
TNF-Mediated Inhibition of Classical Swine Fever Virus Replication Is IRF1-, NF-κB- and JAK/STAT Signaling-Dependent

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells
2.2. Viruses
2.3. Reagents
2.4. Antiviral TNF Assay and JAK/STAT Compound Library Screening
2.5. RNA Isolation and Quantification
2.6. NF-κB Promoter Reporter Assay
2.7. Generation of IFNAR1, IRF3 and IRF1 Gene Knockout PEDSV.15 Cell Lines Using CRISPR/Cas9 Gene Editing
2.8. Western Blot Analyses
3. Results
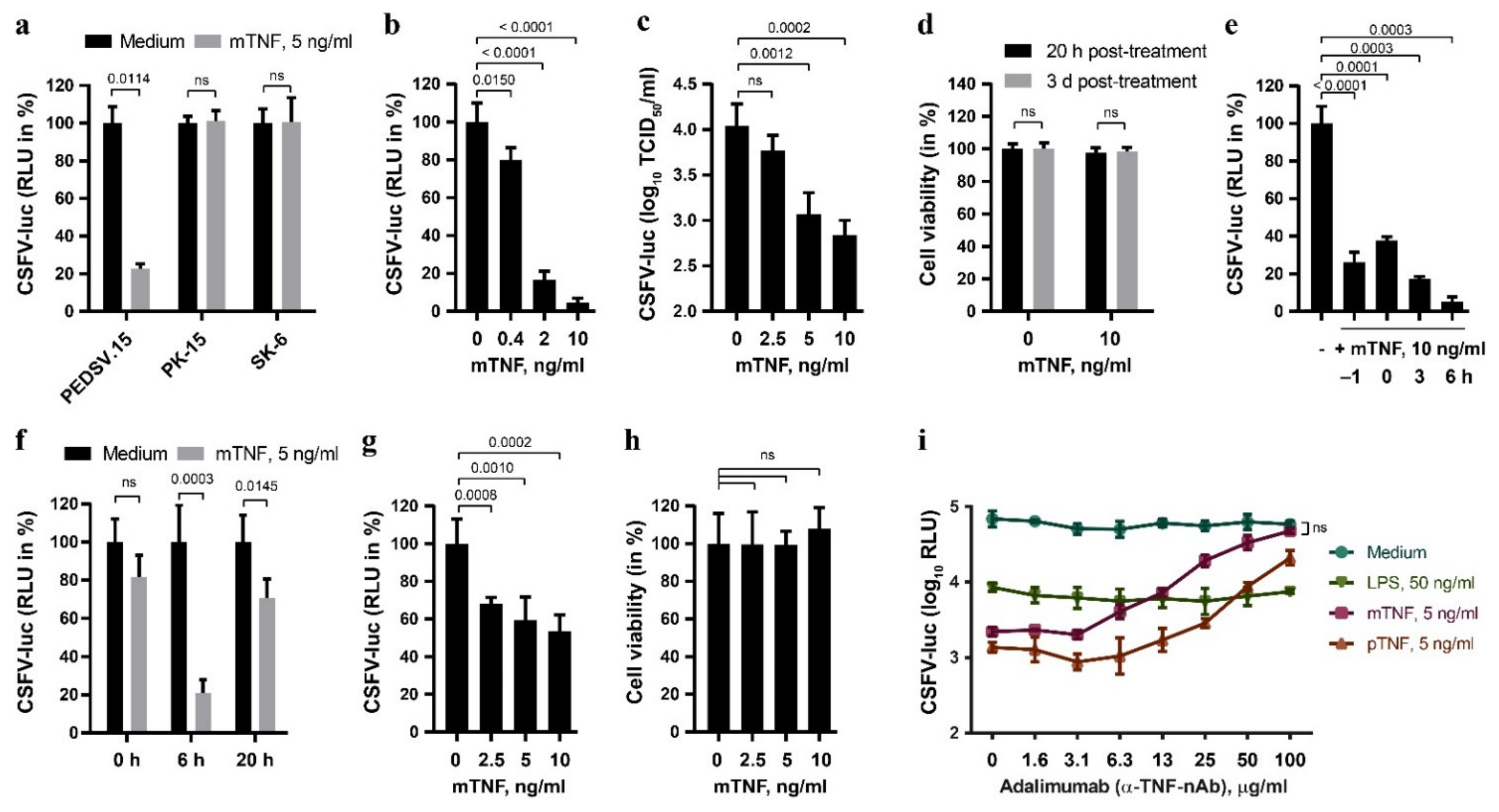
3.1. TNF Inhibits CSFV Replication in Porcine PEDSV.15 Cells and MDM, but Not in the PK-15 and SK-6 Cell Lines
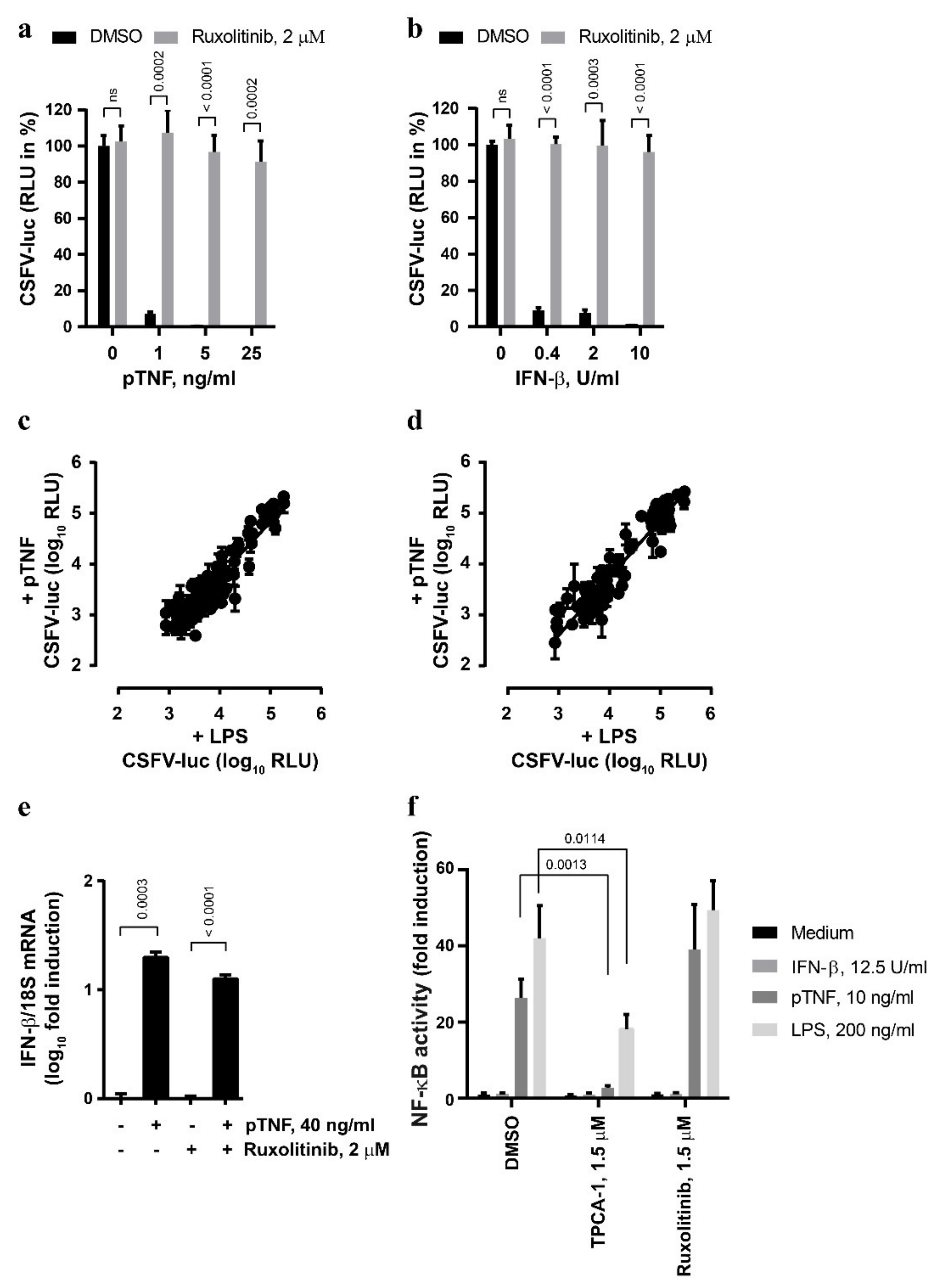
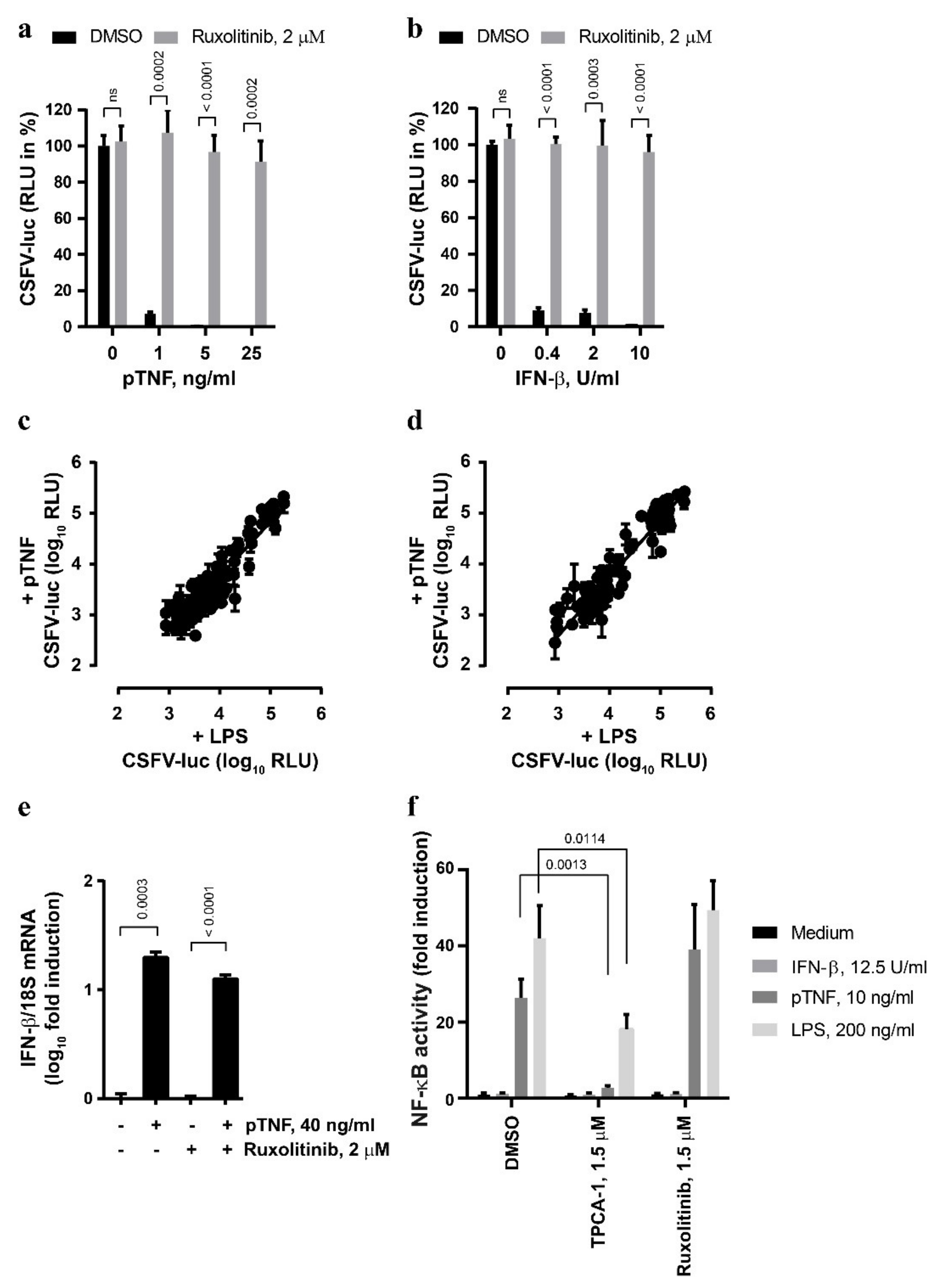
3.2. The Anti-CSFV Activity of TNF Involves JAK/STAT Signaling
3.3. The Anti-CSFV Activity of TNF Requires the Type I IFN Receptor, While IRF3 Is Dispensable
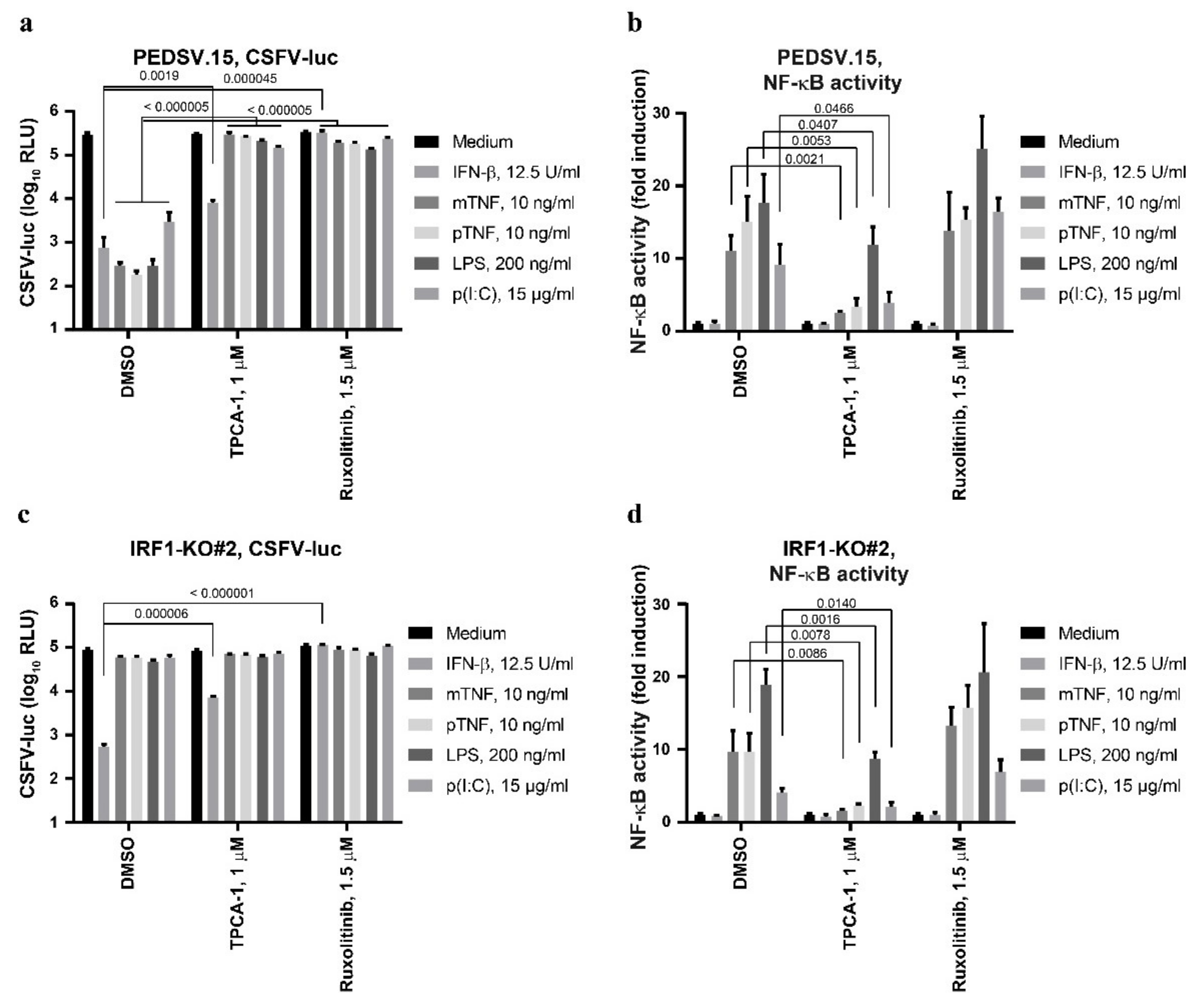
3.4. The Anti-CSFV Activities of LPS and TNF Are IRF1-Dependent
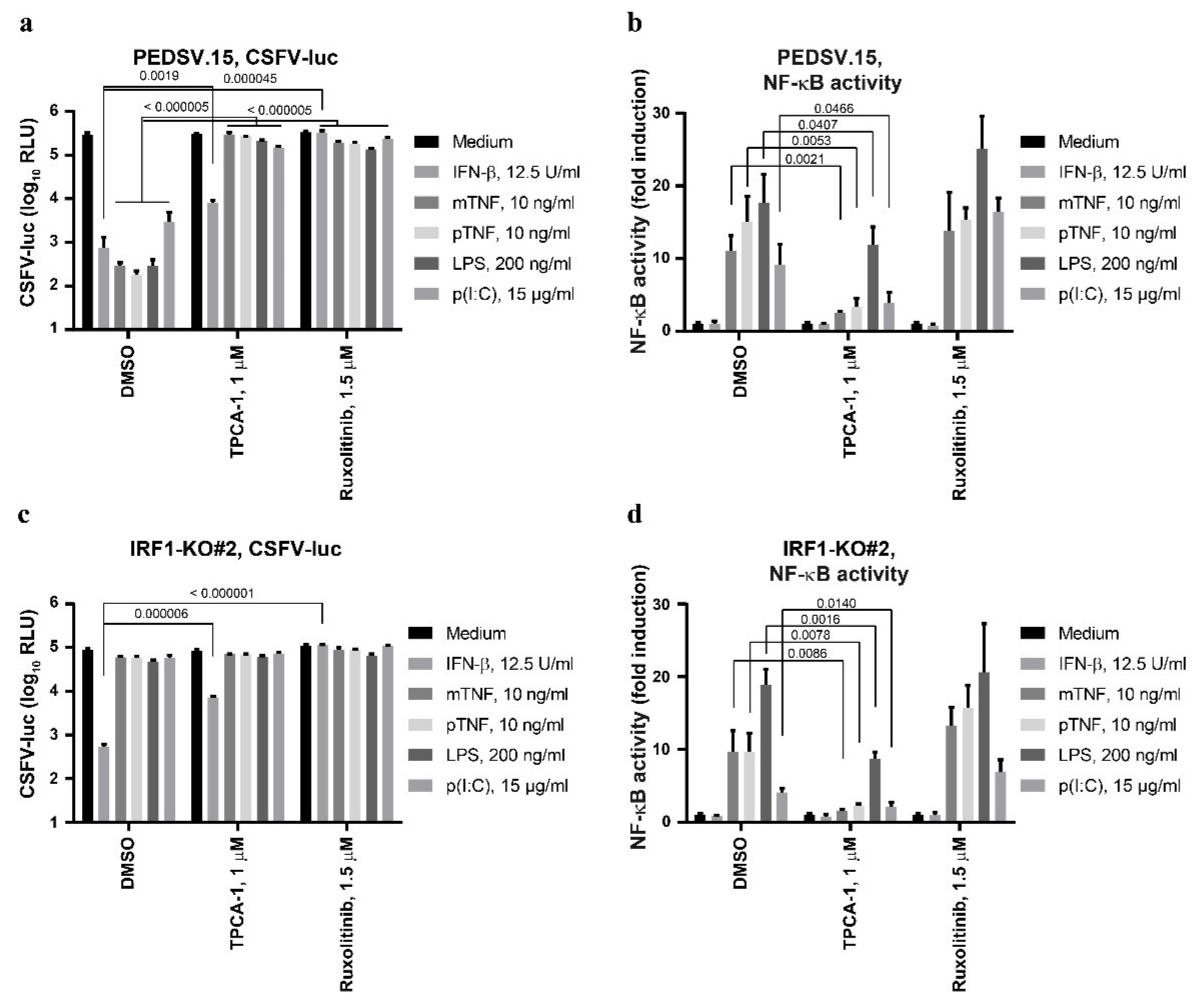
3.5. The Anti-CSFV Activity of TNF Is NF-κB-Dependent, but NF-κB Can Function Independently of IRF1
3.6. CSFV Infection Does Not Interfere with TNF- and LPS-Mediated IFN-β mRNA Induction in PEDSV.15 Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Takeuchi, O.; Akira, S. Innate immunity to virus infection. Immunol. Rev. 2009, 227, 75–86. [Google Scholar] [CrossRef]
- Mesev, E.V.; LeDesma, R.A.; Ploss, A. Decoding type I and III interferon signalling during viral infection. Nat. Microbiol. 2019, 4, 914–924. [Google Scholar] [CrossRef]
