489–504. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, P.G.E. Issues in the treatment of neurological conditions caused by reactivation of varicella-zoster virus (VZV). Neurotherapeutics 2016, 13, 509–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kennedy, P.G.E. Zoster sine herpete: It would be rash to ignore it. Neurology 2011, 76, 416–417. [Google Scholar] [CrossRef]
- Braspenning, S.E.; Sadaoka, T.; Breuer, J.; Verjans, G.M.; Ouwendijk, W.J.; Depledge, D.P. Decoding the architecture of the varicella-zoster virus transcriptome. mBio 2020, 11, 5. [Google Scholar] [CrossRef]
- Heinz, J.; Kennedy, P.G.E.; Mogensen, T.H. The Role of Autophagy in Varicella Zoster Virus Infection. Viruses 2021, 13, 1053. [Google Scholar] [CrossRef]
- Kennedy, P.G.E.; Mogensen, T.H. Varicella-Zoster virus infection of neurons derived from neural stem cells. Viruses 2021, 13, 485. [Google Scholar] [CrossRef]
- Wang, K.; Lau, T.Y.; Morales, M.; Mont, E.K.; Straus, S.E. Laser-capture microdissection: Refining estimates of the quantity and distribution of latent herpes simplex virus 1 and varicella-zoster virus DNA in human trigeminal Ganglia at the single-cell level. J. Virol. 2005, 79, 14079–14087. [Google Scholar] [CrossRef] [Green Version]
- LaGuardia, J.J.; Cohrs, R.J.; Gilden, D.H. Prevalence of varicella-zoster virus DNA in dissociated human trigeminal ganglion neurons and nonneuronal cells. J. Virol. 1999, 73, 8571–8577. [Google Scholar] [CrossRef] [Green Version]
- Schmidbauer, M.; Budka, H.; Pilz, P.; Kurata, T.; Hondo, R. Presence, distribution and spread of productive varicella zoster virus infection in nervous tissues. Brain 1992, 115 Pt 2, 383–398. [Google Scholar] [CrossRef]
- Eshleman, E.; Shahzad, A.; Cohrs, R.J. Varicella zoster virus latency. Futur. Virol. 2011, 6, 341–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corral, C.; Quereda, C.; Muriel, A.; Martínez-Ulloa, P.-L.; González-Gómez, F.-J.; Corral, Í. Clinical spectrum and prognosis of neurological complications of reactivated varicella-zoster infection: The role of immunosuppression. J. Neurovirol. 2020, 26, 696–703. [Google Scholar] [CrossRef]
- Gilden, D.; Mahalingam, R.; Cohrs, R.; Kleinschmidt-DeMasters, B.; Forghani, B. The Protean Manifestations of Varicella-Zoster Virus Vasculopathy. J. Neurovirol. 2002, 8 (Suppl. 2), 75–79. [Google Scholar] [CrossRef]
- Black, F.L.; Woodall, J.P.; Evans, A.S.; Liebhaber, H.; Henle, G. Prevalence of antibody against viruses in the Tiriyo, an isolated Amazon tribe. Am. J. Epidemiol. 1970, 91, 430–438. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Tscharke, D.C. Herpes Simplex Virus Latency Is Noisier the Closer We Look. J. Virol. 2020, 94, e01701–e01719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Depledge, D.P.; Sadaoka, T.; Ouwendijk, W.J.D. Molecular Aspects of Varicella-Zoster Virus Latency. Viruses 2018, 10, 349. [Google Scholar] [CrossRef] [Green Version]
- Kennedy, P.G.E.; Rovnak, J.; Badani, H.; Cohrs, R.J. A comparison of herpes simplex virus type 1 and varicella-zoster virus latency and reactivation. J. Gen. Virol. 2015, 96, 1581–1602. [Google Scholar] [CrossRef] [PubMed]
- Cohrs, R.J.; Barbour, M.; Gilden, D.H. Varicella-zoster virus (VZV) transcription during latency in human ganglia: Detection of transcripts to genes 21, 29, 62, and 63 in a cDNA library enriched for VZV RNA. J. Virol. 1996, 70, 2789–2796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kennedy, P.G.E.; Grinfield, E.; Bell, J.E. Varicella-zoster virus gene expression in latently infected and explanted human ganglia. J. Virol. 2000, 74, 11893–11898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohrs, R.J.; Gilden, D.H.; Kinchington, P.R.; Grinfield, E.; Kennedy, P.G. Varicella-Zoster Virus Gene 66 Transcription and Translation in Latently Infected Human Ganglia. J. Virol. 2003, 77, 6660–6665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Depledge, D.P.; Ouwendijk, W.; Sadaoka, T.; Braspenning, S.E.; Mori, Y.; Cohrs, R.J.; Verjans, G.M.G.M.; Breuer, J. A spliced latency associated VZV transcript maps antisense to the viral transactivator gene 61. Nat. Commun. 2018, 9, 1167. [Google Scholar] [CrossRef]
