Genetic Diversity Analysis of Coxsackievirus A8 Circulating in China and Worldwide Reveals a Highly Divergent Genotype

 ,
,  , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Virus Isolation
2.2. CV-A8 Whole-Genome Sequencing
2.3. Dataset Construction
2.4. Phylogenetic and Evolutionary Analyses of CV-A8 VP1 Sequences
2.5. Whole-Genome and Recombination Analyses
2.6. Nucleotide Sequence Accession Numbers
2.7. Ethics Statement
3. Results
3.1. Operational Mechanism of HFMD Surveillance Network and CV-A8 Dataset Overview
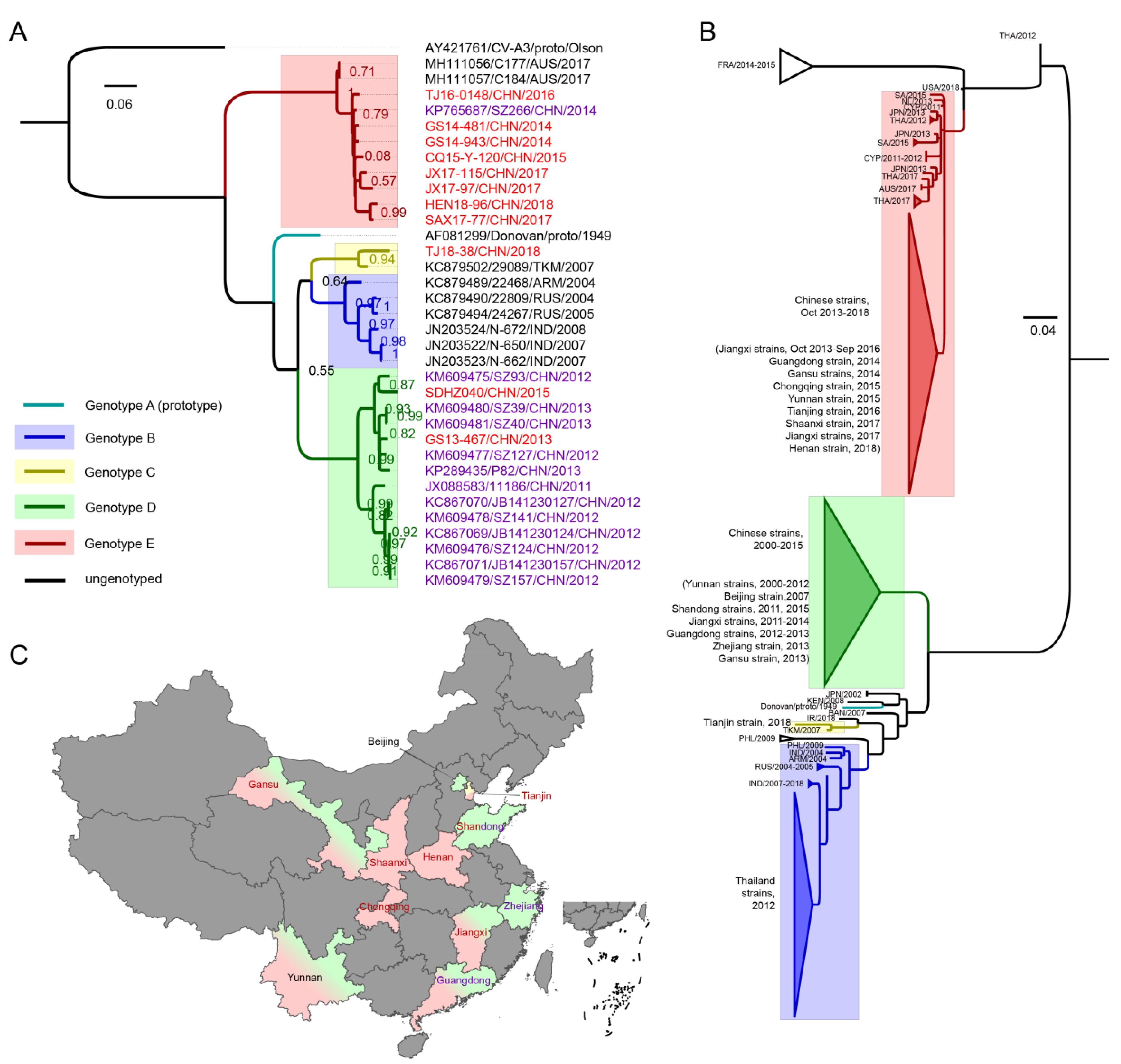
3.2. VP1 Phylogenetic Analysis and Genotyping of CV-A8
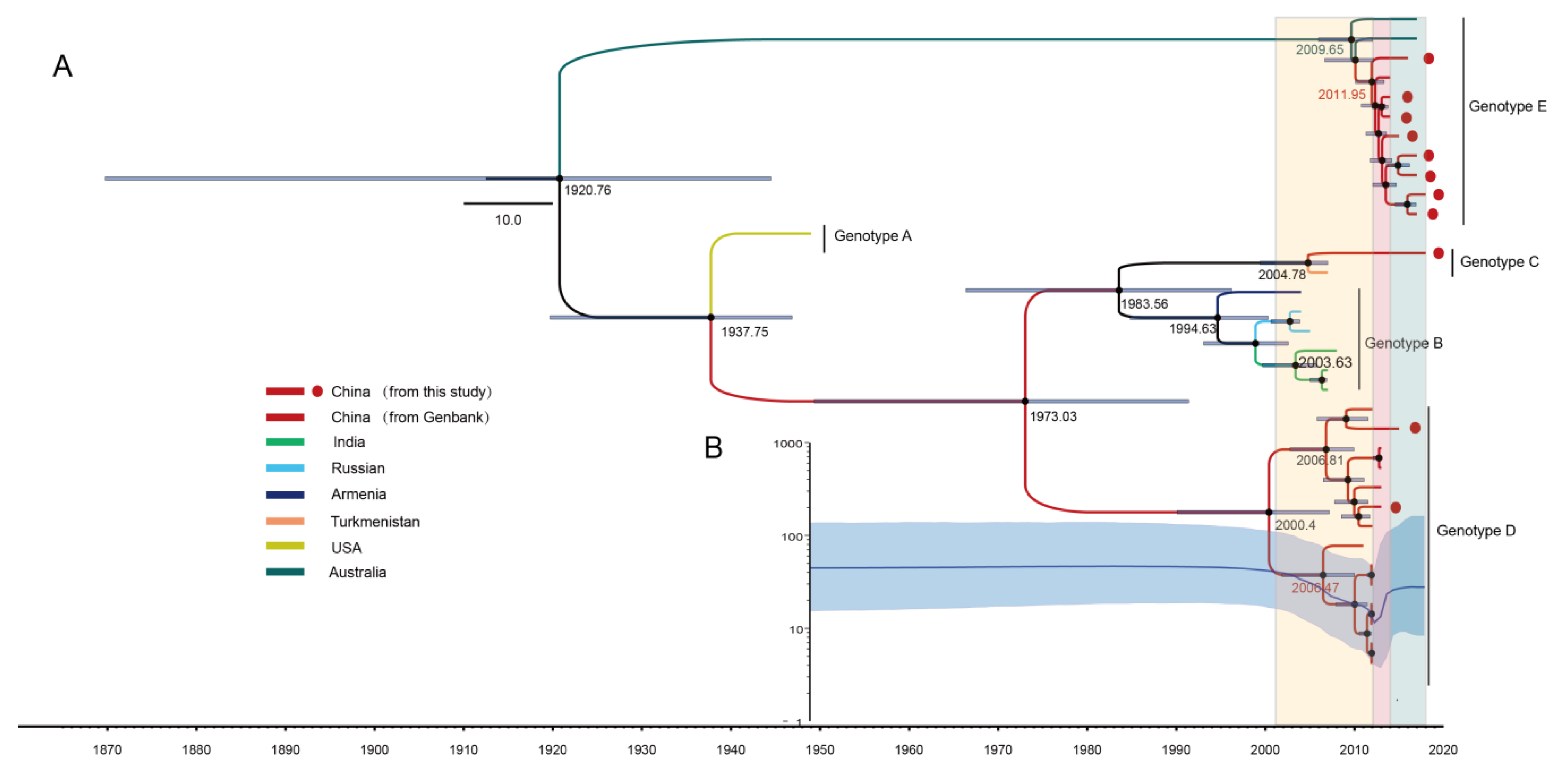
3.3. The Evolutionary Dynamics of CV-A8 Genotypes
3.4. Amino Acid Characterization of the CV-A8 VP1 Capsid
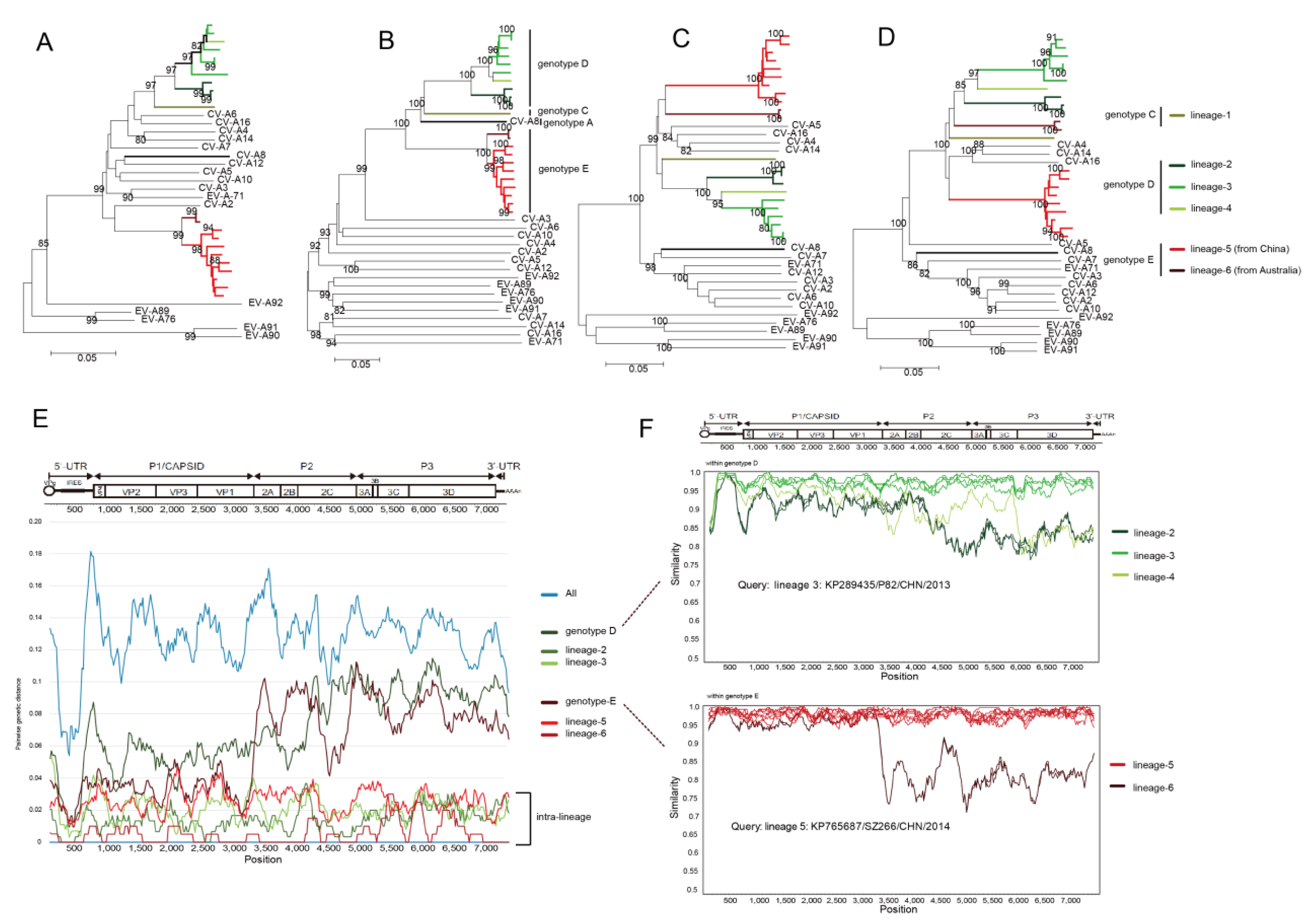
3.5. Whole-Genomic Diversity of CV-A8
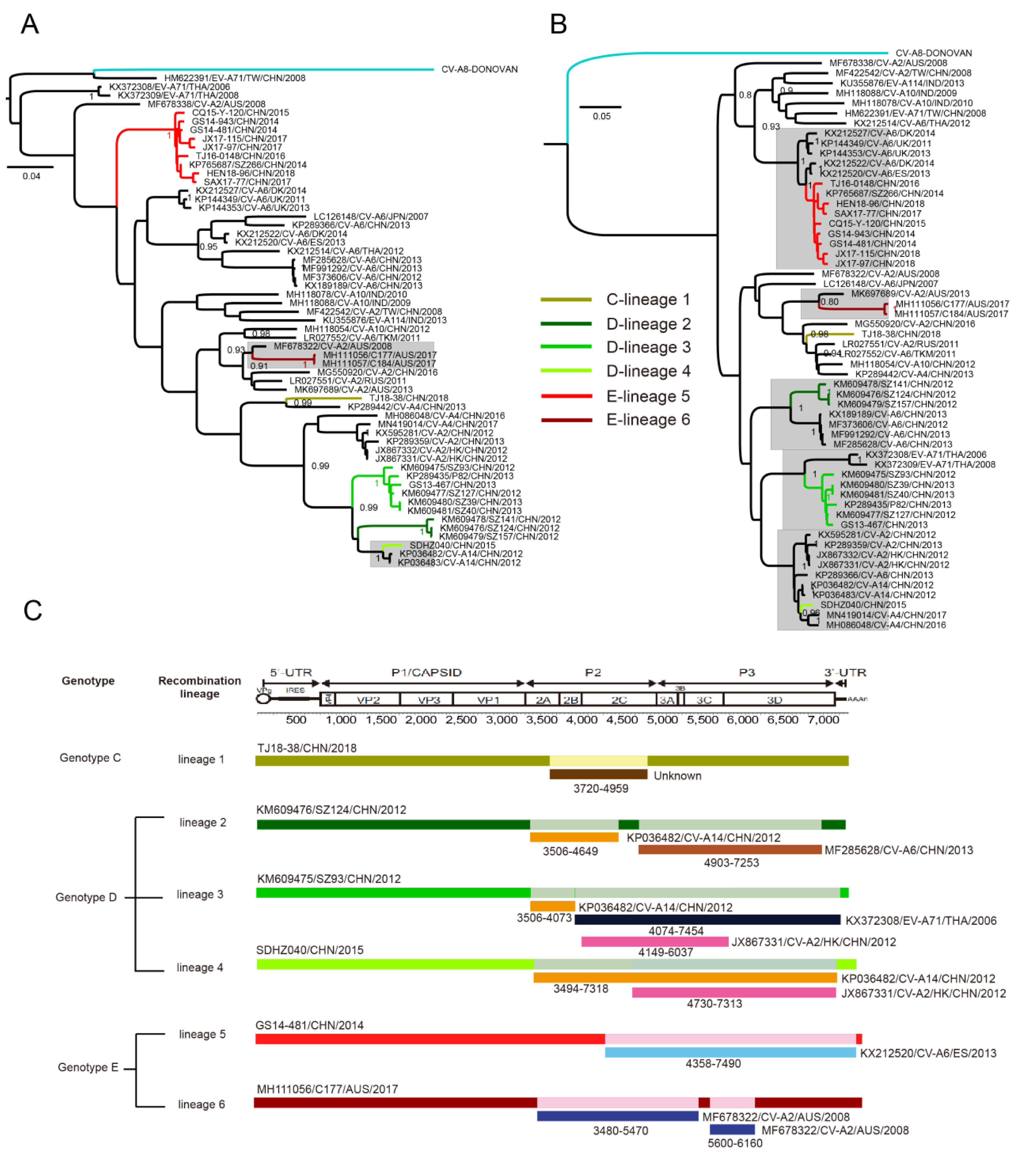
3.6. Multiple Recombination Events Were Detected among CV-A8 Variants
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Oberste, M.S.; Penaranda, S.; Maher, K.; Pallansch, M.A. Complete genome sequences of all members of the species human enterovirus a. J. Gen. Virol. 2004, 85, 1597–1607. [Google Scholar] [CrossRef] [PubMed]
- Zell, R.; Delwart, E.; Gorbalenya, A.E.; Hovi, T.; King, A.M.Q.; Knowles, N.J.; Lindberg, A.M.; Pallansch, M.A.; Palmenberg, A.C.; Reuter, G.; et al. ICTV Virus Taxonomy Profile: Picornaviridae. J. Gen. Virol. 2017, 98, 2421–2422. [Google Scholar] [CrossRef]
- Brown, B.A.; Oberste, M.S.; Alexander, J.P., Jr.; Kennett, M.L.; Pallansch, M.A. Molecular epidemiology and evolution of enterovirus 71 strains isolated from 1970 to 1998. J. Virol. 1999, 73, 9969–9975. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, D.; Yan, D.; Zhu, S.; Liu, J.; Wang, H.; Zhao, S.; Yu, D.; Nan, L.; An, J.; et al. Molecular evidence of persistent epidemic and evolution of subgenotype b1 coxsackievirus a16-associated hand, foot, and mouth disease in China. J. Clin. Microbiol. 2010, 48, 619–622. