Molecular Characterisation of a Novel and Highly Divergent Passerine Adenovirus 1


Abstract
1. Introduction
2. Materials and Methods
2.1. Source of Sample and Extraction of DNA
2.2. Library Construction and Sequencing
2.3. Genome Assembly
2.4. Genome Annotation and Bioinformatics
2.5. Comparative Genomics
2.6. Phylogenetic Analyses
3. Results
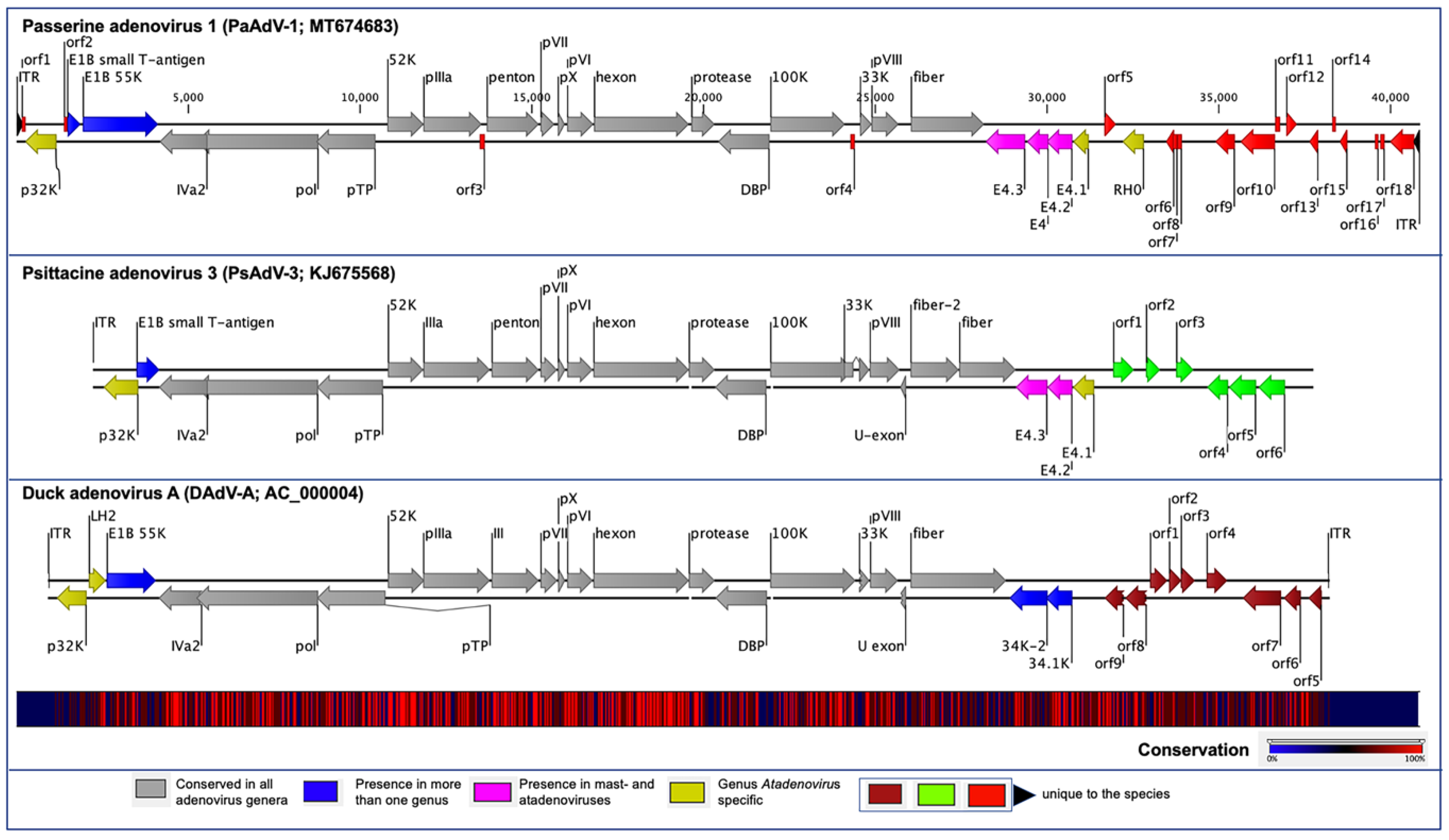
3.1. Genome of PaAdV-1
3.2. Genome Annotation and Comparative Analyses of PaAdV-1
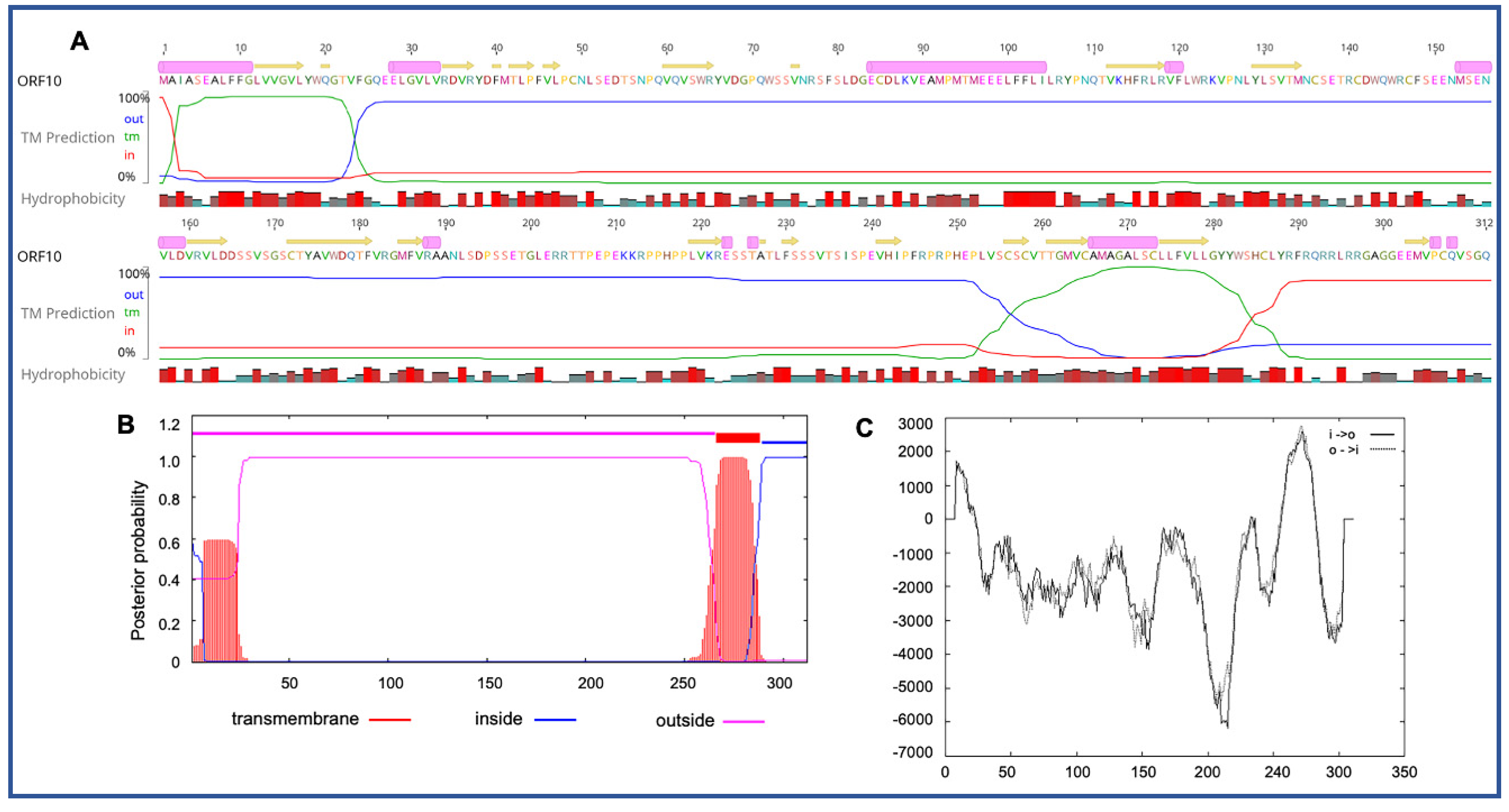
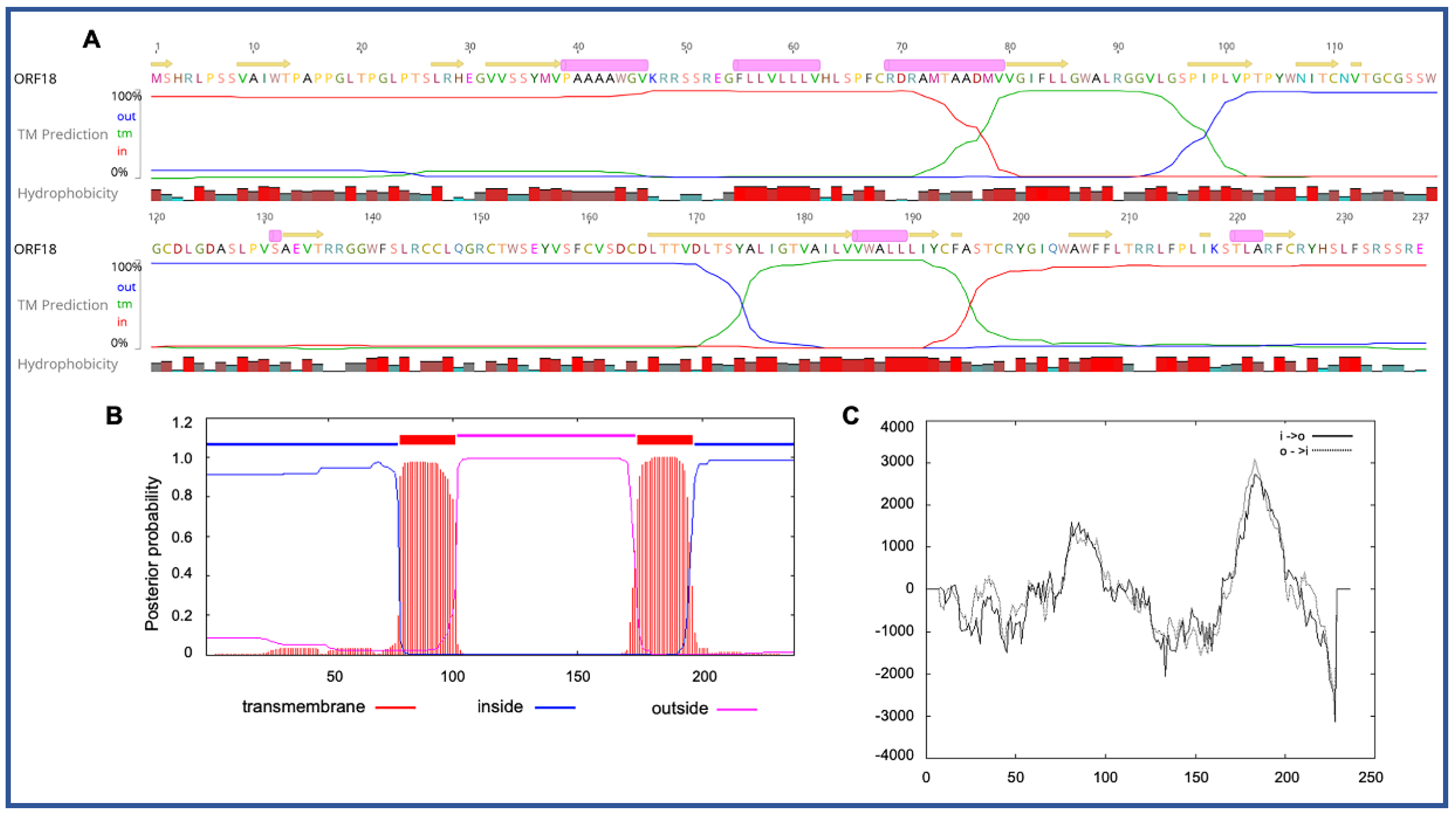
3.3. Unique ORFs in PaAdV-1
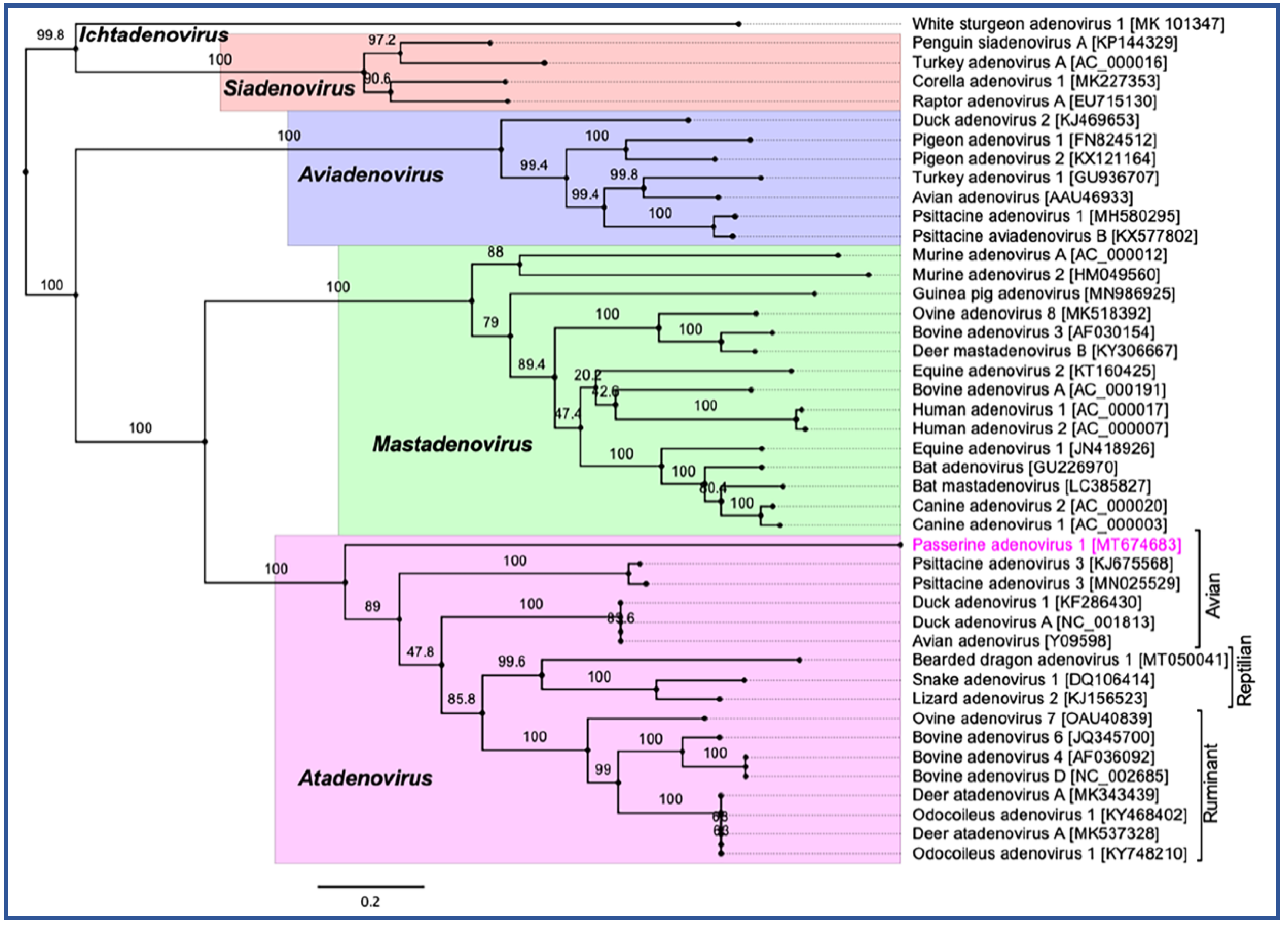
3.4. Evolutionary Relationships of PaAdV-1
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Data Availability
References
- ICTV. Virus taxonomy, ninth report of the International Committee on Taxonomy of Viruses. In Virus Taxonomy; King, A.M.Q., Adams, M.J., Carstens, E.B., Lefkowitz, E.J., Eds.; Elsevier: San Diego, CA, USA, 2012; pp. 125–141. [Google Scholar]
- Benkö, M.; Harrach, B. A proposal for a new (third) genus within the family Adenoviridae. Arch. Virol. 1998, 143, 829–837. [Google Scholar] [CrossRef]
- Dán, A.; Russell, W.C.; Ruzsics, Z.; Harrach, B.; Benkö, M. Analysis of the hexon gene sequence of bovine adenovirus type 4 provides further support for a new adenovirus genus (Atadenovirus). J. Gen. Virol. 1998, 79, 1453–1460. [Google Scholar] [CrossRef] [PubMed]
- Vaz, F.F.; Raso, T.F.; E Agius, J.; Hunt, T.; Leishman, A.; Eden, J.-S.; Phalen, D.N. Opportunistic sampling of wild native and invasive birds reveals a rich diversity of adenoviruses in Australia. Virus Evol. 2020, 6. [Google Scholar] [CrossRef] [PubMed]