- Ouwendijk, W.J.D.; Depledge, D.P.; Rajbhandari, L.; Lenac Roviš, T.; Jonjic, S.; Breuer, J.; Venkatesan, A.; Verjans, G.M.G.M.; Sadaoka, T. Varicella-zoster virus VLT-ORF63 fusion transcript induces broad viral gene expression during reactivation from neuronal latency. Nat. Commun. 2020, 11, 6324. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, B.M.; Bloom, D.C.; Cohrs, R.J.; Gilden, D.H.; Kennedy, P.G.E. Herpes simplex virus-1 and varicella-zoster virus latency in ganglia. J. Neurovirol. 2003, 9, 194–204. [Google Scholar] [CrossRef] [PubMed]
- Theil, D.; Paripovic, I.; Derfuss, T.; Herberger, S.; Strupp, M.; Arbusow, V.; Brandt, T. Dually infected (HSV-1/VZV) single neurons in human trigeminal ganglia. Ann. Neurol. 2003, 54, 678–682. [Google Scholar] [CrossRef] [PubMed]
- Häfelfinger, R.; Burgener, A.V.; Osthoff, M.; Potlukova, E. Simultaneous VZV and HSV-1 Reactivation after Minor Head Injury. Eur. J. Case Rep. Intern. Med. 2020, 7, 001746. [Google Scholar]
- Norberg, P. Divergence and genotyping of human alpha-herpesviruses: An overview. Infect. Genet. Evol. 2010, 10, 14–25. [Google Scholar] [CrossRef]
- Grose, C. Pangaea and the Out-of-Africa Model of Varicella-Zoster Virus Evolution and Phylogeography. J. Virol. 2012, 86, 9558–9565. [Google Scholar] [CrossRef] [Green Version]
- Mahalingam, R.; Gershon, A.; Gershon, M.; Cohen, J.I.; Arvin, A.; Zerboni, L.; Zhu, H.; Gray, W.; Messaoudi, I.; Traina-Dorge, V. Current In Vivo Models of Varicella-Zoster Virus Neurotropism. Viruses 2019, 11, 502. [Google Scholar] [CrossRef] [Green Version]
- Harson, R.; Grose, C. Egress of varicella-zoster virus from the melanoma cell: A tropism for the melanocyte. J. Virol. 1995, 69, 4994–5010. [Google Scholar] [CrossRef] [Green Version]
- Baird, N.L.; Zhu, S.; Pearce, C.M.; Viejo-Borbolla, A. Current In Vitro Models to Study Varicella Zoster Virus Latency and Reactivation. Viruses 2019, 11, 103. [Google Scholar] [CrossRef] [Green Version]
- Markus, A.; Lebenthal-Loinger, I.; Yang, I.H.; Kinchington, P.R.; Goldstein, R.S. An in vitro model of latency and reactivation of varicella zoster virus in human stem cell-derived neurons. PLoS Pathog. 2015, 11, e1004885. [Google Scholar] [CrossRef] [Green Version]
- Sadaoka, T.; Schwartz, C.L.; Rajbhandari, L.; Venkatesan, A.; Cohen, J.I. J Human Embryonic Stem Cell-Derived Neurons Are Highly Permissive for Varicella-Zoster Virus Lytic Infection. Virology 2017, 92, e01108–e01117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Martin-Caraballo, M.; Hsia, S.V. Modulation of Voltage-Gated Sodium Channel Activity in Human Dorsal Root Ganglion Neurons by Herpesvirus Quiescent Infection. J. Virol. 2020, 94, e01823-19. [Google Scholar] [CrossRef] [PubMed]
- Azarkh, Y.; Bos, N.; Gilden, D.H.; Cohrs, R.J. Human trigeminal ganglionic explants as a model to study alphaherpesvirus reactivation. J. Neurovirol. 2012, 18, 456–461. [Google Scholar] [CrossRef] [Green Version]
- Goodwin, T.J.; McCarthy, M.; Cohrs, R.J.; Kaufer, B.B. 3D tissue-like assemblies: A novel approach to investigate virus-cell interactions. Methods 2015, 90, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Pugazhenthi, S.; Nair, S.; Velmurugan, K.; Liang, Q.; Mahalingam, R.; Cohrs, R.J.; Nagel, M.A.; Gilden, D. Varicella-zoster virus infection of differentiated human neural stem cells. J. Virol. 2011, 85, 6678–6686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carpenter, J.E.; Hutchinson, J.A.; Jackson, W.; Grose, C. Egress of light particles among filopodia on the surface of Varicella-Zoster virus-infected cells. J. Virol. 2008, 82, 2821–2835. [Google Scholar] [CrossRef] [Green Version]
