Replication Compartments of DNA Viruses in the Nucleus: Location, Location, Location



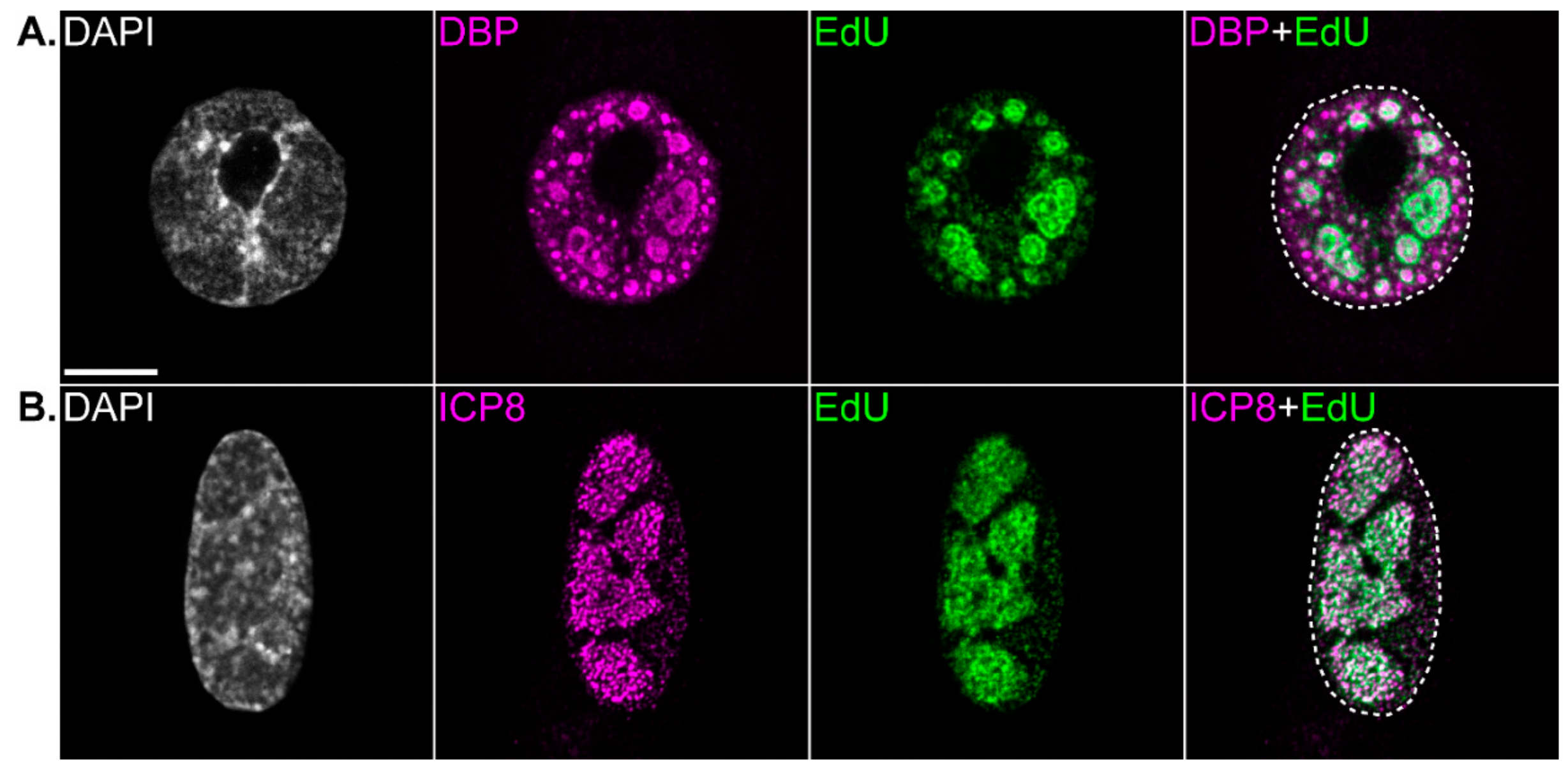{kind=link}
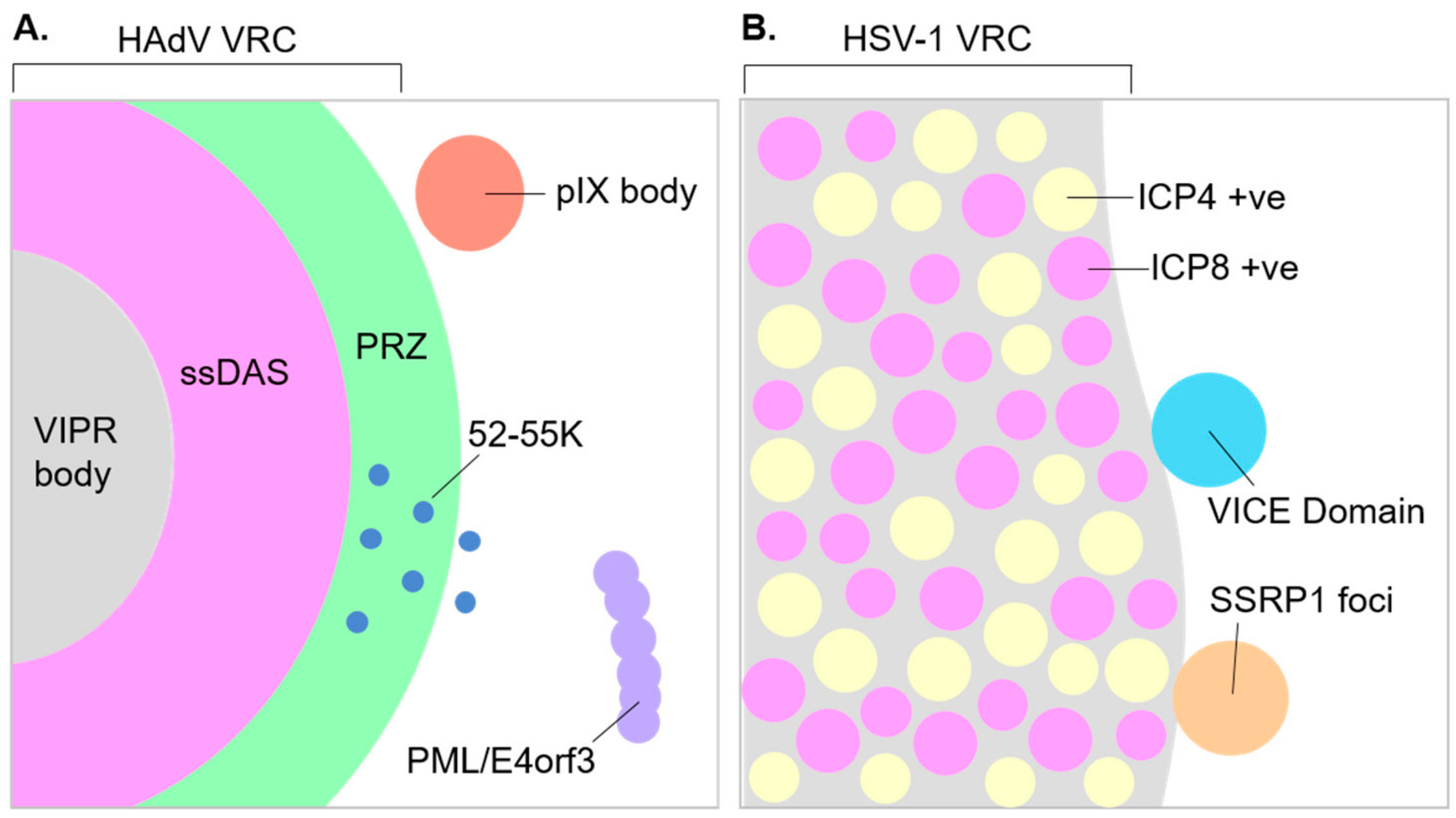{kind=link}
Abstract
1. Introduction
2. The Initiation and Formation of Replication Compartments
3. Interplay between Viral Replication Compartment Formation and Promyelocytic Leukemia Protein Nuclear Bodies
4. What Dictates the Number of Replication Compartments that Form in Each Infected Nucleus?
5. Viral Processes that Initiate Replication Compartment Formation
6. Biophysical Processes in Replication Compartment Formation
7. The Composition of Replication Compartments
8. Viral Processes that Take Place at Replication Compartments
9. DNA Replication
10. Transcription
11. Virion Production at Replication Compartments
12. Structure of Replication Centers and Organization of Viral Processes
13. Virus-Induced Assemblies Proximal to Replication Compartments
14. Conclusions and Future Perspectives
Funding
Acknowledgments
Conflicts of Interest
References
- Fay, N.; Panté, N. Nuclear entry of DNA viruses. Front. Microbiol. 2015, 6, 467. [Google Scholar] [CrossRef] [PubMed]
- Greber, U.F.; Fassati, A. Nuclear import of viral DNA genomes. Traffic 2003, 4, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, T.; Nagata, K.; Wodrich, H. The Role of Nuclear Antiviral Factors against Invading DNA Viruses: The Immediate Fate of Incoming Viral Genomes. Viruses 2016, 8, 290. [Google Scholar] [CrossRef] [PubMed]
- Diner, B.A.; Lum, K.K.; Cristea, I.M. The emerging role of nuclear viral DNA sensors. J. Biol. Chem. 2015, 290, 26412–26421. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Ni, G.; Damania, B. Innate Sensing of DNA Virus Genomes. Annu. Rev. Virol. 2018, 5, 341–362. [Google Scholar] [CrossRef] [PubMed]
- Everett, R.D. Interactions between DNA viruses, ND10 and the DNA damage response. Cell. Microbiol. 2006, 8, 365–374. [Google Scholar] [CrossRef]
- Weitzman, M.D.; Ornelles, D.A. Inactivating intracellular antiviral responses during adenovirus infection. Oncogene 2005, 24, 7686–7696. [Google Scholar] [CrossRef]
- Weitzman, M.D.; Fradet-Turcotte, A. Virus DNA Replication and the Host DNA Damage Response. Annu. Rev. Virol. 2018, 5, 141–164. [Google Scholar] [CrossRef]