- Ganges, L.; Crooke, H.R.; Bohorquez, J.A.; Postel, A.; Sakoda, Y.; Becher, P.; Ruggli, N. Classical swine fever virus: The past, present and future. Virus Res. 2020, 289, 198151. [Google Scholar] [CrossRef] [PubMed]
- Tautz, N.; Tews, B.A.; Meyers, G. The Molecular Biology of Pestiviruses. Adv. Virus Res. 2015, 93, 47–160. [Google Scholar] [CrossRef] [PubMed]
- Cai, B.; Bai, Q.; Chi, X.; Goraya, M.U.; Wang, L.; Wang, S.; Chen, B.; Chen, J.L. Infection with Classical Swine Fever Virus Induces Expression of Type III Interferons and Activates Innate Immune Signaling. Front. Microbiol. 2017, 8, 2558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goraya, M.U.; Ziaghum, F.; Chen, S.; Raza, A.; Chen, Y.; Chi, X. Role of innate immunity in pathophysiology of classical swine fever virus infection. Microb. Pathog. 2018, 119, 248–254. [Google Scholar] [CrossRef]
- Summerfield, A.; Ruggli, N. Immune Responses Against Classical Swine Fever Virus: Between Ignorance and Lunacy. Front. Vet. Sci. 2015, 2, 10. [Google Scholar] [CrossRef] [Green Version]
- Bauhofer, O.; Summerfield, A.; Sakoda, Y.; Tratschin, J.D.; Hofmann, M.A.; Ruggli, N. Classical swine fever virus Npro interacts with interferon regulatory factor 3 and induces its proteasomal degradation. J. Virol. 2007, 81, 3087–3096. [Google Scholar] [CrossRef] [Green Version]
- Petro, T.M. IFN Regulatory Factor 3 in Health and Disease. J. Immunol. 2020, 205, 1981–1989. [Google Scholar] [CrossRef]
- Aggarwal, B.B. Signalling pathways of the TNF superfamily: A double-edged sword. Nat. Rev. Immunol. 2003, 3, 745–756. [Google Scholar] [CrossRef]
- Wajant, H.; Pfizenmaier, K.; Scheurich, P. Tumor necrosis factor signaling. Cell Death Differ. 2003, 10, 45–65. [Google Scholar] [CrossRef] [Green Version]
