SARS-CoV-2 RNA Extraction Using Magnetic Beads for Rapid Large-Scale Testing by RT-qPCR and RT-LAMP

 , , , , , , , and add
Show full author list
, , , , , , , and add
Show full author list
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
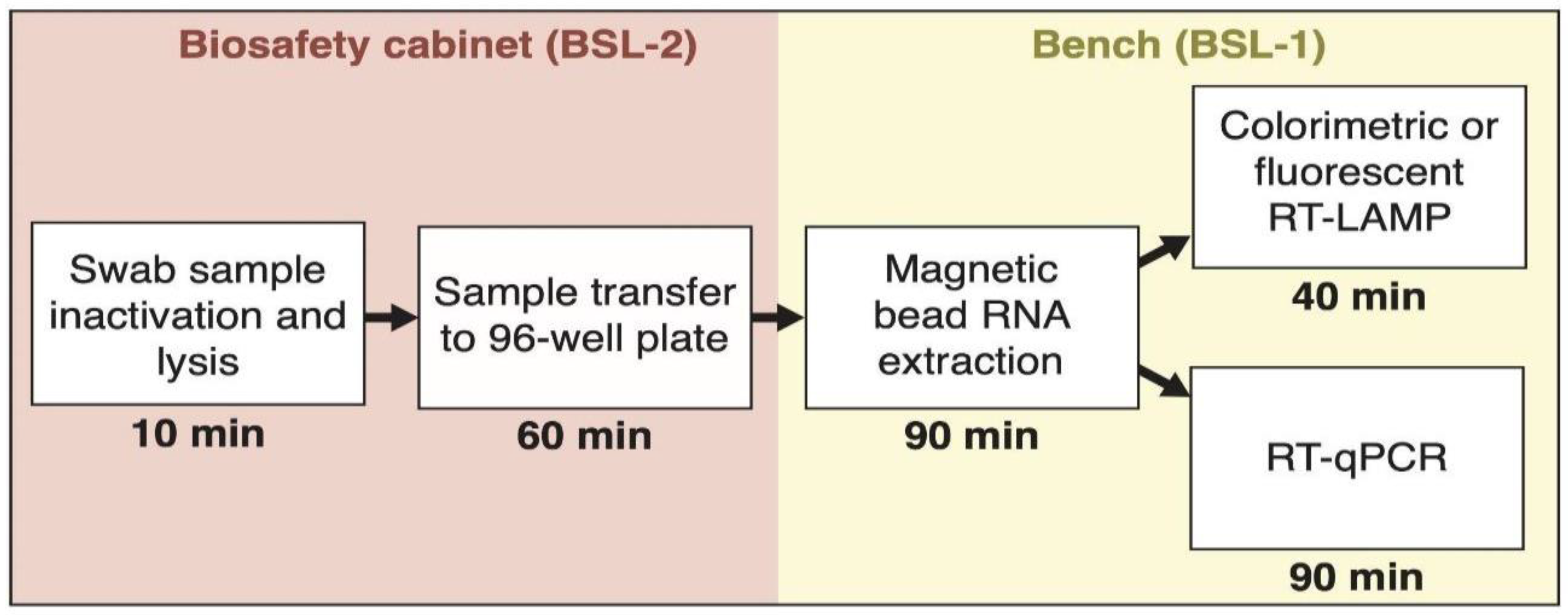
2.1. Clinical Samples and Sample Lysis
2.2. RNA Extraction Using Magnetic Beads in a 96-Well Plate Format
2.3. RNA Extraction Using QIAcube
2.4. SARS-CoV-2 RNA Detection by RT-qPCR
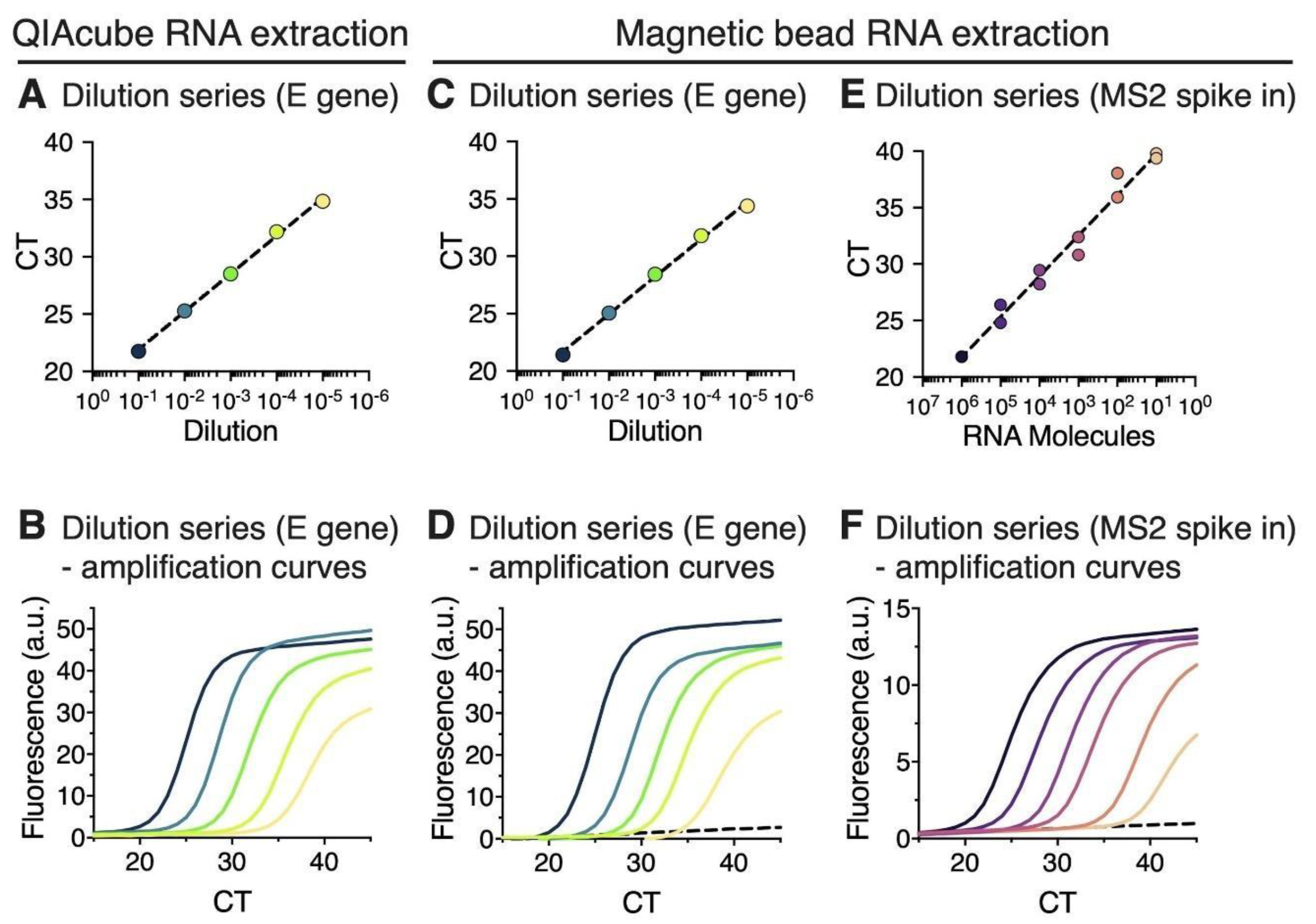