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Zhang, Y.; Ji, T.; Gu, X.; Yang, Q.; Zhu, S.; Xu, W.; Xu, Y.; Shi, Y.; Huang, X.; et al. Persistent circulation of coxsackievirus a6 of genotype d3 in mainland of China between 2008 and 2015. Sci. Rep. 2017, 7, 5491. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Gu, X.; Zhang, Y.; Wei, H.; Li, Q.; Fan, H.; Xu, Y.; Li, J.; Tan, Z.; Song, Y.; et al. Persistent circulation of genotype d coxsackievirus a2 in mainland of China since 2008. PLoS ONE 2018, 13, e0204359. [Google Scholar] [CrossRef] [PubMed]
- Oberste, M.S.; Maher, K.; Kilpatrick, D.R.; Pallansch, M.A. Molecular evolution of the human enteroviruses: Correlation of serotype with vp1 sequence and application to picornavirus classification. J. Virol. 1999, 73, 1941–1948. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Li, J.; Yao, M.X.; Zhang, Y.W.; Hu, T.; Carr, M.J.; Duchene, S.; Zhang, X.C.; Zhang, Z.J.; Zhou, H.; et al. Genome analysis of coxsackievirus a4 isolates from hand, foot, and mouth disease cases in Shandong, China. Front. Microbiol. 2019, 10, 1001. [Google Scholar] [CrossRef]
- Patil, P.R.; Chitambar, S.D.; Gopalkrishna, V. Molecular surveillance of non-polio enterovirus infections in patients with acute gastroenteritis in western India: 2004–2009. J. Med. Virol. 2015, 87, 154–161. [Google Scholar] [CrossRef]
- Bingjun, T.; Yoshida, H.; Yan, W.; Lin, L.; Tsuji, T.; Shimizu, H.; Miyamura, T. Molecular typing and epidemiology of non-polio enteroviruses isolated from yunnan province, the people’s republic of China. J. Med. Virol. 2008, 80, 670–679. [Google Scholar] [CrossRef]
- Chen, L.; Yang, H.; Wang, C.; Yao, X.J.; Zhang, H.L.; Zhang, R.L.; He, Y.Q. Genomic characteristics of coxsackievirus a8 strains associated with hand, foot, and mouth disease and herpangina. Arch. Virol. 2016, 161, 213–217. [Google Scholar] [CrossRef]
- Rao, C.D.; Yergolkar, P.; Shankarappa, K.S. Antigenic diversity of enteroviruses associated with nonpolio acute flaccid paralysis, India, 2007–2009. Emerg. Infect. Dis. 2012, 18, 1833–1840. [Google Scholar] [CrossRef] [PubMed]