- Khatoon, E.; Barman, N.N.; Deka, M.; Hussain, M.D.I.; Borah, P.; Kumar, S. Cytokine responses in pigs after natural infection with classical swine fever virus. Acta Virol. 2019, 63, 60–69. [Google Scholar] [CrossRef]
- Von Rosen, T.; Lohse, L.; Nielsen, J.; Uttenthal, A. Classical swine fever virus infection modulates serum levels of INF-alpha, IL-8 and TNF-alpha in 6-month-old pigs. Res. Vet. Sci. 2013, 95, 1262–1267. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Sun, Y.; Meng, X.Y.; Li, L.F.; Li, Y.; Luo, Y.; Wang, W.; Yu, S.; Yin, C.; Li, S.; et al. Comprehensive evaluation of the host responses to infection with differentially virulent classical swine fever virus strains in pigs. Virus Res. 2018, 255, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Liu, X.; Xu, S.; Chen, D.; Zhou, L.; Ge, X.; Han, J.; Guo, X.; Yang, H. TNF-alpha induced by porcine reproductive and respiratory syndrome virus inhibits the replication of classical swine fever virus C-strain. Vet. Microbiol. 2019, 234, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Wang, J.; He, W.R.; Feng, S.; Li, Y.; Wang, X.; Liao, Y.; Qin, H.Y.; Li, L.F.; Dong, H.; et al. Thioredoxin 2 Is a Novel E2-Interacting Protein That Inhibits the Replication of Classical Swine Fever Virus. J. Virol. 2015, 89, 8510–8524. [Google Scholar] [CrossRef] [Green Version]
- Venkatesh, D.; Ernandez, T.; Rosetti, F.; Batal, I.; Cullere, X.; Luscinskas, F.W.; Zhang, Y.; Stavrakis, G.; Garcia-Cardena, G.; Horwitz, B.H.; et al. Endothelial TNF receptor 2 induces IRF1 transcription factor-dependent interferon-beta autocrine signaling to promote monocyte recruitment. Immunity 2013, 38, 1025–1037. [Google Scholar] [CrossRef] [Green Version]