- Pancholi, N.J.; Price, A.M.; Weitzman, M.D. Take your PIKK: tumour viruses and DNA damage response pathways. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2017, 372. [Google Scholar] [CrossRef]
- Luftig, M.A. Viruses and the DNA Damage Response: Activation and Antagonism. Annu. Rev. Virol. 2014, 1, 605–625. [Google Scholar] [CrossRef]
- Schmid, M.; Speiseder, T.; Dobner, T.; Gonzalez, R.A. DNA virus replication compartments. J. Virol. 2014, 88, 1404–1420. [Google Scholar] [CrossRef]
- De Bruyn Kops, A.; Knipe, D.M. Preexisting nuclear architecture defines the intranuclear location of herpesvirus DNA replication structures. J. Virol. 1994, 68, 3512–3526. [Google Scholar] [CrossRef]
- Jiang, M.; Entezami, P.; Gamez, M.; Stamminger, T.; Imperiale, M.J. Functional reorganization of promyelocytic leukemia nuclear bodies during BK virus infection. MBio 2011, 2, e00281-10. [Google Scholar] [CrossRef] [PubMed]
- Charman, M.; Herrmann, C.; Weitzman, M.D. Viral and Cellular Interactions During Adenovirus DNA Replication. FEBS Lett. 2019, 593, 3531–3550. [Google Scholar] [CrossRef] [PubMed]
- Strang, B.L. Viral and cellular subnuclear structures in human cytomegalovirus-infected cells. J. Gen. Virol. 2015, 96, 239–253. [Google Scholar] [CrossRef] [PubMed]
- Ishov, A.M.; Maul, G.G. The periphery of nuclear domain 10 (ND10) as site of DNA virus deposition. J. Cell Biol. 1996, 134, 815–826. [Google Scholar] [CrossRef] [PubMed]
- Doucas, V.; Ishov, A.M.; Romo, A.; Juguilon, H.; Weitzman, M.D.; Evans, R.M.; Maul, G.G. Adenovirus replication is coupled with the dynamic properties of the PML nuclear structure. Genes Dev. 1996, 10, 196–207. [Google Scholar] [CrossRef] [PubMed]
- Reyes, E.D.; Kulej, K.; Pancholi, N.J.; Akhtar, L.N.; Avgousti, D.C.; Kim, E.T.; Bricker, D.K.; Spruce, L.A.; Koniski, S.A.; Seeholzer, S.H.; et al. Identifying Host Factors Associated with DNA Replicated During Virus Infection. Mol. Cell Proteomics 2017, 16, 2079–2097. [Google Scholar] [CrossRef]
- Young, P.J.; Jensen, K.T.; Burger, L.R.; Pintel, D.J.; Lorson, C.L. Minute virus of mice NS1 interacts with the SMN protein, and they colocalize in novel nuclear bodies induced by parvovirus infection. J. Virol. 2002, 76, 3892–3904. [Google Scholar] [CrossRef]
- Bashir, T.; Rommelaere, J.; Cziepluch, C. In vivo accumulation of cyclin A and cellular replication factors in autonomous parvovirus minute virus of mice-associated replication bodies. J. Virol. 2001, 75, 4394–4398. [Google Scholar] [CrossRef]