- Opanda, S.M.; Wamunyokoli, F.; Khamadi, S.; Coldren, R.; Bulimo, W.D. Genotyping of enteroviruses isolated in kenya from pediatric patients using partial vp1 region. SpringerPlus 2016, 5, 158. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Benschop, K.S.M.; van der Avoort, H.G.; Jusic, E.; Vennema, H.; van Binnendijk, R.; Duizer, E. Polio and measles down the drain: Environmental enterovirus surveillance in the netherlands, 2005 to 2015. Appl. Environ. Microbiol. 2017, 83, e00558-17. [Google Scholar] [CrossRef] [PubMed]
- Damsky, W.E.; Leventhal, J.S.; Khalil, D.; Vesely, M.D.; Craiglow, B.G.; Milstone, L.M.; Choate, K.A. Recurrent coxsackievirus infection in a patient with lamellar ichthyosis. Pediatr. Dermatol. 2016, 33, e140–e142. [Google Scholar] [CrossRef]
- Apostol, L.N.; Imagawa, T.; Suzuki, A.; Masago, Y.; Lupisan, S.; Olveda, R.; Saito, M.; Omura, T.; Oshitani, H. Genetic diversity and molecular characterization of enteroviruses from sewage-polluted urban and rural rivers in the philippines. Virus Genes 2012, 45, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Puenpa, J.; Mauleekoonphairoj, J.; Linsuwanon, P.; Suwannakarn, K.; Chieochansin, T.; Korkong, S.; Theamboonlers, A.; Poovorawan, Y. Prevalence and characterization of enterovirus infections among pediatric patients with hand foot mouth disease, herpangina and influenza like illness in Thailand, 2012. PLoS ONE 2014, 9, e98888. [Google Scholar] [CrossRef]
- He, Y.Q.; Chen, L.; Xu, W.B.; Yang, H.; Wang, H.Z.; Zong, W.P.; Xian, H.X.; Chen, H.L.; Yao, X.J.; Hu, Z.L.; et al. Emergence, circulation, and spatiotemporal phylogenetic analysis of coxsackievirus a6- and coxsackievirus a10-associated hand, foot, and mouth disease infections from 2008 to 2012 in Shenzhen, China. J. Clin. Microbiol. 2013, 51, 3560–3566. [Google Scholar] [CrossRef]
- Xu, W.; Zhang, Y. Isolation and characterization of vaccine-derived polioviruses, relevance for the global polio eradication initiative. Methods Mol. Biol. 2016, 1387, 213–226. [Google Scholar]