- Yarilina, A.; Ivashkiv, L.B. Type I interferon: A new player in TNF signaling. Curr. Dir. Autoimmun. 2010, 11, 94–104. [Google Scholar] [CrossRef] [Green Version]
- Yarilina, A.; Park-Min, K.H.; Antoniv, T.; Hu, X.; Ivashkiv, L.B. TNF activates an IRF1-dependent autocrine loop leading to sustained expression of chemokines and STAT1-dependent type I interferon-response genes. Nat. Immunol. 2008, 9, 378–387. [Google Scholar] [CrossRef]
- Feng, H.; Zhang, Y.B.; Gui, J.F.; Lemon, S.M.; Yamane, D. Interferon regulatory factor 1 (IRF1) and anti-pathogen innate immune responses. PLoS Pathog. 2021, 17, e1009220. [Google Scholar] [CrossRef]
- Bonelli, M.; Dalwigk, K.; Platzer, A.; Olmos Calvo, I.; Hayer, S.; Niederreiter, B.; Holinka, J.; Sevelda, F.; Pap, T.; Steiner, G.; et al. IRF1 is critical for the TNF-driven interferon response in rheumatoid fibroblast-like synoviocytes: JAKinibs suppress the interferon response in RA-FLSs. Exp. Mol. Med. 2019, 51, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.Q.; Li, X.N.; Liang, J.J.; Cai, X.B.; Tao, Q.; Li, Y.X.; Qin, Q.; Xu, S.P.; Luo, T.R. IRF1 up-regulates isg15 gene expression in dsRNA stimulation or CSFV infection by targeting nucleotides -487 to -325 in the 5′ flanking region. Mol. Immunol. 2018, 94, 153–165. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Chang, H.; Yang, X.; Zhao, Y.; Chen, L.; Wang, X.; Liu, H.; Wang, C.; Zhao, J. Antiviral Activity of Porcine Interferon Regulatory Factor 1 against Swine Viruses in Cell Culture. Viruses 2015, 7, 5908–5918. [Google Scholar] [CrossRef] [PubMed]