2.5. Estimation of the Detection Limit of Magnetic Bead RNA Extraction Using MS2 RNA Spike-In
2.6. SARS-CoV-2 RNA Detection by Colorimetric RT-LAMP
2.7. SARS-CoV-2 RNA Detection by Fluorescent RT-LAMP
2.8. Ethics Statement
3. Results
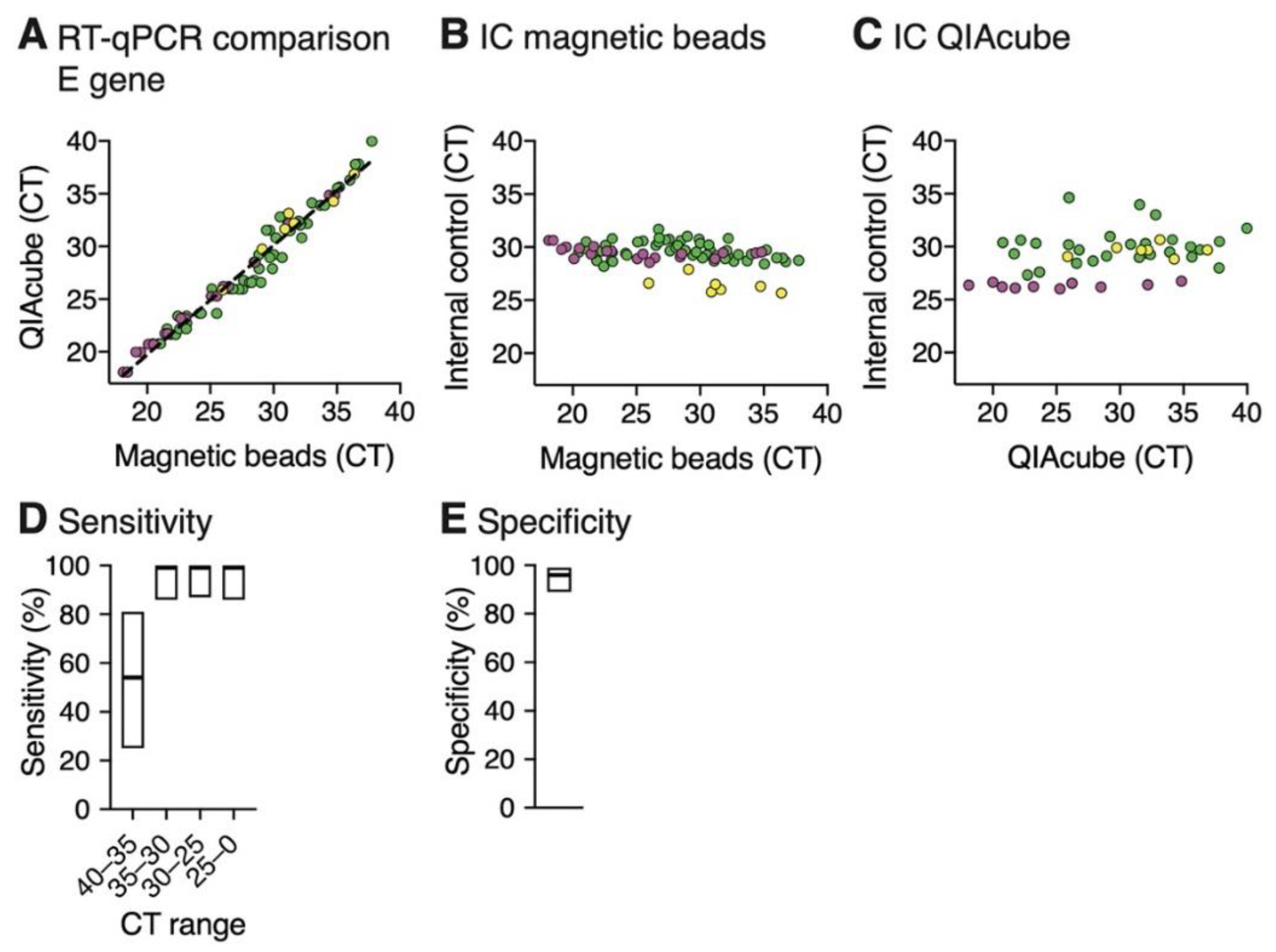
3.1. RNA Extraction Using Magnetic Beads Yields RT-qPCR Results That Are Comparable to a Commercial Extraction Kit
3.2. RT-qPCR Performed on Magnetic Bead Extracted RNA Shows High Detection Sensitivity and Specificity