- Kumar, S.; Stecher, G.; Tamura, K. Mega7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Yang, C.F.; Naguib, T.; Yang, S.J.; Nasr, E.; Jorba, J.; Ahmed, N.; Campagnoli, R.; van der Avoort, H.; Shimizu, H.; Yoneyama, T.; et al. Circulation of endemic type 2 vaccine-derived poliovirus in egypt from 1983 to 1993. J. Virol. 2003, 77, 8366–8377. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. Raxml version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. Jmodeltest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef]
- Suchard, M.A.; Lemey, P.; Baele, G.; Ayres, D.L.; Drummond, A.J.; Rambaut, A. Bayesian phylogenetic and phylodynamic data integration using beast 1.10. Virus Evol. 2018, 4, vey016. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. Swiss-model: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef]
- Rozas, J.; Ferrer-Mata, A.; Sanchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sanchez-Gracia, A. Dnasp 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Salminen, M.O.; Carr, J.K.; Burke, D.S.; McCutchan, F.E. Identification of breakpoints in intergenotypic recombinants of HIV type 1 by bootscanning. Aids Res. Hum. Retrovir. 1995, 11, 1423–1425. [Google Scholar] [CrossRef]
- Martin, D.P.; Murrell, B.; Khoosal, A.; Muhire, B. Detecting and analyzing genetic recombination using rdp4. Methods Mol. Biol. 2017, 1525, 433–460. [Google Scholar]
- Zhao, Y.; Zhou, D.; Ni, T.; Karia, D.; Kotecha, A.; Wang, X.; Rao, Z.; Jones, E.Y.; Fry, E.E.; Ren, J.; et al. Hand-foot-and-mouth disease virus receptor kremen1 binds the canyon of coxsackie virus a10. Nat. Commun. 2020, 11, 38. [Google Scholar] [CrossRef]
- Bouslama, L.; Nasri, D.; Chollet, L.; Belguith, K.; Bourlet, T.; Aouni, M.; Pozzetto, B.; Pillet, S. Natural recombination event within the capsid genomic region leading to a chimeric strain of human enterovirus b. J. Virol. 2007, 81, 8944–8952. [Google Scholar] [CrossRef][Green Version]
- Zhang, Y.; Yan, D.; Zhu, S.; Nishimura, Y.; Ye, X.; Wang, D.; Jorba, J.; Zhu, H.; An, H.; Shimizu, H.; et al. An insight into recombination with enterovirus species c and nucleotide g-480 reversion from the viewpoint of neurovirulence of vaccine-derived polioviruses. Sci. Rep. 2015, 5, 17291. [Google Scholar] [CrossRef] [PubMed]
