Evolutionary Study of the Crassphage Virus at Gene Level

 , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Sequences Selection, Retrieval, and Data Preparation
2.2. Phylogenetic Tree Reconstruction and Comparison
2.3. Quantifying Sequence Conservation
2.4. Function Prediction and Distant Similarity Searches
2.5. Data Visualization and Availability
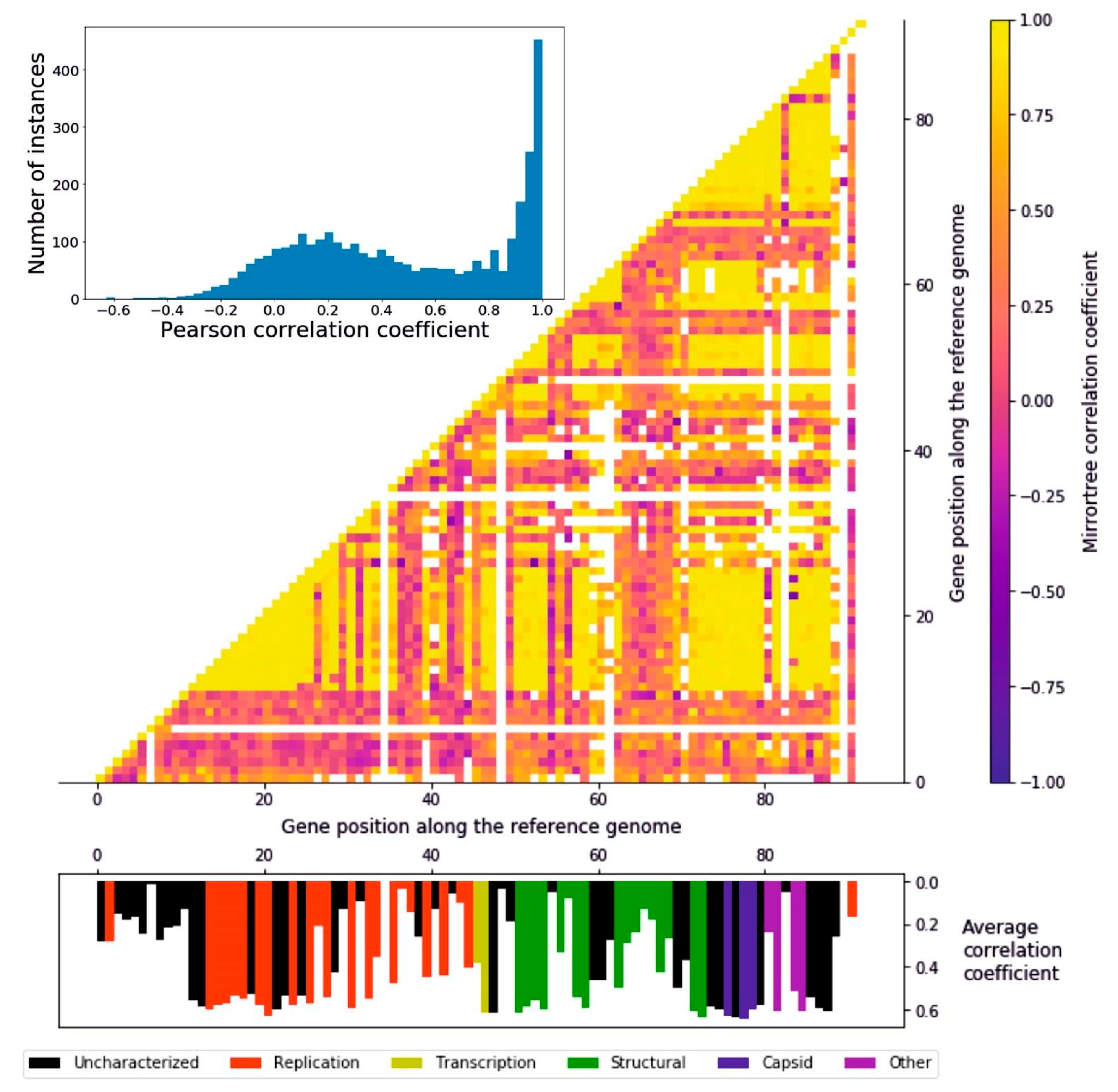
3. Results
3.1. Protein Identification and Clustering
3.2. Protein Conservation
3.3. Tree Comparison: Consistency of Evolutionary Signal
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Handelsman, J.; Rondon, M.R.; Brady, S.F.; Clardy, J.; Goodman, R.M. Molecular biological access to the chemistry of unknown soil microbes: A new frontier for natural products. Chem. Biol. 1998, 5, R245–R249. [Google Scholar] [CrossRef]
- Koonin, E.V. Environmental microbiology and metagenomics: The Brave New World is here, what’s next? Environ. Microbiol. 2018, 20, 4210–4212. [Google Scholar] [CrossRef]
- Koonin, E.V.; Dolja, V.V. Metaviromics: A tectonic shift in understanding virus evolution. Virus Res. 2018, 246, A1–A3. [Google Scholar] [CrossRef]
- Al-Shayeb, B.; Sachdeva, R.; Chen, L.-X.; Ward, F.; Munk, P.; Devoto, A.; Castelle, C.J.; Olm, M.R.; Bouma-Gregson, K.; Amano, Y.; et al. Clades of huge phages from across Earth’s ecosystems. Nature 2020, 578, 425–431. [Google Scholar] [CrossRef] [PubMed]
- Schulz, F.; Roux, S.; Paez-Espino, D.; Jungbluth, S.; Walsh, D.A.; Denef, V.J.; McMahon, K.D.; Konstantinidis, K.T.; Eloe-Fadrosh, E.A.; Kyrpides, N.C.; et al. Giant virus diversity and host interactions through global metagenomics. Nature 2020, 578, 432–436. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Gao, M. Jumbo Bacteriophages: An Overview. Front. Microbiol. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Devoto, A.E.; Santini, J.M.; Olm, M.R.; Anantharaman, K.; Munk, P.; Tung, J.; Archie, E.A.; Turnbaugh, P.J.; Seed, K.D.; Blekhman, R.; et al. Megaphages infect Prevotella and variants are widespread in gut microbiomes. Nat. Microbiol. 2019, 4, 693–700. [Google Scholar] [CrossRef]
- Simmonds, P.; Adams, M.J.; Benkő, M.; Breitbart, M.; Brister, J.R.; Carstens, E.B.; Davison, A.J.; Delwart, E.; Gorbalenya, A.E.; Harrach, B.; et al. Virus taxonomy in the age of metagenomics. Nat. Rev. Microbiol. 2017, 15, 161–168. [Google Scholar] [CrossRef]