- Cao, T.; Li, X.; Xu, Y.; Zhang, S.; Wang, Z.; Shan, Y.; Sun, J.; Fang, W.; Li, X. Npro of Classical Swine Fever Virus Suppresses Type III Interferon Production by Inhibiting IRF1 Expression and Its Nuclear Translocation. Viruses 2019, 11, 998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seebach, J.D.; Schneider, M.K.; Comrack, C.A.; LeGuern, A.; Kolb, S.A.; Knolle, P.A.; Germana, S.; DerSimonian, H.; LeGuern, C.; Sachs, D.H. Immortalized bone-marrow derived pig endothelial cells. Xenotransplantation 2001, 8, 48–61. [Google Scholar] [CrossRef] [PubMed]
- Sautter, C.A.; Auray, G.; Python, S.; Liniger, M.; Summerfield, A. Phenotypic and functional modulations of porcine macrophages by interferons and interleukin-4. Dev. Comp. Immunol. 2018, 84, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Ruggli, N.; Tratschin, J.D.; Mittelholzer, C.; Hofmann, M.A. Nucleotide sequence of classical swine fever virus strain Alfort/187 and transcription of infectious RNA from stably cloned full-length cDNA. J. Virol. 1996, 70, 3478–3487. [Google Scholar] [CrossRef] [Green Version]
- Suter, R.; Summerfield, A.; Thomann-Harwood, L.J.; McCullough, K.C.; Tratschin, J.D.; Ruggli, N. Immunogenic and replicative properties of classical swine fever virus replicon particles modified to induce IFN-alpha/beta and carry foreign genes. Vaccine 2011, 29, 1491–1503. [Google Scholar] [CrossRef]
- Mayer, D.; Thayer, T.M.; Hofmann, M.A.; Tratschin, J.D. Establishment and characterisation of two cDNA-derived strains of classical swine fever virus, one highly virulent and one avirulent. Virus Res. 2003, 98, 105–116. [Google Scholar] [CrossRef]