- Lukashev, A.N.; Lashkevich, V.A.; Koroleva, G.A.; Ilonen, J.; Hinkkanen, A.E. Recombination in uveitis-causing enterovirus strains. J. Gen. Virol. 2004, 85, 463–470. [Google Scholar] [CrossRef]
- Zhang, Y.; Tan, X.J.; Wang, H.Y.; Yan, D.M.; Zhu, S.L.; Wang, D.Y.; Ji, F.; Wang, X.J.; Gao, Y.J.; Chen, L.; et al. An outbreak of hand, foot, and mouth disease associated with subgenotype c4 of human enterovirus 71 in Shandong, China. J. Clin. Virol. 2009, 44, 262–267. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Tan, X.; Cui, A.; Mao, N.; Xu, S.; Zhu, Z.; Zhou, J.; Shi, J.; Zhao, Y.; Wang, X.; et al. Complete genome analysis of the c4 subgenotype strains of enterovirus 71: Predominant recombination c4 viruses persistently circulating in China for 14 years. PLoS ONE 2013, 8, e56341. [Google Scholar] [CrossRef] [PubMed]
- Shulman, L.M.; Manor, Y.; Sofer, D. Poliovirus vaccine and vaccine-derived polioviruses. N. Engl. J. Med. 2010, 363, 1870, author reply 1870–1871. [Google Scholar]
- Minor, P. Vaccine-derived poliovirus (vdpv): Impact on poliomyelitis eradication. Vaccine 2009, 27, 2649–2652. [Google Scholar] [CrossRef]
- Gaunt, E.; Harvala, H.; Osterback, R.; Sreenu, V.B.; Thomson, E.; Waris, M.; Simmonds, P. Genetic characterization of human coxsackievirus a6 variants associated with atypical hand, foot and mouth disease: A potential role of recombination in emergence and pathogenicity. J. Gen. Virol. 2015, 96, 1067–1079. [Google Scholar] [CrossRef]
- Song, Y.; Zhang, Y.; Han, Z.; Xu, W.; Xiao, J.; Wang, X.; Wang, J.; Yang, J.; Yu, Q.; Yu, D.; et al. Genetic recombination in fast-spreading coxsackievirus a6 variants: A potential role in evolution and pathogenicity. Virus Evol. 2020. [Google Scholar] [CrossRef]
- Feng, X.; Guan, W.; Guo, Y.; Yu, H.; Zhang, X.; Cheng, R.; Wang, Z.; Zhang, Z.; Zhang, J.; Li, H.; et al. A novel recombinant lineage’s contribution to the outbreak of coxsackievirus a6-associated hand, foot and mouth disease in Shanghai, China, 2012–2013. Sci. Rep. 2015, 5, 11700. [Google Scholar] [CrossRef]