- Cziepluch, C.; Lampel, S.; Grewenig, A.; Grund, C.; Lichter, P.; Rommelaere, J. H-1 parvovirus-associated replication bodies: a distinct virus-induced nuclear structure. J. Virol. 2000, 74, 4807–4815. [Google Scholar] [CrossRef] [PubMed]
- Lallemand-Breitenbach, V.; de Thé, H. PML nuclear bodies: from architecture to function. Curr. Opin. Cell Biol. 2018, 52, 154–161. [Google Scholar] [CrossRef] [PubMed]
- Scherer, M.; Stamminger, T. Emerging Role of PML Nuclear Bodies in Innate Immune Signaling. J. Virol. 2016, 90, 5850–5854. [Google Scholar] [CrossRef] [PubMed]
- Everett, R.D.; Chelbi-Alix, M.K. PML and PML nuclear bodies: implications in antiviral defence. Biochimie 2007, 89, 819–830. [Google Scholar] [CrossRef]
- Everett, R.D. The spatial organization of DNA virus genomes in the nucleus. PLoS Pathog. 2013, 9, e1003386. [Google Scholar] [CrossRef]
- Everett, R.D.; Boutell, C.; Hale, B.G. Interplay between viruses and host sumoylation pathways. Nat. Rev. Microbiol. 2013, 11, 400–411. [Google Scholar] [CrossRef]
- Cuchet-Lourenço, D.; Boutell, C.; Lukashchuk, V.; Grant, K.; Sykes, A.; Murray, J.; Orr, A.; Everett, R.D. SUMO pathway dependent recruitment of cellular repressors to herpes simplex virus type 1 genomes. PLoS Pathog. 2011, 7, e1002123. [Google Scholar] [CrossRef]
- Brown, J.R.; Conn, K.L.; Wasson, P.; Charman, M.; Tong, L.; Grant, K.; McFarlane, S.; Boutell, C. SUMO Ligase Protein Inhibitor of Activated STAT1 (PIAS1) Is a Constituent Promyelocytic Leukemia Nuclear Body Protein That Contributes to the Intrinsic Antiviral Immune Response to Herpes Simplex Virus 1. J. Virol. 2016, 90, 5939–5952. [Google Scholar] [CrossRef]
- Alandijany, T.; Roberts, A.P.E.; Conn, K.L.; Loney, C.; McFarlane, S.; Orr, A.; Boutell, C. Distinct temporal roles for the promyelocytic leukaemia (PML) protein in the sequential regulation of intracellular host immunity to HSV-1 infection. PLoS Pathog. 2018, 14, e1006769. [Google Scholar]
- Lukashchuk, V.; Everett, R.D. Regulation of ICP0-Null Mutant Herpes Simplex Virus Type 1 Infection by ND10 Components ATRX and hDaxx. J. Virol. 2010, 84, 4026–4040. [Google Scholar] [CrossRef]
- Alandijany, T. Host Intrinsic and Innate Intracellular Immunity During Herpes Simplex Virus Type 1 (HSV-1) Infection. Front. Microbiol. 2019, 10, 2611. [Google Scholar] [CrossRef] [PubMed]