- Pénzes, J.J.; Menéndez-Conejero, R.; Condezo, G.N.; Ball, I.; Papp, T.; Doszpoly, A.; Paradela, A.D.; Pérez-Berná, A.J.; López-Sanz, M.; Nguyen, T.H.; et al. Molecular Characterization of a Lizard Adenovirus Reveals the First Atadenovirus with Two Fiber Genes and the First Adenovirus with Either One Short or Three Long Fibers per Penton. J. Virol. 2014, 88, 11304–11314. [Google Scholar] [CrossRef] [PubMed]
- Farkas, S.L.; Benkő, M.; Élő, P.; Ursu, K.; Dán, Á.; Ahne, W.; Harrach, B. Genomic and phylogenetic analyses of an adenovirus isolated from a corn snake (Elaphe guttata) imply a common origin with members of the proposed new genus Atadenovirus The GenBank accession number of the sequence reported in this paper is AY082603. J. Gen. Virol. 2002, 83, 2403–2410. [Google Scholar] [CrossRef] [PubMed]
- Harrach, B. Adenoviruses: General Features. In Reference Module in Biomedical Sciences; Elsevier: Amsterdam, The Netherlands, 2014. [Google Scholar]
- Vrati, S.; Brookes, D.; Strike, P.; Khatri, A.; Boyle, D.; Both, G. Unique Genome Arrangement of an Ovine Adenovirus: Identification of New Proteins and Proteinase Cleavage Sites. Virology 1996, 220, 186–199. [Google Scholar] [CrossRef]
- Miller, M.M.; Cornish, T.E.; Creekmore, T.E.; Fox, K.; Laegreid, W.; McKenna, J.; Vasquez, M.; Woods, L.W. Whole-genome sequences of Odocoileus hemionus deer adenovirus isolates from deer, moose and elk are highly conserved and support a new species in the genus Atadenovirus. J. Gen. Virol. 2017, 98, 2320–2328. [Google Scholar] [CrossRef]
- Pénzes, J.J.; Szirovicza, L.; Harrach, B. The complete genome sequence of bearded dragon adenovirus 1 harbors three genes encoding proteins of the C-type lectin-like domain superfamily. Infect. Genet. Evol. 2020, 83, 104321. [Google Scholar] [CrossRef]