- Wang, M.; Liniger, M.; Munoz-Gonzalez, S.; Bohorquez, J.A.; Hinojosa, Y.; Gerber, M.; Lopez-Soria, S.; Rosell, R.; Ruggli, N.; Ganges, L. A Polyuridine Insertion in the 3′ Untranslated Region of Classical Swine Fever Virus Activates Immunity and Reduces Viral Virulence in Piglets. J. Virol. 2020, 94, e01214-19. [Google Scholar] [CrossRef] [Green Version]
- Greiser-Wilke, I.; Moennig, V.; Coulibaly, C.O.; Dahle, J.; Leder, L.; Liess, B. Identification of conserved epitopes on a hog cholera virus protein. Arch. Virol. 1990, 111, 213–225. [Google Scholar] [CrossRef]
- Mittelholzer, C.; Moser, C.; Tratschin, J.D.; Hofmann, M.A. Generation of cytopathogenic subgenomic RNA of classical swine fever virus in persistently infected porcine cell lines. Virus Res. 1997, 51, 125–137. [Google Scholar] [CrossRef]
- Von Niederhausern, B.; Bertoni, G.; Hertig, C.; Pfister, H.; Peterhans, E.; Pauli, U. Cloning and expression in mammalian cells of porcine tumor necrosis factor alpha: Examination of biological properties. Vet. Immunol. Immunopathol. 1993, 38, 57–74. [Google Scholar] [CrossRef]
- Alves, M.P.; Guzylack-Piriou, L.; Juillard, V.; Audonnet, J.C.; Doel, T.; Dawson, H.; Golde, W.T.; Gerber, H.; Peduto, N.; McCullough, K.C.; et al. Innate immune defenses induced by CpG do not promote vaccine-induced protection against foot-and-mouth disease virus in pigs. Clin. Vaccine Immunol. 2009, 16, 1151–1157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montague, T.G.; Cruz, J.M.; Gagnon, J.A.; Church, G.M.; Valen, E. CHOPCHOP: A CRISPR/Cas9 and TALEN web tool for genome editing. Nucleic Acids Res. 2014, 42, W401–W407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruggli, N.; Bird, B.H.; Liu, L.; Bauhofer, O.; Tratschin, J.D.; Hofmann, M.A. Npro