- Landolfo, S.; De Andrea, M.; Dell’Oste, V.; Gugliesi, F. Intrinsic host restriction factors of human cytomegalovirus replication and mechanisms of viral escape. World J. Virol. 2016, 5, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Tsai, K.; Chan, L.; Gibeault, R.; Conn, K.; Dheekollu, J.; Domsic, J.; Marmorstein, R.; Schang, L.M.; Lieberman, P.M. Viral reprogramming of the Daxx histone H3.3 chaperone during early Epstein-Barr virus infection. J. Virol. 2014, 88, 14350–14363. [Google Scholar] [CrossRef] [PubMed]
- Tsai, K.; Thikmyanova, N.; Wojcechowskyj, J.A.; Delecluse, H.-J.; Lieberman, P.M. EBV Tegument Protein BNRF1 Disrupts DAXX-ATRX to Activate Viral Early Gene Transcription. PLOS Pathog. 2011, 7, e1002376. [Google Scholar] [CrossRef] [PubMed]
- Everett, R.D.; Murray, J. ND10 components relocate to sites associated with herpes simplex virus type 1 nucleoprotein complexes during virus infection. J. Virol. 2005, 79, 5078–5089. [Google Scholar] [CrossRef] [PubMed]
- Lanfranca, M.P.; Mostafa, H.H.; Davido, D.J. HSV-1 ICP0: An E3 Ubiquitin Ligase That Counteracts Host Intrinsic and Innate Immunity. Cells 2014, 3, 438–454. [Google Scholar] [CrossRef]
- Smith, M.C.; Boutell, C.; Davido, D.J. HSV-1 ICP0: paving the way for viral replication. Future Virol. 2011, 6, 421–429. [Google Scholar] [CrossRef]
- Maul, G.G.; Guldner, H.H.; Spivack, J.G. Modification of discrete nuclear domains induced by herpes simplex virus type 1 immediate early gene 1 product (ICP0). J. Gen. Virol. 1993, 74, 2679–2690. [Google Scholar] [CrossRef]
- Boutell, C.; Sadis, S.; Everett, R.D. Herpes simplex virus type 1 immediate-early protein ICP0 and is isolated RING finger domain act as ubiquitin E3 ligases in vitro. J. Virol. 2002, 76, 841–850. [Google Scholar] [CrossRef]
- Boutell, C.; Everett, R.D. Regulation of alphaherpesvirus infections by the ICP0 family of proteins. J. Gen. Virol. 2013, 94, 465–481. [Google Scholar] [CrossRef]
- Parkinson, J.; Everett, R.D. Alphaherpesvirus proteins related to herpes simplex virus type 1 ICP0 affect cellular structures and proteins. J. Virol. 2000, 74, 10006–10017. [Google Scholar] [CrossRef] [PubMed]