- Bauer, D.W.; Huffman, J.B.; Homa, F.L.; Evilevitch, A. Herpes Virus Genome, The Pressure Is On. J. Am. Chem. Soc. 2013, 135, 11216–11221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauer, D.W.; Li, D.; Huffman, J.; Homa, F.L.; Wilson, K.; Leavitt, J.C.; Casjens, S.R.; Baines, J.; Evilevitch, A. Exploring the Balance between DNA Pressure and Capsid Stability in Herpesviruses and Phages. J. Virol. 2015, 89, 9288–9298. [Google Scholar] [CrossRef] [Green Version]
- Stavropoulos, T.A.; Strathdee, C.A. An enhanced packaging system for helper-dependent herpes simplex virus vectors. J. Virol. 1998, 72, 7137–7143. [Google Scholar] [CrossRef] [Green Version]
- Goodwin, T.J.; McCarthy, M.; Osterrieder, N.; Cohrs, R.J.; Kaufer, B.B. Three-dimensional normal human neural progenitor tissue-like assemblies: A model of persistent varicella-zoster virus infection. PLoS Pathog. 2013, 8, e1003512. [Google Scholar] [CrossRef] [Green Version]
- Richards, A.L.; Sollars, P.J.; Smith, G.A. New tools to convert bacterial artificial chromosomes to a self-excising design and their application to a herpes simplex virus type 1 infectious clone. BMC Biotechnol. 2016, 16, 64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kent, J.R.; Zeng, P.Y.; Atanasiu, D.; Gardner, J.; Fraser, N.W.; Berger, S.L. During lytic infection herpes simplex virus type 1 is associated with histones bearing modifications that correlate with active transcription. J. Virol. 2004, 78, 10178–10186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conn, K.L.; Hendzel, M.J.; Schang, L.M. Linker histones are mobilized during infection with herpes simplex virus type 1. J. Virol. 2008, 82, 8629–8646. [Google Scholar] [CrossRef] [Green Version]
- Lacasse, J.J.; Schang, L.M. During lytic infections, herpes simplex virus type 1 DNA is in complexes with the properties of unstable nucleosomes. J. Virol. 2010, 84, 1920–1933. [Google Scholar] [CrossRef] [Green Version]
- Narayanan, A.; Ruyechan, W.T.; Kristie, T.M. The coactivator host cell factor-1 mediates Set1 and MLL1 H3K4 trimethylation at herpesvirus immediate early promoters for initiation of infection. Proc. Natl. Acad. Sci. USA 2007, 104, 10835–10840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wysocka, J.; Herr, W. The herpes simplex virus VP16-induced complex: The makings of a regulatory switch. Trends Biochem. Sci. 2003, 28, 294–304. [Google Scholar] [CrossRef]
- Hu, M.; Depledge, D.P.; Flores Cortes, E.; Breuer, J.; Schang, L.M. Chromatin dynamics and the transcriptional competence of HSV-1 genomes during lytic infections. PLoS Pathog. 2019, 15, e1008076. [Google Scholar] [CrossRef]
- Ou, Y.; Davis, K.A.; Traina-Dorge, V.; Gray, W.L. Simian varicella virus expresses a latency-associated transcript that is antisense to open reading frame 61 (ICP0) mRNA in neural ganglia of latently infected monkeys. J. Virol. 2007, 81, 8149–8156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukerjee, R.; Kang, W.; Suri, V.; Fraser, N.W. A non-consensus branch point plays an important role in determining the stability of the 2-kb LAT intron during acute and latent infections of herpes simplex virus type-1. Virology 2004, 324, 340–349. [Google Scholar] [CrossRef] [PubMed]
- Phelan, D.; Barrozo, E.R.; Bloom, D.C. HSV1 latent transcription and non-coding RNA: A critical retrospective. J. Neuroimmunol. 2017, 308, 65–101. [Google Scholar] [CrossRef] [PubMed]