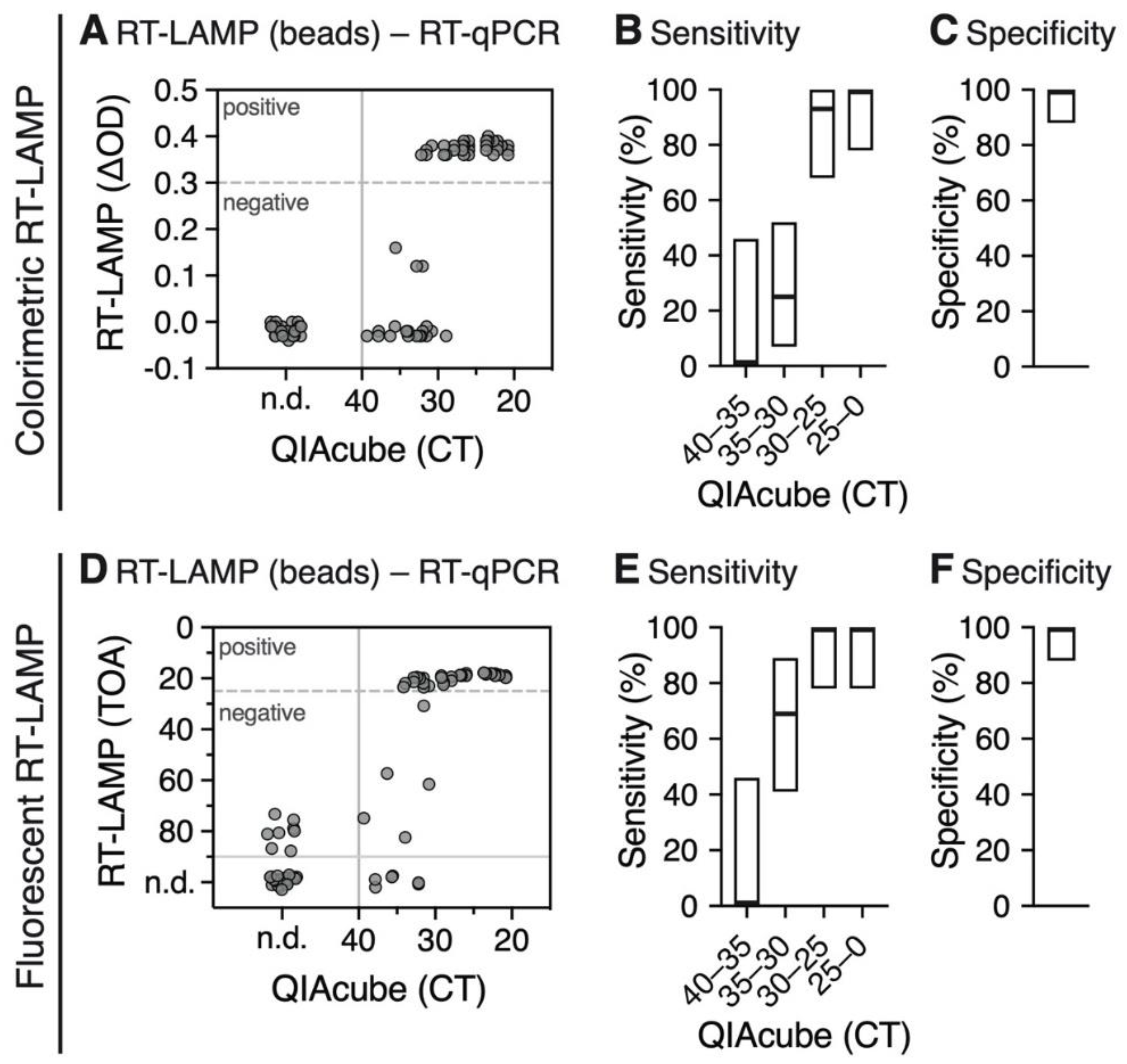
3.3. RNA Extraction Using Magnetic Beads is Compatible with the RT-LAMP Assay
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Vogels, C.B.F.; Brito, A.F.; Wyllie, A.L.; Fauver, J.R.; Ott, I.M.; Kalinich, C.C.; Petrone, M.E.; Casanovas-Massana, A.; Muenker, M.C.; Moore, A.J.; et al. Analytical Sensitivity and Efficiency Comparisons of SARS-CoV-2 RT–qPCR Primer–Probe Sets. Nat. Microbiol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Corman, V.M.; Landt, O.; Kaiser, M.; Molenkamp, R.; Meijer, A.; Chu, D.K.; Bleicker, T.; Brünink, S.; Schneider, J.; Schmidt, M.L.; et al. Detection of 2019 Novel Coronavirus (2019-nCoV) by Real-Time RT-PCR. Eurosurveillance 2020, 25, 2000045. [Google Scholar] [CrossRef]
- RNA Extraction Kits for COVID-19 Tests Are in Short Supply in US. The Scientist. Available online: /www.the-scientist.com/news-opinion/rna-extraction-kits-for-covid-19-tests-are-in-short-supply-in-us-67250 (accessed on 6 August 2020).