- Reichelt, M.; Joubert, L.; Perrino, J.; Koh, A.L.; Phanwar, I.; Arvin, A.M. 3D reconstruction of VZV infected cell nuclei and PML nuclear cages by serial section array scanning electron microscopy and electron tomography. PLoS Pathog. 2012, 8, e1002740. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Oliver, S.L.; Sommer, M.; Rajamani, J.; Reichelt, M.; Arvin, A.M. Disruption of PML nuclear bodies is mediated by ORF61 SUMO-interacting motifs and required for varicella-zoster virus pathogenesis in skin. PLoS Pathog. 2011, 7, e1002157. [Google Scholar] [CrossRef] [PubMed]
- Orazio, N.I.; Naeger, C.M.; Karlseder, J.; Weitzman, M.D. The adenovirus E1b55K/E4orf6 complex induces degradation of the Bloom helicase during infection. J. Virol. 2011, 85, 1887–1892. [Google Scholar] [CrossRef] [PubMed]
- Schreiner, S.; Wimmer, P.; Sirma, H.; Everett, R.D.; Blanchette, P.; Groitl, P.; Dobner, T. Proteasome-dependent degradation of Daxx by the viral E1B-55K protein in human adenovirus-infected cells. J. Virol. 2010, 84, 7029–7038. [Google Scholar] [CrossRef]
- Schreiner, S.; Bürck, C.; Glass, M.; Groitl, P.; Wimmer, P.; Kinkley, S.; Mund, A.; Everett, R.D.; Dobner, T. Control of human adenovirus type 5 gene expression by cellular Daxx/ATRX chromatin-associated complexes. Nucleic Acids Res. 2013, 41, 3532–3550. [Google Scholar] [CrossRef]
- Leppard, K.N.; Everett, R.D. The adenovirus type 5 E1b 55K and E4 Orf3 proteins associate in infected cells and affect ND10 components. J. Gen. Virol. 1999, 80, 997–1008. [Google Scholar] [CrossRef]
- Carvalho, T.; Seeler, J.S.; Ohman, K.; Jordan, P.; Pettersson, U.; Akusjärvi, G.; Carmo-Fonseca, M.; Dejean, A. Targeting of adenovirus E1A and E4-ORF3 proteins to nuclear matrix-associated PML bodies. J. Cell Biol. 1995, 131, 45–56. [Google Scholar] [CrossRef]
- Stracker, T.H.; Lee, D.V.; Carson, C.T.; Araujo, F.D.; Ornelles, D.A.; Weitzman, M.D. Serotype-Specific Reorganization of the Mre11 Complex by Adenoviral E4orf3 Proteins. J. Virol. 2005, 79, 6664–6673. [Google Scholar] [CrossRef]
- Higginbotham, J.M.; O’Shea, C.C. Adenovirus E4-ORF3 Targets PIAS3 and Together with E1B-55K Remodels SUMO Interactions in the Nucleus and at Virus Genome Replication Domains. J. Virol. 2015, 89, 10260–10272. [Google Scholar] [CrossRef]
- Berscheminski, J.; Wimmer, P.; Brun, J.; Ip, W.H.; Groitl, P.; Horlacher, T.; Jaffray, E.; Hay, R.T.; Dobner, T.; Schreiner, S. Sp100 isoform-specific regulation of human adenovirus 5 gene expression. J. Virol. 2014, 88, 6076–6092. [Google Scholar] [CrossRef] [PubMed]