- Umbach, J.L.; Nagel, M.A.; Cohrs, R.J.; Gilden, D.H.; Cullen, B.R. Analysis of human alphaherpesvirus microRNA expression in latently infected human trigeminal ganglia. J. Virol. 2009, 83, 10677–10683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.H.; Lee, L.Y.; Garber, D.A.; Schaffer, P.A.; Knipe, D.M.; Coen, D.M. Neither LAT nor open reading frame P mutations increase expression of spliced or intron-containing ICP0 transcripts in mouse ganglia latently infected with herpes simplex virus. J. Virol. 2002, 76, 4764–4772. [Google Scholar] [CrossRef] [Green Version]
- Burton, E.A.; Hong, C.S.; Glorioso, J.C. The stable 2.0-kilobase intron of the herpes simplex virus type 1 latency-associated transcript does not function as an antisense repressor of ICP0 in nonneuronal cells. J. Virol. 2003, 77, 3516–3530. [Google Scholar] [CrossRef] [Green Version]
- Amelio, A.L.; Giordani, N.V.; Kubat, N.J.; O’Neil, J.E.; Bloom, D.C. Deacetylation of the herpes simplex virus type 1 latency-associated transcript (LAT) enhancer and a decrease in LAT abundance precede an increase in ICP0 transcriptional permissiveness at early times postexplant. J. Virol. 2006, 80, 2063–2068. [Google Scholar] [CrossRef] [Green Version]
- Berger, S.L.; Kouzarides, T.; Shiekhattar, R.; Shilatifard, A. An operational definition of epigenetics. Genes Dev. 2009, 23, 781–783. [Google Scholar] [CrossRef] [Green Version]
- Jones, J.O.; Arvin, A.M. Viral and cellular gene transcription in fibroblasts infected with small plaque mutants of varicella-zoster virus. Antivir. Res. 2005, 68, 56–65. [Google Scholar] [CrossRef]
- Kuny, C.V.; Szpara, M.L. Alphaherpesvirus Genomics: Past, Present and Future. Curr. Issues Mol. Biol. 2021, 42, 41–80. [Google Scholar]
- Zhong, R.; Roeder, R.G.; Heintz, N. The primary structure and expression of four cloned human histone genes. Nucleic Acids Res. 1983, 11, 7409–7425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birchmeier, C.; Folk, W.; Birnstiel, M.L. The terminal RNA stem-loop structure and 80 bp of spacer DNA are required for the formation of 3’ termini of sea urchin H2A Mrna. Cell 1983, 35, 433–440. [Google Scholar] [CrossRef]
- Dominski, Z.; Marzluff, W.F. Formation of the 3’ end of histone Mrna. Gene 1999, 239, 1–14. [Google Scholar] [CrossRef]
- Jenuwein, T.; Allis, C.D. Translating the histone code. Science 2001, 293, 1074–1080. [Google Scholar] [CrossRef] [Green Version]
- Taylor, B.C.; Young, N.L. Combinations of histone post-translational modifications. Biochem. J. 2021, 478, 511–532. [Google Scholar] [CrossRef]
- Gary, L.; Gilden, D.H.; Cohrs, R.J. Epigenetic Regulation of Varicella-Zoster Virus Open Reading Frames 62 and 63 in Latently Infected Human Trigeminal Ganglia. J. Virol. 2006, 80, 4921–4926. [Google Scholar] [CrossRef] [Green Version]
- Kubat, N.J.; Tran, R.K.; McAnany, P.; Bloom, D.C. Specific histone tail modification and not DNA methylation is a determinant of herpes simplex virus type 1 latent gene expression. J. Virol. 2004, 78, 1139–1149. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.-Y.; Zhou, C.; Johnson, K.E.; Colgrove, R.C.; Coen, D.M.; Knipe, D.M. Herpesviral latency-associated transcript gene promotes assembly of heterochromatin on viral lytic-gene promoters in latent infection. Proc. Natl. Acad. Sci. USA 2005, 102, 16055–16059. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.Y.; Mandarino, A.; Chao, M.V.; Mohr, I.; Wilson, A.C. Transient reversal of episome silencing precedes VP16-dependent transcription during reactivation of latent HSV-1 in neurons. PLoS Pathog. 2012, 8, e1002540. [Google Scholar] [CrossRef]
- Kobayashi, M.; Kim, J.-Y.; Camarena, V.; Roehm, P.C.; Chao, M.V.; Wilson, A.C.; Mohr, I. A primary neuron culture system for the study of herpes simplex virus latency and reactivation. J. Vis. Exp. 2012, 62, e3823. [Google Scholar] [CrossRef]