- Aitken, J.; The Crick COVID-19 Consortium; Ambrose, K.; Barrell, S.; Beale, R.; Bineva-Todd, G.; Biswas, D.; Byrne, R.; Caidan, S.; Cherepanov, P.; et al. Scalable and Robust SARS-CoV-2 Testing in an Academic Center. Nat. Biotechnol. 2020, 38, 927–931. [Google Scholar] [CrossRef] [PubMed]
- Guruceaga, X.; Sierra, A.; Marino, D.; Santin, I.; Nieto-Garai, J.A.; Bilbao, J.R.; Lorizate, M.; Aspichueta, P.; coBIG (COVID19 Basque Inter-institutional Group); Mayor, U. Fast SARS-CoV-2 Detection Protocol Based on RNA Precipitation and RT-qPCR in Nasopharyngeal Swab Samples. medRxiv 2008. [Google Scholar] [CrossRef]
- Zhao, Z.; Cui, H.; Song, W.; Ru, X.; Zhou, W.; Yu, X. A Simple Magnetic Nanoparticles-Based Viral RNA Extraction Method for Efficient Detection of SARS-CoV-2. bioRxiv 2020. [Google Scholar] [CrossRef]
- Thi, V.L.D.; Herbst, K.; Boerner, K.; Meurer, M.; Kremer, L.P.; Kirrmaier, D.; Freistaedter, A.; Papagiannidis, D.; Galmozzi, C.; Stanifer, M.L.; et al. A Colorimetric RT-LAMP Assay and LAMP-Sequencing for Detecting SARS-CoV-2 RNA in Clinical Samples. Sci. Transl. Med. 2020, eabc7075. [Google Scholar] [CrossRef]
- Lever, M.A.; Torti, A.; Eickenbusch, P.; Michaud, A.B.; Antl-Temkiv, T.Å.; Jã¸rgensen, B.B.; Šantl-Temkiv, T.; Jørgensen, B.B. A Modular Method for the Extraction of DNA and RNA, and the Separation of DNA Pools from Diverse Environmental Sample Types. Front. Microbiol. 2015, 6, 476. [Google Scholar] [CrossRef]
- Chomczynski, P.; Sacchi, N. The Single-Step Method of RNA Isolation by Acid Guanidinium Thiocyanate–Phenol–Chloroform Extraction: Twenty-Something Years on. Nat. Protoc. 2006, 1, 581–585. [Google Scholar] [CrossRef]
- Boom, R.; Sol, C.J.; Salimans, M.M.; Jansen, C.L.; Dillen, P.M.W.-V.; Van Der Noordaa, J. Rapid and Simple Method for Purification of Nucleic Acids. J. Clin. Microbiol. 1990, 28, 495–503. [Google Scholar] [CrossRef]
- Vandeventer, P.E.; Lin, J.S.; Zwang, T.; Nadim, A.; Johal, M.S.; Niemz, A. Multiphasic DNA Adsorption to Silica Surfaces under Varying Buffer, pH, and Ionic Strength Conditions. J. Phys. Chem. B 2012, 116, 5661–5670. [Google Scholar] [CrossRef]
- Borodina, T.A.; Lehrach, H.; Soldatov, A.V. DNA Purification on Homemade Silica Spin-Columns. Anal. Biochem. 2003, 321, 135–137. [Google Scholar] [CrossRef]
- Holmberg, R.; Gindlesperger, A.; Stokes, T.; Brady, D.; Thakore, N.; Belgrader, P.; Cooney, C.G.; Chandler, D.P. High-throughput, Automated Extraction of DNA and RNA from Clinical Samples Using TruTip Technology on Common Liquid Handling Robots. J. Vis. Exp. 2013, 76, e50356. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.H.; Gessert, S.F.; Chen, Y.; Sergeev, N.V.; Haghiri, B. Preparation of Iron Oxide Silica Particles for Zika Viral RNA Extraction. Heliyon 2018, 4, e00572. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Li, R.; Chen, Y.; Pan, P.; Tong, W.; Dong, X.; Chen, Y.; Yu, D. Integrated DNA and RNA Extraction Using Magnetic Beads from Viral Pathogens Causing Acute Respiratory Infections. Sci. Rep. 2017, 7, 45199. [Google Scholar] [CrossRef]
- Notomi, T.; Okayama, H.; Masubuchi, H.; Yonekawa, T.; Watanabe, K.; Amino, N.; Hase, T. Loop-Mediated Isothermal Amplification of DNA. Nucleic Acids Res. 2000, 28, E63. [Google Scholar] [CrossRef]