- To, K.K.-W.; Tse, H.; Chan, W.-M.; Choi, G.K.Y.; Zhang, A.J.X.; Sridhar, S.; Wong, S.C.Y.; Chan, J.F.; Chan, A.S.F.; Woo, P.C.Y.; et al. A Novel Psittacine Adenovirus Identified During an Outbreak of Avian Chlamydiosis and Human Psittacosis: Zoonosis Associated with Virus-Bacterium Coinfection in Birds. PLOS Neglected Trop. Dis. 2014, 8, e3318. [Google Scholar] [CrossRef]
- Duarte, M.A.; Silva, J.M.F.; Brito, C.R.; Teixeira, D.S.; Melo, F.; Ribeiro, B.M.; Nagata, T.; Campos, F.S. Faecal Virome Analysis of Wild Animals from Brazil. Viruses 2019, 11, 803. [Google Scholar] [CrossRef]
- Needle, D.B.; Wise, A.G.; Gregory, C.R.; Maes, R.K.; Sidor, I.F.; Ritchie, B.W.; Agnew, D. Necrotizing Ventriculitis in Fledgling Chimney Swifts (Chaetura Pelagica) Associated With a Novel Adenovirus, Chimney Swift Adenovirus-1 (CsAdV-1). Veter. Pathol. 2019, 56, 907–914. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, A.P.J.; Rangel, M.C.V.; Vidovszky, M.Z.; Rossi, J.L., Jr.; Vicentini, F.; Harrach, B.; Kaján, G.L. Identification of two novel adenoviruses in smooth-billed ani and tropical screech owl. PLoS ONE 2020, 15, e0229415. [Google Scholar] [CrossRef] [PubMed]
- Harrach, B.; Meehan, B.M.; Benkő, M.; Adair, B.M.; Todd, D. Close Phylogenetic Relationship between Egg Drop Syndrome Virus, Bovine Adenovirus Serotype 7, and Ovine Adenovirus Strain 287. Virology 1997, 229, 302–306. [Google Scholar] [CrossRef][Green Version]
- Hess, M.; Blöcker, H.; Brandt, P. The Complete Nucleotide Sequence of the Egg Drop Syndrome Virus: An Intermediate between Mastadenoviruses and Aviadenoviruses. Virology 1997, 238, 145–156. [Google Scholar] [CrossRef]