- Wilson, A.C.; Mohr, I. A cultured affair: HSV latency and reactivation in neurons. Trends Microbiol. 2012, 20, 604–611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, T.; Zhou, G.; Roizman, B. HSV-1 gene expression from reactivated ganglia is disordered and concurrent with suppression of latency-associated transcript and miRNAs. Proc. Natl. Acad. Sci. USA 2011, 108, 18820–18824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohrs, R.J.; Gilden, D.H. Prevalence and Abundance of Latently Transcribed Varicella-Zoster Virus Genes in Human Ganglia. J. Virol. 2007, 81, 2950–2956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohrs, R.J.; Srock, K.; Barbour, M.B.; Owens, G.; Mahalingam, R.; E Devlin, M.; Wellish, M.; Gilden, D.H. Varicella-zoster virus (VZV) transcription during latency in human ganglia: Construction of a cDNA library from latently infected human trigeminal ganglia and detection of a VZV transcript. J. Virol. 1994, 68, 7900–7908. [Google Scholar] [CrossRef] [Green Version]
- Nagel, M.A.; Choe, A.; Traktinskiy, I.; Cordery-Cotter, R.; Gilden, D.; Cohrs, R.J. Varicella-Zoster Virus Transcriptome in Latently Infected Human Ganglia. J. Virol. 2010, 85, 2276–2287. [Google Scholar] [CrossRef] [Green Version]
- Avgousti, D.C.; Weitzman, M.D. Stress Flips a Chromatin Switch to Wake Up Latent Virus. Cell Host Microbe 2015, 18, 639–641. [Google Scholar] [CrossRef] [Green Version]
- Cliffe, A.R.; Arbuckle, J.H.; Vogel, J.L.; Geden, M.J.; Rothbart, S.B.; Cusack, C.L.; Strahl, B.D.; Kristie, T.M.; Deshmukh, M. Neuronal Stress Pathway Mediating a Histone Methyl/Phospho Switch Is Required for Herpes Simplex Virus Reactivation. Cell Host Microbe 2015, 18, 649–658. [Google Scholar] [CrossRef] [Green Version]
- Sabbattini, P.; Sjoberg, M.; Nikic, S.; Frangini, A.; Holmqvist, P.-H.; Kunowska, N.; Carroll, T.; Brookes, E.; Arthur, S.; Pombo, A.; et al. An H3K9/S10 methyl-phospho switch modulates Polycomb and Pol II binding at repressed genes during differentiation. Mol. Biol. Cell 2014, 25, 904–915. [Google Scholar] [CrossRef] [Green Version]
- Arbuckle, J.H.; Gardina, P.J.; Gordon, D.N.; Hickman, H.; Yewdell, J.W.; Pierson, T.C.; Myers, T.G.; Kristie, T.M. Inhibitors of the Histone Methyltransferases EZH2/1 Induce a Potent Antiviral State and Suppress Infection by Diverse Viral Pathogens. mBio 2017, 8, e01141-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harancher, M.R.; Packard, J.E.; Cowan, S.P.; DeLuca, N.A.; Dembowski, J.A. Antiviral Properties of the LSD1 Inhibitor SP-2509. J. Virol. 2020, 94, e00974-20. [Google Scholar] [CrossRef] [PubMed]
- Nevels, M.; Nitzsche, A.; Paulus, C. How to control an infectious bead string: Nucleosome-based regulation and targeting of herpesvirus chromatin. Rev. Med Virol. 2011, 21, 154–180. [Google Scholar] [CrossRef]
- Schang, L.M.; Hu, M.; Cortes, E.F.; Sun, K. Chromatin-mediated epigenetic regulation of HSV-1 transcription as a potential target in antiviral therapy. Antivir. Res. 2021, 192, 105103. [Google Scholar] [CrossRef]
- Hackenberg, M.; Previti, C.; Luque-Escamilla, P.L.; Carpena, P.; Martínez-Aroza, J.; Oliver, J.L. CpGcluster: A distance-based algorithm for CpG-island detection. BMC Bioinform. 2006, 7, 446. [Google Scholar] [CrossRef] [Green Version]
- Antequera, F.; Boyes, J.; Bird, A. High levels of de novo methylation and altered chromatin structure at CpG islands in cell lines. Cell 1990, 62, 503–514. [Google Scholar] [CrossRef]
- Youssoufian, H.; Hammer, S.M.; Hirsch, M.S.; Mulder, C. Methylation of the viral genome in an in vitro model of herpes simplex virus latency. Proc. Natl. Acad. Sci. USA 1982, 79, 2207–2210. [Google Scholar] [CrossRef] [Green Version]
- Dressler, G.R.; Rock, D.L.; Fraser, N.W. Latent herpes simplex virus type 1 DNA is not extensively methylated in vivo. J. Gen. Virol. 1987, 68 Pt 6, 1761–1765. [Google Scholar] [CrossRef]