- Zhang, Y.; Odiwuor, N.; Xiong, J.; Sun, L.; Nyaruaba, R.O.; Wei, H.; Tanner, N.A. Rapid Molecular Detection of SARS-CoV-2 (COVID-19) Virus RNA Using Colorimetric LAMP. medRxiv 2020. [Google Scholar] [CrossRef]
- Tanner, N.A.; Zhang, Y.; Evans, T.C.J. Visual Detection of Isothermal Nucleic Acid Amplification Using pH-Sensitive Dyes. Biotechniques 2015, 58, 59–68. [Google Scholar] [CrossRef]
- Jones, G.; Patel, N.; Levy, M.; Storeygard, A.; Balk, D.; Gittleman, J.L.; Daszak, P. Global Trends in Emerging Infectious Diseases. Nature 2008, 451, 990–993. [Google Scholar] [CrossRef]
- Smith, K.F.; Goldberg, M.E.; Rosenthal, S.; Carlson, L.; Chen, J.; Chen, C.; Ramachandran, S. Global Rise in Human Infectious Disease Outbreaks. J. R. Soc. Interf. 2014, 11, 20140950. [Google Scholar] [CrossRef]
- Lindahl, J.; Grace, D. The Consequences of Human Actions on Risks for Infectious Diseases: A Review. Infect. Ecol. Epidemiol. 2015, 5, 30048. [Google Scholar] [CrossRef]
- Oberacker, P.; Stepper, P.; Bond, D.; Hipp, K.; Hore, T.; Jurkowski, T. Simple Synthesis of Functionalized Paramagnetic Beads for Nucleic Acid Purification and Manipulation. BIO-PROTOCOL 2019, 9, 9. [Google Scholar] [CrossRef]
- Wee, S.K.; Sivalingam, S.P.; Yap, E.P.H. Rapid Direct Nucleic Acid Amplification Test without RNA Extraction for SARS-CoV-2 Using a Portable PCR Thermocycler. Genes 2020, 11, 664. [Google Scholar] [CrossRef] [PubMed]
- Smyrlaki, I.; Ekman, M.; Vondracek, M.; Papanicoloau, N.; Lentini, A.; Aarum, J.; Muradrasoli, S.; Albert, J.; Högberg, B.; Reinius, B. Massive and Rapid COVID-19 Testing Is Feasible by Extraction-Free SARS-CoV-2 RT-qPCR. medRxiv 2020. [Google Scholar] [CrossRef]
- Rabe, B.A.; Cepko, C. SARS-CoV-2 Detection Using an Isothermal Amplification Reaction and a Rapid, Inexpensive Protocol for Sample Inactivation and Purification. medRxiv 2020. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Klein, S.; Müller, T.G.; Khalid, D.; Sonntag-Buck, V.; Heuser, A.-M.; Glass, B.; Meurer, M.; Morales, I.; Schillak, A.; Freistaedter, A.; et al. SARS-CoV-2 RNA Extraction Using Magnetic Beads for Rapid Large-Scale Testing by RT-qPCR and RT-LAMP. Viruses 2020, 12, 863. https://doi.org/10.3390/v12080863
Klein S, Müller TG, Khalid D, Sonntag-Buck V, Heuser A-M, Glass B, Meurer M, Morales I, Schillak A, Freistaedter A, et al. SARS-CoV-2 RNA Extraction Using Magnetic Beads for Rapid Large-Scale Testing by RT-qPCR and RT-LAMP. Viruses. 2020; 12(8):863. https://doi.org/10.3390/v12080863
Chicago/Turabian StyleKlein, Steffen, Thorsten G. Müller, Dina Khalid, Vera Sonntag-Buck, Anke-Mareil Heuser, Bärbel Glass, Matthias Meurer, Ivonne Morales, Angelika Schillak, Andrew Freistaedter, and et al. 2020. "SARS-CoV-2 RNA Extraction Using Magnetic Beads for Rapid Large-Scale Testing by RT-qPCR and RT-LAMP" Viruses 12, no. 8: 863. https://doi.org/10.3390/v12080863
APA StyleKlein, S., Müller, T. G., Khalid, D., Sonntag-Buck, V., Heuser, A.-M., Glass, B., Meurer, M., Morales, I., Schillak, A., Freistaedter, A., Ambiel, I., Winter, S. L., Zimmermann, L., Naumoska, T., Bubeck, F., Kirrmaier, D., Ullrich, S., Barreto Miranda, I., Anders, S., ... Chlanda, P. (2020). SARS-CoV-2 RNA Extraction Using Magnetic Beads for Rapid Large-Scale Testing by RT-qPCR and RT-LAMP. Viruses, 12(8), 863. https://doi.org/10.3390/v12080863