of classical swine fever virus is an antagonist of double-stranded RNA-mediated apoptosis and IFN-alpha/beta induction. Virology 2005, 340, 265–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mease, P.J. Adalimumab: An anti-TNF agent for the treatment of psoriatic arthritis. Expert Opin. Biol. Ther. 2005, 5, 1491–1504. [Google Scholar] [CrossRef]
- Villarino, A.V.; Kanno, Y.; O’Shea, J.J. Mechanisms and consequences of Jak-STAT signaling in the immune system. Nat. Immunol. 2017, 18, 374–384. [Google Scholar] [CrossRef]
- Honda, K.; Takaoka, A.; Taniguchi, T. Type I interferon [corrected] gene induction by the interferon regulatory factor family of transcription factors. Immunity 2006, 25, 349–360. [Google Scholar] [CrossRef] [Green Version]
- Choi, C.; Hwang, K.K.; Chae, C. Classical swine fever virus induces tumor necrosis factor-alpha and lymphocyte apoptosis. Arch. Virol. 2004, 149, 875–889. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.C.; Yeh, W.C.; Ohashi, P.S. LPS/TLR4 signal transduction pathway. Cytokine 2008, 42, 145–151. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | Target Sequence (gRNA) 1 |
|---|---|
| IFNAR1-target1 | GATAATTGGATAAAGTTGCCTGG |
| IFNAR-target2 | CAGGAAACAGCACTTCTCCGTGG |
| IRF3-target1 | GCCGCAAGCCGTGCTTCCAAGGG |
| IRF3-target2 | TAGATCTTGTGTGGGTCGTGGGG |
| IRF1-target2 | GCTCAGCTGTGCGGGTGTACCGG |
| Oligonucleotide | Sequence (5′-3′) 1 |
|---|---|
| IFNAR1_CC9_1F | caccgGATAATTGGATAAAGTTGCC |
| IFNAR1_CC9_1R | aaacGGCAACTTTATCCAATTATCc |
| IFNAR1_CC9_2F | caccgCAGGAAACAGCACTTCTCCG |
| IFNAR1_CC9_2R | aaacCGGAGAAGTGCTGTTTCCTGc |