- Gasparovic, M.L.; Maginnis, M.S.; O’Hara, B.A.; Dugan, A.S.; Atwood, W.J. Modulation of PML protein expression regulates JCV infection. Virology 2009, 390, 279–288. [Google Scholar] [CrossRef]
- Tang, Q.; Bell, P.; Tegtmeyer, P.; Maul, G.G. Replication but not transcription of simian virus 40 DNA is dependent on nuclear domain 10. J. Virol. 2000, 74, 9694–9700. [Google Scholar] [CrossRef] [PubMed]
- Boichuk, S.; Hu, L.; Makielski, K.; Pandolfi, P.P.; Gjoerup, O.V. Functional connection between Rad51 and PML in homology-directed repair. PLoS ONE 2011, 6, e25814. [Google Scholar] [CrossRef] [PubMed]
- Erickson, K.D.; Bouchet-Marquis, C.; Heiser, K.; Szomolanyi-Tsuda, E.; Mishra, R.; Lamothe, B.; Hoenger, A.; Garcea, R.L. Virion assembly factories in the nucleus of polyomavirus-infected cells. PLoS Pathog. 2012, 8, e1002630. [Google Scholar] [CrossRef] [PubMed]
- Bienkowska-Haba, M.; Luszczek, W.; Keiffer, T.R.; Guion, L.G.M.; DiGiuseppe, S.; Scott, R.S.; Sapp, M. Incoming human papillomavirus 16 genome is lost in PML protein-deficient HaCaT keratinocytes. Cell. Microbiol. 2017, 19, e12708. [Google Scholar] [CrossRef] [PubMed]
- Day, P.M.; Baker, C.C.; Lowy, D.R.; Schiller, J.T. Establishment of papillomavirus infection is enhanced by promyelocytic leukemia protein (PML) expression. PNAS 2004, 101, 14252–14257. [Google Scholar] [CrossRef]
- Stepp, W.H.; Meyers, J.M.; McBride, A.A. Sp100 provides intrinsic immunity against human papillomavirus infection. MBio 2013, 4, e00845-13. [Google Scholar] [CrossRef]
- Guion, L.; Bienkowska-Haba, M.; DiGiuseppe, S.; Florin, L.; Sapp, M. PML nuclear body-residing proteins sequentially associate with HPV genome after infectious nuclear delivery. PLOS Pathog. 2019, 15, e1007590. [Google Scholar] [CrossRef]
- Stepp, W.H.; Stamos, J.D.; Khurana, S.; Warburton, A.; McBride, A.A. Sp100 colocalizes with HPV replication foci and restricts the productive stage of the infectious cycle. PLoS Pathog. 2017, 13, e1006660. [Google Scholar] [CrossRef]
- Kobiler, O.; Brodersen, P.; Taylor, M.P.; Ludmir, E.B.; Enquist, L.W. Herpesvirus replication compartments originate with single incoming viral genomes. MBio 2011, 2, e00278-11. [Google Scholar] [CrossRef] [PubMed]