- Madeira, F.; Park, Y.M.; Lee, J.; Buso, N.; Gur, T.; Madhusoodanan, N.; Basutkar, P.; Tivey, A.R.N.; Potter, S.C.; Finn, R.D.; et al. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 2019, 47, W636–W641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phillips, J.E.; Corces, V.G. CTCF: Master weaver of the genome. Cell 2009, 137, 1194–1211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghirlando, R.; Felsenfeld, G. 2016. CTCF: Making the right connections. Genes Dev. 2016, 30, 881–891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- West, A.G.; Gaszner, M.; Felsenfeld, G. Insulators: Many functions, many mechanisms. Genes Dev. 2002, 16, 271–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amelio, A.L.; McAnany, P.K.; Bloom, D.C. A chromatin insulator-like element in the herpes simplex virus type 1 latency-associated transcript region binds CCCTC-binding factor and displays enhancer-blocking and silencing activities. J. Virol. 2006, 80, 2358–2368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Q.; Lin, L.; Smith, S.; Huang, J.; Berger, S.L.; Zhou, J. CTCF-dependent chromatin boundary element between the latency-associated transcript and ICP0 promoters in the herpes simplex virus type 1 genome. J. Virol. 2007, 81, 5192–5201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stevens, H.C.; Fiskerstrand, C.; Bubb, V.J.; Dalziel, R.; Quinn, J.P. A regulatory domain spanning the repeat sequence RE1 from herpes simplex virus type 1 has cell specific differential functions in trigeminal neurons and fibroblasts. FEBS Lett. 2009, 583, 3335–3338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.S.; Raja, P.; Pan, D.; Pesola, J.M.; Coen, D.M.; Knipe, D.M. CCCTC-Binding Factor Acts as a Heterochromatin Barrier on Herpes Simplex Viral Latent Chromatin and Contributes to Poised Latent Infection. mBio 2018, 9, e02372-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ertel, M.K.; Cammarata, A.L.; Hron, R.J.; Neumann, D.M. CTCF occupation of the herpes simplex virus 1 genome is disrupted at early times postreactivation in a transcription-dependent manner. J. Virol. 2012, 86, 12741–12759. [Google Scholar] [CrossRef] [Green Version]
- Washington, S.D.; Edenfield, S.I.; Lieux, C.; Watson, Z.L.; Taasan, S.M.; Dhummakupt, A.; Bloom, D.C.; Neumann, D.M. Depletion of the Insulator Protein CTCF Results in Herpes Simplex Virus 1 Reactivation In Vivo. J. Virol. 2018, 92, e00173-18. [Google Scholar] [CrossRef] [Green Version]
- Washington, S.D.; Musarrat, F.; Ertel, M.K.; Backes, G.L.; Neumann, D.M. CTCF Binding Sites in the Herpes Simplex Virus 1 Genome Display Site-Specific CTCF Occupation, Protein Recruitment, and Insulator Function. J. Virol. 2018, 92, e00156-18. [Google Scholar] [CrossRef] [Green Version]
- Ziebarth, J.D.; Bhattacharya, A.; Cui, Y. CTCFBSDB 2.0: A database for CTCF-binding sites and genome organization. Nucleic Acids Res. 2013, 41, D188–D194. [Google Scholar] [CrossRef] [Green Version]
- Bao, L.; Zhou, M.; Cui, Y. CTCFBSDB: A CTCF-binding site database for characterization of vertebrate genomic insulators. Nucleic Acids Res. 2008, 36, D83–D87. [Google Scholar] [CrossRef] [Green Version]
- Washington, S.D.; Singh, P.; Johns, R.N.; Edwards, T.G.; Mariani, M.; Frietze, S.; Bloom, D.C.; Neumann, D.M. The CCCTC Binding Factor, CTRL2, Modulates Heterochromatin Deposition and the Establishment of Herpes Simplex Virus 1 Latency In Vivo. J. Virol. 2019, 93, e00415–e00419. [Google Scholar] [CrossRef] [Green Version]
- Jouanguy, E.; Béziat, V.; Mogensen, T.H.; Casanova, J.-L.; Tangye, S.G.; Zhang, S.-Y. Human inborn errors of immunity to herpes viruses. Curr. Opin. Immunol. 2020, 62, 106–122. [Google Scholar] [CrossRef]