| IRF3_CC9_1F | caccgGCCGCAAGCCGTGCTTCCAA |
| IRF3_CC9_1R | aaacTTGGAAGCACGGCTTGCGGCc |
| IRF3_CC9_2F | caccgTAGATCTTGTGTGGGTCGTG |
| IRF3_CC9_2R | aaacCACGACCCACACAAGATCTAc |
| IRF1_CC9_2F | caccgGCTCAGCTGTGCGGGTGTAC |
| IRF1_CC9_2R | aaacGTACACCCGCACAGCTGAGCc |
| Oligonucleotide | Sequence (5′-3′) (gRNA) |
|---|---|
| IFNAR1-gF | TTGGTATGTGTGCATTGAAAGA |
| IFNAR1-gR | ATGAGCTTGGGAAGTGAACTGT |
| IRF3-gF | CTGATATCTCAGCTGAACCAGG |
| IRF3-gR2 | GGTATCAGAGGTACTGTATC |
| IRF1-gF | TGTGTATAGGCAGGCATACGAG |
| IRF1-gR | ACTGAGGCTTGCTGGATGTATT |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liniger, M.; Gerber, M.; Renzullo, S.; García-Nicolás, O.; Ruggli, N. TNF-Mediated Inhibition of Classical Swine Fever Virus Replication Is IRF1-, NF-κB- and JAK/STAT Signaling-Dependent. Viruses 2021, 13, 2017. https://doi.org/10.3390/v13102017
Liniger M, Gerber M, Renzullo S, García-Nicolás O, Ruggli N. TNF-Mediated Inhibition of Classical Swine Fever Virus Replication Is IRF1-, NF-κB- and JAK/STAT Signaling-Dependent. Viruses. 2021; 13(10):2017. https://doi.org/10.3390/v13102017
Chicago/Turabian StyleLiniger, Matthias, Markus Gerber, Sandra Renzullo, Obdulio García-Nicolás, and Nicolas Ruggli. 2021. "TNF-Mediated Inhibition of Classical Swine Fever Virus Replication Is IRF1-, NF-κB- and JAK/STAT Signaling-Dependent" Viruses 13, no. 10: 2017. https://doi.org/10.3390/v13102017
APA StyleLiniger, M., Gerber, M., Renzullo, S., García-Nicolás, O., & Ruggli, N. (2021). TNF-Mediated Inhibition of Classical Swine Fever Virus Replication Is IRF1-, NF-κB- and JAK/STAT Signaling-Dependent. Viruses, 13(10), 2017. https://doi.org/10.3390/v13102017