- Phalen, D.N.; Agius, J.; Vaz, F.F.; Eden, J.-S.; Setyo, L.C.; Donahoe, S. A survey of a mixed species aviary provides new insights into the pathogenicity, diversity, evolution, host range, and distribution of psittacine and passerine adenoviruses. Avian Pathol. 2019, 48, 437–443. [Google Scholar] [CrossRef] [PubMed]
- Rinder, M.; Schmitz, A.; Baas, N.; Korbel, R. Molecular identification of novel and genetically diverse adenoviruses in Passeriform birds. Virus Genes 2020, 56, 316–324. [Google Scholar] [CrossRef]
- Karen, A.F.; Levi, A.; Laura, H.-H.; Myrna, M. A Mortality Event in Elk (Cervus elaphus) Calves Associated with Malnutrition, Pasteurellosis, and Deer Adenovirus in Colorado, USA. J. Wildl. Dis. 2017, 53, 674–676. [Google Scholar]
- Thomson, D.; Meers, J.; Harrach, B. Molecular confirmation of an adenovirus in brushtail possums (Trichosurus vulpecula). Virus Res. 2002, 83, 189–195. [Google Scholar] [CrossRef]
- Gál, J.; Mándoki, M.; Sós, E.; Kertész, P.; Koroknai, V.; Banyai, K.; Farkas, S.L. Novel adenovirus detected in kowari (Dasyuroides byrnei) with pneumonia. Acta Microbiol. et Immunol. Hung. 2017, 64, 81–90. [Google Scholar] [CrossRef][Green Version]
- Garcia-Morante, B.; Pénzes, J.J.; Costa, T.; Martorell, J.; Martinez, J. Hyperplastic stomatitis and esophagitis in a tortoise (Testudo graeca) associated with an adenovirus infection. J. Veter. Diagn. Investig. 2016, 28, 579–583. [Google Scholar] [CrossRef]
- Fu, G.; Chen, H.; Yu, H.; Cheng, L.; Fu, Q.; Shi, S.; Wan, C.; Chen, C.; Lin, J. Full Genome Sequence of Egg Drop Syndrome Virus Strain FJ12025 Isolated from Muscovy Duckling. Genome Announc. 2013, 1, e00623-13. [Google Scholar] [CrossRef] [PubMed]
- Davison, A.J.; Benkő, M.; Harrach, B. Genetic content and evolution of adenoviruses. J. Gen. Virol. 2003, 84, 2895–2908. [Google Scholar] [CrossRef] [PubMed]