- Yang, S.; Wu, J.; Ding, C.; Cui, Y.; Zhou, Y.; Li, Y.; Deng, M.; Wang, C.; Xu, K.; Ren, J.; et al. Epidemiological features of and changes in incidence of infectious diseases in China in the first decade after the sars outbreak: An observational trend study. Lancet Infect. Dis. 2017, 17, 716–725. [Google Scholar] [CrossRef]
- Ji, T.; Han, T.; Tan, X.; Zhu, S.; Yan, D.; Yang, Q.; Song, Y.; Cui, A.; Zhang, Y.; Mao, N.; et al. Surveillance, epidemiology, and pathogen spectrum of hand, foot, and mouth disease in mainland of China from 2008 to 2017. Biosaf. Health 2019, 1, 32–40. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genbank No. | Strain ID | Source | Length | Isolated Province (China) | Isolated Country | Isolated Year | Genotype |
|---|---|---|---|---|---|---|---|
| AF081299 | Donovan | Genbank | Full-length | NA | United States | 1949 | A |
| KC879489 | 22468 | Genbank | VP1 | NA | Armenia | 2004 | B |
| KC879490 | 22809 | Genbank | VP1 | NA | Russia | 2004 | B |
| KC879494 | 24267 | Genbank | VP1 | NA | Russia | 2005 | B |
| JN203522 | N-650 | Genbank | VP1 | NA | Inida | 2007 | B |
| JN203523 | N-662 | Genbank | VP1 | NA | Inida | 2007 | B |
| KC879502 | 29089 | Genbank | VP1 | NA | Turkmenistan | 2007 | C |
| JN203524 | N-672 | Genbank | VP1 | NA | Inida | 2008 | B |
| JX088583 | 11186 | Genbank | VP1 | Shandong | China | 2011 | D |
| KC867069 | JB141230124 | Genbank | VP1 | Guangdong | China | 2012 | D |
| KC867070 | JB141230127 | Genbank | VP1 | Guangdong | China | 2012 | D |
| KC867071 | JB141230157 | Genbank | VP1 | Guangdong | China | 2012 | D |
| KM609475 | SZ93 | Genbank | Full-length | Guangdong | China | 2012 | D |
| KM609476 | SZ124 | Genbank | Full-length | Guangdong | China | 2012 | D |
| KM609477 | SZ127 | Genbank | Full-length | Guangdong | China | 2012 | D |
| KM609478 | SZ141 | Genbank | Full-length | Guangdong | China | 2012 | D |
| KM609479 | SZ157 | Genbank | Full-length | Guangdong | China | 2012 | D |