- Ansari, R.; Rosen, L.B.; Lisco, A.; Gilden, D.; Holland, S.M.; Zerbe, C.S.; A Bonomo, R.; I Cohen, J. Primary and Acquired Immunodeficiencies Associated With Severe Varicella-Zoster Virus Infections. Clin. Infect. Dis. 2020. [Google Scholar] [CrossRef]
- Duncan, C.J.; Hambleton, S. Varicella zoster virus immunity: A primer. J. Infect. 2015, 71 (Suppl. 1), S47–S53. [Google Scholar] [CrossRef]
- Brunell, P.A.; Ross, A.; Miller, L.H.; Kuo, B. Prevention of Varicella by Zoster Immune Globulin. N. Engl. J. Med. 1969, 280, 1191–1194. [Google Scholar] [CrossRef]
- Brunell, P.A.; Gershon, A.A.; Hughes, W.T.; Riley, H.D., Jr.; Smith, J. Prevention of varicella in high-risk children: A collaborative study. Pediatrics 1972, 50, 718–722. [Google Scholar]
- Brunell, P.A.; Gershon, A.A. Passive Immunization against Varicella-Zoster Infections and Other Modes of Therapy. J. Infect. Dis. 1973, 127, 415–423. [Google Scholar] [CrossRef]
- Gershon, A.A.; Steinberg, S.; Brunell, P.A. Zoster Immune Globulin. A further assessment. N. Engl. J. Med. 1974, 290, 243–245. [Google Scholar] [CrossRef] [PubMed]
- Sen, N.; Sung, P.; Panda, A.; Arvin, A.M. Distinctive Roles for Type I and Type II Interferons and Interferon Regulatory Factors in the Host Cell Defense against Varicella-Zoster Virus. J. Virol. 2018, 92, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bayat, A.; Burbelo, P.D.; Browne, S.K.; Quinlivan, M.; Martinez, B.; Holland, S.M.; Buvanendran, A.; Kroin, J.S.; Mannes, A.J.; Breuer, J.; et al. Anti-cytokine autoantibodies in postherpetic neuralgia. J. Transl. Med. 2015, 13, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mogensen, K.E.; Daubas, P.; Gresser, I.; Sereni, D.; Varet, B. Patient with circulating antibodies to alpha-interferon. Lancet 1981, 318, 1227–1228. [Google Scholar] [CrossRef]
- Hüfner, K.; Derfuss, T.; Herberger, S.; Sunami, K.; Russell, S.; Sinicina, I.; Arbusow, V.; Strupp, M.; Brandt, T.; Theil, D. Latency of alpha-herpes viruses is accompanied by a chronic inflammation in human trigeminal ganglia but not in dorsal root ganglia. J. Neuropathol. Exp. Neurol. 2006, 65, 1022–1030. [Google Scholar] [CrossRef] [Green Version]
- Sutherland, J.P.; Steain, M.; Buckland, M.E.; Rodriguez, M.; Cunningham, A.L.; Slobedman, B.; Abendroth, A. Persistence of a T Cell Infiltrate in Human Ganglia Years After Herpes Zoster and During Post-herpetic Neuralgia. Front. Microbiol. 2019, 10, 2117. [Google Scholar] [CrossRef]
- Biron, C.A.; Byron, K.S.; Sullivan, J.L. Severe herpesvirus infections in an adolescent without natural killer cells. N. Engl. J. Med. 1989, 320, 1731–1735. [Google Scholar] [CrossRef] [PubMed]
- Abdollahpour, H.; Appaswamy, G.; Kotlarz, D.; Diestelhorst, J.; Beier, R.; Schäffer, A.A.; Gertz, E.M.; Schambach, A.; Kreipe, H.H.; Pfeifer, D.; et al. The phenotype of human STK4 deficiency. Blood 2012, 119, 3450–3457. [Google Scholar] [CrossRef]
- Ogunjimi, B.; Zhang, S.-Y.; Sørensen, K.B.; Skipper, K.; Carter-Timofte, M.; Kerner, G.; Luecke, S.; Prabakaran, T.; Cai, Y.; Meester, J.; et al. Inborn errors in RNA polymerase III underlie severe varicella zoster virus infections. J. Clin. Investig. 2017, 127, 3543–3556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ablasser, A.; Bauernfeind, F.; Hartmann, G.; Latz, E.; Fitzgerald, K.; Hornung, V. RIG-I-dependent sensing of poly(dA:dT) through the induction of an RNA polymerase III–transcribed RNA intermediate. Nat. Immunol. 2009, 10, 1065–1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carter-Timofte, M.E.; Paludan, S.R.; Mogensen, T.H. RNA Polymerase III as a Gatekeeper to Prevent Severe VZV Infections. Trends Mol. Med. 2018, 24, 904–915. [Google Scholar] [CrossRef]