- Sarker, S.; Batinovic, S.; Talukder, S.; Das, S.; Park, F.; Petrovski, S.; Forwood, J.K.; Helbig, K.J.; Raidal, S.R. Molecular characterisation of a novel pathogenic avipoxvirus from the Australian magpie (Gymnorhina tibicen). Virology 2019, 540, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Sarker, S.; Das, S.; Lavers, J.; Hutton, I.; Helbig, K.J.; Imbery, J.; Upton, C.; Raidal, S.R. Genomic characterization of two novel pathogenic avipoxviruses isolated from pacific shearwaters (Ardenna spp.). BMC Genom. 2017, 18, 298. [Google Scholar] [CrossRef]
- Sarker, S.; Roberts, H.K.; Tidd, N.; Ault, S.; Ladmore, G.; Peters, A.; Forwood, J.; Helbig, K.J.; Raidal, S.R. Molecular and microscopic characterization of a novel Eastern grey kangaroopox virus genome directly from a clinical sample. Sci. Rep. 2017, 7, 16472. [Google Scholar] [CrossRef]
- Sutherland, M.; Sarker, S.; Vaz, P.K.; Legione, A.R.; Devlin, J.M.; MacWhirter, P.L.; Whiteley, P.L.; Raidal, S.R. Disease surveillance in wild Victorian cacatuids reveals co-infection with multiple agents and detection of novel avian viruses. Veter. Microbiol. 2019, 235, 257–264. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Boratyn, G.M.; Camacho, C.; Cooper, P.S.; Coulouris, G.; Fong, A.; Ma, N.; Madden, T.L.; Matten, W.T.; McGinnis, S.D.; Merezhuk, Y.; et al. BLAST: A more efficient report with usability improvements. Nucleic Acids Res. 2013, 41, W29–W33. [Google Scholar] [CrossRef]
- Tcherepanov, V.; Ehlers, A.; Upton, C. Genome Annotation Transfer Utility (GATU): Rapid annotation of viral genomes using a closely related reference genome. BMC Genom. 2006, 7, 150. [Google Scholar] [CrossRef]