- Dutilh, B.E.; Cassman, N.; McNair, K.; Sanchez, S.E.; Silva, G.G.Z.; Boling, L.; Barr, J.J.; Speth, D.R.; Seguritan, V.; Aziz, R.K.; et al. A highly abundant bacteriophage discovered in the unknown sequences of human faecal metagenomes. Nat. Commun. 2014, 5, 4498. [Google Scholar] [CrossRef]
- Koonin, E.V.; Yutin, N. The crAss-like Phage Group: How Metagenomics Reshaped the Human Virome. Trends Microbiol. 2020, 28, 349–359. [Google Scholar] [CrossRef]
- Edwards, R.A.; Vega, A.A.; Norman, H.M.; Ohaeri, M.; Levi, K.; Dinsdale, E.A.; Cinek, O.; Aziz, R.K.; McNair, K.; Barr, J.J.; et al. Global phylogeography and ancient evolution of the widespread human gut virus crAssphage. Nat. Microbiol. 2019, 4, 1727–1736. [Google Scholar] [CrossRef] [PubMed]
- Dutilh, B.E.; Schmieder, R.; Nulton, J.; Felts, B.; Salamon, P.; Edwards, R.A.; Mokili, J.L. Reference-independent comparative metagenomics using cross-assembly: crAss. Bioinformatics 2012, 28, 3225–3231. [Google Scholar] [CrossRef] [PubMed]
- Yutin, N.; Makarova, K.S.; Gussow, A.B.; Krupovic, M.; Segall, A.; Edwards, R.A.; Koonin, E.V. Discovery of an expansive bacteriophage family that includes the most abundant viruses from the human gut. Nat. Microbiol. 2018, 3, 38–46. [Google Scholar] [CrossRef]
- Guerin, E.; Shkoporov, A.; Stockdale, S.R.; Clooney, A.G.; Ryan, F.J.; Sutton, T.D.S.; Draper, L.A.; Gonzalez-Tortuero, E.; Ross, R.P.; Hill, C. Biology and Taxonomy of crAss-like Bacteriophages, the Most Abundant Virus in the Human Gut. Cell Host Microbe 2018, 24, 653–664.e6. [Google Scholar] [CrossRef] [PubMed]
- Balcazar, J.L. Bacteriophages as Vehicles for Antibiotic Resistance Genes in the Environment. PLoS Pathog. 2014, 10, e1004219. [Google Scholar] [CrossRef]
- Penadés, J.R.; Chen, J.; Quiles-Puchalt, N.; Carpena, N.; Novick, R.P. Bacteriophage-mediated spread of bacterial virulence genes. Curr. Opin. Microbiol. 2015, 23, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Carding, S.R.; Davis, N.; Hoyles, L. Review article: The human intestinal virome in health and disease. Aliment. Pharmacol. Ther. 2017, 46, 800–815. [Google Scholar] [CrossRef]
- de la Cuesta-Zuluaga, J.; Corrales-Agudelo, V.; Velásquez-Mejía, E.P.; Carmona, J.A.; Abad, J.M.; Escobar, J.S. Gut microbiota is associated with obesity and cardiometabolic disease in a population in the midst of Westernization. Sci. Rep. 2018, 8, 11356. [Google Scholar] [CrossRef]