| KM609480 | SZ39 | Genbank | Full-length | Guangdong | China | 2013 | D |
| KM609481 | SZ40 | Genbank | Full-length | Guangdong | China | 2013 | D |
| KP289435 | P82 | Genbank | Full-length | Zhejiang | China | 2013 | D |
| MT648779 | GS-13-467 | This study | Full-length | Guangdong | China | 2013 | D |
| KP765687 | SZ266 | Genbank | Full-length | Guangdong | China | 2014 | E |
| MT648780 | GS-14-481 | This study | Full-length | Gansu | China | 2014 | E |
| MT648781 | GS-14-943 | This study | Full-length | Gansu | China | 2014 | E |
| MT648778 | CQ-15-Y-120 | This study | Full-length | Chongqing | China | 2015 | E |
| MT648783 | SD-HZ040 | This study | Full-length | Shandong | China | 2015 | D |
| MT648785 | TJ-16-0148 | This study | Full-length | Tianjin | China | 2016 | E |
| MH111056 | C177 | Genbank | Full-length | NA | Australia | 2017 | E |
| MH111057 | C184 | Genbank | Full-length | NA | Australia | 2017 | E |
| MT648784 | SaX-17-77 | This study | Full-length | Shaanxi | China | 2017 | E |
| MT648787 | JX-17-115 | This study | Full-length | Jiangxi | China | 2017 | E |
| MT648788 | JX-17-97 | This study | Full-length | Jiangxi | China | 2017 | E |
| MT648782 | HeN-18-96 | This study | Full-length | Henan | China | 2018 | E |
| MT648786 | TJ-18-38 | This study | Full-length | Tianjin | China | 2018 | C |
| Genotype | Polymorphic Sites | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 25 | 30 | 61 | * 100 | * 103 | * 240 | * 241 | 279 | 282 | |
| BC-Loop | HI-loop | ||||||||
| A | D | N | N | Q | F | A | T | T | T |
| B | . | . | . | R | . | . | . | . | A |
| . | . | . | K | . | . | . | . | A | |
| . | . | . | . | . | . | . | . | A | |
| . | . | . | . | . | . | . | . | A | |
| . | . | . | . | . | . | . | . | A | |
| . | . | . | . | . | . | . | . | V | |
| C | . | . | . | . | . | . | . | . | . |
| . | . | . | . | . | . | . | . | . | |
| D | S | . | . | R | . | . | . | . | . |
| S | . | . | . | . | . | . | . | . | |
| S | . | . | . | . | . | I | . | . | |
| S | . | . | . | . | . | . | . | . | |