- Benson, D.A.; Clark, K.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Sayers, E.W. GenBank. Nucleic Acids Res. 2013, 42, D32–D37. [Google Scholar] [CrossRef]
- Krogh, A.; Larsson, B.; Von Heijne, G.; Sonnhammer, E.L.L. Predicting transmembrane protein topology with a hidden markov model: Application to complete genomes11Edited by F. Cohen. J. Mol. Boil. 2001, 305, 567–580. [Google Scholar] [CrossRef] [PubMed]
- Tusnády, G.E.; Simon, I. The HMMTOP transmembrane topology prediction server. Bioinformatics 2001, 17, 849–850. [Google Scholar] [CrossRef]
- Hofmann, K.; Stoffel, W. TMBASE—A database of membrane spanning protein segments. Biol. Chem. Hoppe-Seyler 1993, 166, 374. [Google Scholar]
- Zimmermann, L.; Stephens, A.; Nam, S.-Z.; Rau, D.; Kübler, J.; Lozajic, M.; Gabler, F.; Söding, J.; Lupas, A.N.; Alva, V. A Completely Reimplemented MPI Bioinformatics Toolkit with a New HHpred Server at its Core. J. Mol. Boil. 2018, 430, 2237–2243. [Google Scholar] [CrossRef]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J.E. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; Beer, T.A.P.D.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef]
- Armenteros, J.J.A.; Tsirigos, K.D.; Sønderby, C.K.; Petersen, T.N.; Winther, O.; Brunak, S.; Von Heijne, G.; Nielsen, H. SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat. Biotechnol. 2019, 37, 420–423. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Guindon, S.; Dufayard, J.-F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing the Performance of PhyML 3.0. Syst. Boil. 2010, 59, 307–321. [Google Scholar] [CrossRef]
- Molecular Evolution, Phylogenetics and Epidemiology. FigTree. Available online: http://tree.bio.ed.ac.uk/software/figtree/ (accessed on 2 December 2011).