- Kashyap, P.C.; Quigley, E.M. Therapeutic implications of the gastrointestinal microbiome. Curr. Opin. Pharmacol. 2018, 38, 90–96. [Google Scholar] [CrossRef]
- Akhter, S.; Aziz, R.K.; Edwards, R.A. PhiSpy: A novel algorithm for finding prophages in bacterial genomes that combines similarity- and composition-based strategies. Nucleic Acids Res. 2012, 40, e126. [Google Scholar] [CrossRef]
- Juan, D.; Pazos, F.; Valencia, A. High-confidence prediction of global interactomes based on genome-wide coevolutionary networks. Proc. Natl. Acad. Sci. USA 2008, 105, 934–939. [Google Scholar] [CrossRef] [PubMed]
- Burton, R.S.; Rawson, P.D.; Edmands, S. Genetic architecture of physiological phenotypes: Empirical evidence for coadapted gene complexes. Am. Zool. 1999, 39, 451–462. [Google Scholar] [CrossRef][Green Version]
- Pazos, F.; Valencia, A. Protein co-evolution, co-adaptation and interactions. EMBO J. 2008, 27, 2648–2655. [Google Scholar] [CrossRef] [PubMed]
- Van Rossum, G.; Drake, F.L., Jr. Python Reference Manual; Centrum voor Wiskunde en Informatica: Amsterdam, The Netherlands, 1995. [Google Scholar]
- Cock, P.J.A.; Antao, T.; Chang, J.T.; Chapman, B.A.; Cox, C.J.; Dalke, A.; Friedberg, I.; Hamelryck, T.; Kauff, F.; Wilczynski, B.; et al. Biopython: Freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics 2009, 25, 1422–1423. [Google Scholar] [CrossRef]
- Hyatt, D.; Chen, G.-L.; LoCascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Hoang, D.T.; Chernomor, O.; von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the Ultrafast Bootstrap Approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef] [PubMed]
- Larsson, A. AliView: A fast and lightweight alignment viewer and editor for large datasets. Bioinformatics 2014, 30, 3276–3278. [Google Scholar] [CrossRef] [PubMed]
- Pazos, F.; Valencia, A. Similarity of phylogenetic trees as indicator of protein–protein interaction. Protein Eng. Des. Sel. 2001, 14, 609–614. [Google Scholar] [CrossRef] [PubMed]
- Jones, E.; Oliphant, T.; Peterson, P. SciPy: Open Source Scientific Tools for Python. 2001. Available online: https://www.scienceopen.com/document?vid=ab12905a-8a5b-43d8-a2bb-defc771410b9 (accessed on 14 September 2020).