| G | . | . | . | . | . | . | . | . | |
| S | . | . | . | . | . | . | . | . | |
| G | . | . | . | . | . | . | . | . | |
| S | . | . | . | . | . | I | . | . | |
| S | . | . | . | . | . | . | . | . | |
| G | . | . | . | . | . | . | . | . | |
| G | . | . | . | . | . | . | . | . | |
| G | . | . | . | . | . | . | . | . | |
| G | . | . | . | . | . | . | . | . | |
| G | . | . | . | . | . | . | . | . | |
| E | N | D | S | N | Y | T | V | A | D |
| N | D | S | N | Y | T | V | A | D | |
| N | D | S | N | Y | T | V | A | D | |
| N | D | S | N | Y | T | V | A | D | |
| N | D | S | N | Y | T | V | A | D | |
| S | D | S | N | Y | T | V | . | D | |
| S | D | S | N | Y | T | V | . | D | |
| N | D | S | N | Y | T | V | A | N | |
| N | D | S | N | Y | T | V | A | D | |
| N | D | S | N | Y | T | V | A | D | |
| N | D | S | N | Y | T | V | A | D | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, Y.; Wang, D.; Zhang, Y.; Han, Z.; Xiao, J.; Lu, H.; Yan, D.; Ji, T.; Yang, Q.; Zhu, S.; et al. Genetic Diversity Analysis of Coxsackievirus A8 Circulating in China and Worldwide Reveals a Highly Divergent Genotype. Viruses 2020, 12, 1061. https://doi.org/10.3390/v12101061
Song Y, Wang D, Zhang Y, Han Z, Xiao J, Lu H, Yan D, Ji T, Yang Q, Zhu S, et al. Genetic Diversity Analysis of Coxsackievirus A8 Circulating in China and Worldwide Reveals a Highly Divergent Genotype. Viruses. 2020; 12(10):1061. https://doi.org/10.3390/v12101061
Chicago/Turabian StyleSong, Yang, Dongyan Wang, Yong Zhang, Zhenzhi Han, Jinbo Xiao, Huanhuan Lu, Dongmei Yan, Tianjiao Ji, Qian Yang, Shuangli Zhu, and et al. 2020. "Genetic Diversity Analysis of Coxsackievirus A8 Circulating in China and Worldwide Reveals a Highly Divergent Genotype" Viruses 12, no. 10: 1061. https://doi.org/10.3390/v12101061
APA StyleSong, Y., Wang, D., Zhang, Y., Han, Z., Xiao, J., Lu, H., Yan, D., Ji, T., Yang, Q., Zhu, S., & Xu, W. (2020). Genetic Diversity Analysis of Coxsackievirus A8 Circulating in China and Worldwide Reveals a Highly Divergent Genotype. Viruses, 12(10), 1061. https://doi.org/10.3390/v12101061