- Silverman, B.D. Hydrophobicity of transmembrane proteins: Spatially profiling the distribution. Protein Sci. 2003, 12, 586–599. [Google Scholar] [CrossRef]
- Elazar, A.; Weinstein, J.J.; Prilusky, J.; Fleishman, S.J. Interplay between hydrophobicity and the positive-inside rule in determining membrane-protein topology. Proc. Natl. Acad. Sci. USA 2016, 113, 10340–10345. [Google Scholar] [CrossRef]
- Sester, M.; Ruszics, Z.; Mackley, E.; Burgert, H.-G. The transmembrane domain of the adenovirus E3/19K protein acts as an endoplasmic reticulum retention signal and contributes to intracellular sequestration of major histocompatibility complex class I molecules. J. Virol. 2013, 87, 6104–6117. [Google Scholar] [CrossRef]
- Maclachlan, N.J.; Dubovi, E.J. Adenoviridae. In Fenner’s Veterinary Virology, 5th ed.; MacLachlan, N.J., Dubovi, E.J., Eds.; Academic Press: Boston, MA, USA, 2017; pp. 217–227. [Google Scholar]
- Doszpoly, A.; Harrach, B.; LaPatra, S.; Benkő, M. Unconventional gene arrangement and content revealed by full genome analysis of the white sturgeon adenovirus, the single member of the genus Ichtadenovirus. Infect. Genet. Evol. 2019, 75, 103976. [Google Scholar] [CrossRef]
- Rauschhuber, C.; Wolf, A.; Ehrhardt, A. Transcriptional activity of inverted terminal repeats of various human adenovirus serotypes. J. Gen. Virol. 2010, 92, 669–674. [Google Scholar] [CrossRef]
- Oaks, J.L.; Schrenzel, M.; Rideout, B.; Sandfort, C. Isolation and Epidemiology of Falcon Adenovirus. J. Clin. Microbiol. 2005, 43, 3414–3420. [Google Scholar] [CrossRef]
- Gilson, T.; Blanchette, P.; Ballmann, M.Z.; Papp, T.; Pénzes, J.J.; Benkő, M.; Harrach, B.; Branton, P.E. Using the E4orf6-Based E3 Ubiquitin Ligase as a Tool to Analyze the Evolution of Adenoviruses. J. Virol. 2016, 90, 7350–7367. [Google Scholar] [CrossRef]
- Dán, Á.; Élő, P.; Harrach, B.; Zádori, Z.; Benkö, M. Four new inverted terminal repeat sequences from bovine adenoviruses reveal striking differences in the length and content of the ITRs. Virus Genes 2001, 22, 175–179. [Google Scholar] [CrossRef]
- Both, G.W. Identification of a unique family of F-box proteins in atadenoviruses. Virology 2002, 304, 425–433. [Google Scholar] [CrossRef][Green Version]
- Wellehan, J.F.X.; Johnson, A.J.; Harrach, B.; Benkő, M.; Pessier, A.P.; Johnson, C.M.; Garner, M.M.; Childress, A.; Jacobson, E.R. Detection and Analysis of Six Lizard Adenoviruses by Consensus Primer PCR Provides Further Evidence of a Reptilian Origin for the Atadenoviruses. J. Virol. 2004, 78, 13366–13369. [Google Scholar] [CrossRef]
- Harrach, B.; Information, R. Reptile Adenoviruses in Cattle? Acta Veter. Hung. 2000, 48, 485–490. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Fearnside, K.; Sarker, S.; Forwood, J.; Raidal, S.R. A novel pathogenic aviadenovirus from red-bellied parrots (Poicephalus rufiventris) unveils deep recombination events among avian host lineages. Virology 2017, 502, 188–197. [Google Scholar] [CrossRef] [PubMed]
- Sarker, S. Metagenomic Dataset of an Eastern spinebill (Acanthorhynchus tenuirostris). Mendeley. Available online: http://dx.doi.org/10.17632/57zhhhs7xz.1 (accessed on 20 June 2020).