- Shannon, C.E. A Mathematical Theory of Communication. Bell Syst. Tech. J. 1948, 27, 623–656. [Google Scholar] [CrossRef]
- Grazziotin, A.L.; Koonin, E.V.; Kristensen, D.M. Prokaryotic Virus Orthologous Groups (pVOGs): A resource for comparative genomics and protein family annotation. Nucleic Acids Res. 2017, 45, D491–D498. [Google Scholar] [CrossRef]
- Lavezzo, E.; Falda, M.; Fontana, P.; Bianco, L.; Toppo, S. Enhancing protein function prediction with taxonomic constraints—The Argot2.5 web server. Methods 2016, 93, 15–23. [Google Scholar] [CrossRef]
- Yang, J.; Zhang, Y. I-TASSER server: New development for protein structure and function predictions. Nucleic Acids Res. 2015, 43, W174–W181. [Google Scholar] [CrossRef]
- Zhou, N.; Jiang, Y.; Bergquist, T.R.; Lee, A.J.; Kacsoh, B.Z.; Crocker, A.W.; Lewis, K.A.; Georghiou, G.; Nguyen, H.N.; Hamid, M.N.; et al. The CAFA challenge reports improved protein function prediction and new functional annotations for hundreds of genes through experimental screens. Genome Biol. 2019, 20, 244. [Google Scholar] [CrossRef] [PubMed]
- Kryshtafovych, A.; Schwede, T.; Topf, M.; Fidelis, K.; Moult, J. Critical assessment of methods of protein structure prediction (CASP)—Round XIII. Proteins Struct. Funct. Bioinform. 2019, 87, 1011–1020. [Google Scholar] [CrossRef]
- Hunter, J.D. Matplotlib: A 2D Graphics Environment. Comput. Sci. Eng. 2007, 9, 90–95. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL): An online tool for phylogenetic tree display and annotation. Bioinformatics 2007, 23, 127–128. [Google Scholar] [CrossRef]
- Dwidar, M.; Yokobayashi, Y. Riboswitch Signal Amplification by Controlling Plasmid Copy Number. ACS Synth. Biol. 2019, 8, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Reyes, A.; Haynes, M.; Hanson, N.; Angly, F.E.; Heath, A.C.; Rohwer, F.; Gordon, J.I. Viruses in the faecal microbiota of monozygotic twins and their mothers. Nature 2010, 466, 334–338. [Google Scholar] [CrossRef] [PubMed]
- Hendrix, R.W.; Lawrence, J.G.; Hatfull, G.F.; Casjens, S. The origins and ongoing evolution of viruses. Trends Microbiol. 2000, 8, 504–508. [Google Scholar] [CrossRef]
- Adriaenssens, E.M.; Cowan, D.A. Using Signature Genes as Tools To Assess Environmental Viral Ecology and Diversity. Appl. Environ. Microbiol. 2014, 80, 4470–4480. [Google Scholar] [CrossRef]
- Krupovic, M.; Koonin, E.V. Multiple origins of viral capsid proteins from cellular ancestors. Proc. Natl. Acad. Sci. USA 2017, 114, E2401–E2410. [Google Scholar] [CrossRef]
- Szöllősi, G.J.; Tannier, E.; Daubin, V.; Boussau, B. The Inference of Gene Trees with Species Trees. Syst. Biol. 2015, 64, e42–e62. [Google Scholar] [CrossRef]
- Nichols, R. Gene trees and species trees are not the same. Trends Ecol. Evol. 2001, 16, 358–364. [Google Scholar] [CrossRef]
- Pamilo, P.; Nei, M. Relationships between gene trees and species trees. Mol. Biol. Evol. 1988. [Google Scholar] [CrossRef]
- Shkoporov, A.N.; Khokhlova, E.V.; Fitzgerald, C.B.; Stockdale, S.R.; Draper, L.A.; Ross, R.P.; Hill, C. ΦCrAss001 represents the most abundant bacteriophage family in the human gut and infects Bacteroides intestinalis. Nat. Commun. 2018, 9, 4781. [Google Scholar] [CrossRef]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rossi, A.; Treu, L.; Toppo, S.; Zschach, H.; Campanaro, S.; Dutilh, B.E. Evolutionary Study of the Crassphage Virus at Gene Level. Viruses 2020, 12, 1035. https://doi.org/10.3390/v12091035
Rossi A, Treu L, Toppo S, Zschach H, Campanaro S, Dutilh BE. Evolutionary Study of the Crassphage Virus at Gene Level. Viruses. 2020; 12(9):1035. https://doi.org/10.3390/v12091035
Chicago/Turabian StyleRossi, Alessandro, Laura Treu, Stefano Toppo, Henrike Zschach, Stefano Campanaro, and Bas E. Dutilh. 2020. "Evolutionary Study of the Crassphage Virus at Gene Level" Viruses 12, no. 9: 1035. https://doi.org/10.3390/v12091035
APA StyleRossi, A., Treu, L., Toppo, S., Zschach, H., Campanaro, S., & Dutilh, B. E. (2020). Evolutionary Study of the Crassphage Virus at Gene Level. Viruses, 12(9), 1035. https://doi.org/10.3390/v12091035