- Carter-Timofte, M.E.; Hansen, A.F.; Mardahl, M.; Fribourg, S.; Rapaport, F.; Zhang, S.-Y.; Casanova, J.-L.; Paludan, S.R.; Christiansen, M.; Larsen, C.S.; et al. Varicella-zoster virus CNS vasculitis and RNA polymerase III gene mutation in identical twins. Neurol. Neuroimmunol. Neuroinflamm. 2018, 5, e500. [Google Scholar] [CrossRef] [Green Version]
- Carter-Timofte, M.E.; Hansen, A.F.; Christiansen, M.; Paludan, S.R.; Mogensen, T.H. Mutations in RNA Polymerase III genes and defective DNA sensing in adults with varicella-zoster virus CNS infection. Genes Immun. 2018, 20, 214–223. [Google Scholar] [CrossRef]
- Thomsen, M.M.; Tyrberg, T.; Skaalum, K.; Carter-Timofte, M.; Freytag, M.R.; Norberg, P.; Helleberg, M.; Storgaard, M.; Nielsen, H.; Bodilsen, J.; et al. Genetic variants and immune responses in a cohort of patients with varicella zoster virus encephalitis. J. Infect. Dis. 2021. [Google Scholar] [CrossRef]
- Shalabi, M.; Whitley, R.J. Recurrent Benign Lymphocytic Meningitis. Clin. Infect. Dis. 2006, 43, 1194–1197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tedder, D.G.; Ashley, R.; Tyler, K.L.; Levin, M.J. Herpes Simplex Virus Infection as a Cause of Benign Recurrent Lymphocytic Meningitis. Ann. Intern. Med. 1994, 121, 334–338. [Google Scholar] [CrossRef] [PubMed]
- Ohmichi, T.; Takezawa, H.; Fujii, C.; Tomii, Y.; Yoshida, T.; Nakagawa, M. Mollaret cells detected in a patient with varicella-zoster virus meningitis. Clin. Neurol. Neurosurg. 2012, 114, 1086–1087. [Google Scholar] [CrossRef] [PubMed]
- Hait, A.S.; Thomsen, M.M.; Larsen, S.M.; Helleberg, M.; Mardahl, M.; Barfod, T.S.; Christiansen, M.; Brandt, C.; Mogensen, T.H. Whole-Exome Sequencing of Patients With Recurrent HSV-2 Lymphocytic Mollaret Meningitis. J. Infect. Dis. 2021, 223, 1776–1786. [Google Scholar] [CrossRef]
- Hait, A.S.; Olagnier, D.; Sancho-Shimizu, V.; Skipper, K.A.; Helleberg, M.; Larsen, S.M.; Bodda, C.; Moldovan, L.I.; Ren, F.; Brinck Andersen, N.-S.; et al. Defects in LC3B2 and ATG4A underlie HSV2 meningitis and reveal a critical role for autophagy in antiviral defense in humans. Sci. Immunol. 2020, 5, 54. [Google Scholar] [CrossRef]
- Carpenter, J.E.; Jackson, W.; Benetti, L.; Grose, C. Autophagosome formation during varicella-zoster virus infection following endoplasmic reticulum stress and the unfolded protein response. J. Virol. 2011, 85, 9414–9424. [Google Scholar] [CrossRef] [Green Version]
- Buckingham, E.M.; Carpenter, J.E.; Jackson, W.; Zerboni, L.; Arvin, A.M.; Grose, C. Autophagic flux without a block differentiates varicella-zoster virus infection from herpes simplex virus infection. Proc. Natl. Acad. Sci. USA 2015, 112, 256–261. [Google Scholar] [CrossRef] [Green Version]
- Graybill, C.; Morgan, M.J.; Levin, M.J.; Lee, K.S. Varicella-zoster virus inhibits autophagosome-lysosome fusion and the degradation stage of mTOR-mediated autophagic flux. Virology 2018, 522, 220–227. [Google Scholar] [CrossRef]
- Girsch, J.H.; Jackson, W.; Carpenter, J.E.; Moninger, T.O.; Jarosinski, K.W.; Grose, C. Exocytosis of Progeny Infectious Varicella-Zoster Virus Particles via a Mannose-6-Phosphate Receptor Pathway without Xenophagy following Secondary Envelopment. J. Virol. 2020, 94, 16. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kennedy, P.G.E.; Mogensen, T.H.; Cohrs, R.J. Recent Issues in Varicella-Zoster Virus Latency. Viruses 2021, 13, 2018. https://doi.org/10.3390/v13102018
Kennedy PGE, Mogensen TH, Cohrs RJ. Recent Issues in Varicella-Zoster Virus Latency. Viruses. 2021; 13(10):2018. https://doi.org/10.3390/v13102018
Chicago/Turabian StyleKennedy, Peter G. E., Trine H. Mogensen, and Randall J. Cohrs. 2021. "Recent Issues in Varicella-Zoster Virus Latency" Viruses 13, no. 10: 2018. https://doi.org/10.3390/v13102018
APA StyleKennedy, P. G. E., Mogensen, T. H., & Cohrs, R. J. (2021). Recent Issues in Varicella-Zoster Virus Latency. Viruses, 13(10), 2018. https://doi.org/10.3390/v13102018