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PaAdV-1 Synteny | Start (nt) | Stop (nt) | Strand | Size (aa) | PsAdV-3 Synteny |
|---|---|---|---|---|---|
| ORF01 | 150 | 263 | + | 37 | |
| p32K | 250 | 1161 | − | 303 | p32K |
| ORF02 | 1368 | 1469 | + | 33 | |
| E1B protein, small T-antigen | 1466 | 1849 | + | 127 | E1B protein, small T-antigen |
| E1B protein, large T-antigen | 1911 | 4049 | + | 712 | |
| IVa2 protein | 4094 | 5482 | − | 462 | IVa2 protein |
| DNA polymerase | 5164 | 8634 | − | 1156 | DNA polymerase |
| pTP | 8568 | 10,208 | − | 546 | pTP |
| 52K protein | 10,547 | 11,581 | + | 344 | 52K protein |
| pIIIa protein | 11,574 | 13,259 | + | 561 | IIIa protein |
| ORF03 | 13,201 | 13,350 | − | 49 | |
| penton protein | 13,405 | 14,916 | + | 503 | penton protein |
| pVII protein | 14,942 | 15,289 | + | 115 | pVII protein |
| pX protein | 15,384 | 15,587 | + | 67 | pX protein |
| pVI protein | 15,651 | 16,403 | + | 250 | pVI protein |
| hexon protein | 16,421 | 19,150 | + | 909 | hexon protein |
| Protease | 19,221 | 19,901 | + | 226 | protease |
| DBP | 19,999 | 21,486 | − | 495 | DNA-binding protein |
| 100K protein | 21,508 | 23,607 | + | 699 | 100K protein |
| ORF04 | 23,769 | 23,897 | − | 42 | |
| 33K protein | 24,044 | 24,391 | + | 115 | 33K protein |
| pVIII protein | 24,379 | 25,053 | + | 224 | pVIII protein |
| fibre protein | 25,407 | 27,509 | + | 700 | fibre 2 protein |
| E4.3 protein | 27,555 | 28,685 | − | 376 | E4.3 protein |
| E4 protein | 28,737 | 29,360 | − | 207 | |
| E4.2 protein | 29,299 | 30,036 | − | 245 | E4.2 protein |
| E4.1 protein | 30,058 | 30,513 | − | 151 | E4.1 protein |
| ORF05 | 30,907 | 31,185 | + | 92 | |
| RH0 | 31,341 | 31,958 | − | 205 | |
| ORF06 | 32,572 | 32,808 | − | 78 | |
| ORF07 | 32,762 | 32,902 | − | 46 | |
| ORF08 | 32,896 | 33,015 | − | 39 | |
| ORF09 | 33,901 | 34,446 | − | 181 | |
| ORF10 | 34,604 | 35,542 | − | 312 | |
| ORF11 | 35,541 | 35,672 | + | 43 | |
| ORF12 | 35,828 | 36,118 | + | 96 | |
| ORF13 | 36,429 | 36,683 | − | 84 | |
| ORF14 | 37,082 | 37,195 | + | 37 | |
| ORF15 | 37,306 | 37,527 | − | 73 | |
| ORF16 | 38,325 | 38,432 | − | 35 | |
| ORF17 | 38,483 | 38,611 | − | 42 | |
| ORF18 | 38,767 | 39,480 | − | 237 |
| Reference Atadenoviruses | Genome Identity (%) | G+C Content | % Pairwise AA Similarity with PaAdV-1 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| p32K | IVa2 | DNA Pol | pTP | Penton | Hexon | Protease | DBP | Fibre Protein | ||||
| Avian | PaAdV-1 | 53.7 | ||||||||||
| PsAdV-3 | 55.58 | 53.5 | 30.83 | 59.01 | 53.45 | 33.28 | 75.94 | 76.44 | 56.19 | 36.02 | 17.52 | |
| DAdV-A | 54.25 | 45.5 | 32.78 | 64.56 | 52.89 | 32.32 | 73.57 | 80.04 | 53.1 | 36.97 | 25.42 | |
| Ruminant | BAdV-D | 53.38 | 37.1 | 29.73 | 70.35 | 52.73 | 32.37 | 72.69 | 76.09 | 56.19 | 35.77 | 22.38 |
| OAdV-7 | 53.69 | 33.6 | 33.53 | 69.97 | 51.71 | 32.77 | 73.29 | 77.89 | 53.98 | 36.07 | 23.27 | |
| OdAdV-1 | 53.27 | 33.3 | 32.65 | 63.59 | 52.09 | 31.45 | 73.35 | 77.32 | 54.42 | 36.08 | 23.99 | |
| Reptilian | LAdV-2 | 53.43 | 47.8 | 32.47 | 59.23 | 52.39 | 33.38 | 72.91 | 76.64 | 56.64 | 35.45 | 15.65 |
| SnAdV-1 | 55.58 | 56.8 | 33.51 | 57.94 | 50.34 | 32.49 | 72.63 | 75.33 | 58.41 | 34.5 | 20.06 | |
| BDAdV-1 | 55.2 | 56.6 | 33.15 | 58.80 | 50.70 | 33.14 | 69.18 | 75.84 | 58.08 | 31.72 | 24.35 | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Athukorala, A.; Forwood, J.K.; Phalen, D.N.; Sarker, S. Molecular Characterisation of a Novel and Highly Divergent Passerine Adenovirus 1. Viruses 2020, 12, 1036. https://doi.org/10.3390/v12091036
Athukorala A, Forwood JK, Phalen DN, Sarker S. Molecular Characterisation of a Novel and Highly Divergent Passerine Adenovirus 1. Viruses. 2020; 12(9):1036. https://doi.org/10.3390/v12091036
Chicago/Turabian StyleAthukorala, Ajani, Jade K. Forwood, David N. Phalen, and Subir Sarker. 2020. "Molecular Characterisation of a Novel and Highly Divergent Passerine Adenovirus 1" Viruses 12, no. 9: 1036. https://doi.org/10.3390/v12091036
APA StyleAthukorala, A., Forwood, J. K., Phalen, D. N., & Sarker, S. (2020). Molecular Characterisation of a Novel and Highly Divergent Passerine Adenovirus 1. Viruses, 12(9), 1036. https://doi.org/10.3390/v12091036