- Tomer, E.; Cohen, E.M.; Drayman, N.; Afriat, A.; Weitzman, M.D.; Zaritsky, A.; Kobiler, O. Coalescing replication compartments provide the opportunity for recombination between coinfecting herpesviruses. FASEB J. 2019, 33, 9388–9403. [Google Scholar] [CrossRef] [PubMed]
- Sekine, E.; Schmidt, N.; Gaboriau, D.; O’Hare, P. Spatiotemporal dynamics of HSV genome nuclear entry and compaction state transitions using bioorthogonal chemistry and super-resolution microscopy. PLoS Pathog. 2017, 13, e1006721. [Google Scholar] [CrossRef] [PubMed]
- Dembowski, J.A.; DeLuca, N.A. Temporal Viral Genome-Protein Interactions Define Distinct Stages of Productive Herpesviral Infection. MBio 2018, 9, e01182-18. [Google Scholar] [CrossRef] [PubMed]
- Kobiler, O.; Lipman, Y.; Therkelsen, K.; Daubechies, I.; Enquist, L.W. Herpesviruses carrying a Brainbow cassette reveal replication and expression of limited numbers of incoming genomes. Nat Commun. 2010, 1, 146. [Google Scholar] [CrossRef] [PubMed]
- Taylor, T.J.; McNamee, E.E.; Day, C.; Knipe, D.M. Herpes simplex virus replication compartments can form by coalescence of smaller compartments. Virology 2003, 309, 232–247. [Google Scholar] [CrossRef]
- Phelan, A.; Dunlop, J.; Patel, A.H.; Stow, N.D.; Clements, J.B. Nuclear sites of herpes simplex virus type 1 DNA replication and transcription colocalize at early times postinfection and are largely distinct from RNA processing factors. J. Virol. 1997, 71, 1124–1132. [Google Scholar] [CrossRef]
- Maul, G.G. Nuclear domain 10, the site of DNA virus transcription and replication. Bioessays 1998, 20, 660–667. [Google Scholar] [CrossRef]
- Kobiler, O.; Weitzman, M.D. Herpes simplex virus replication compartments: From naked release to recombining together. PLoS Pathog. 2019, 15, e00278-11. [Google Scholar] [CrossRef]
- Lynch, K.L.; Gooding, L.R.; Garnett-Benson, C.; Ornelles, D.A.; Avgousti, D.C. Epigenetics and the dynamics of chromatin during adenovirus infections. FEBS Lett. 2019, 3551–3570. [Google Scholar] [CrossRef]
- Knipe, D.M.; Cliffe, A. Chromatin control of herpes simplex virus lytic and latent infection. Nat. Rev. Microbiol. 2008, 6, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Lieberman, P.M. Chromatin Organization and Virus Gene Expression. J. Cell Physiol. 2008, 216, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Imperiale, M.J. Design stars: how small DNA viruses remodel the host nucleus. Future Virol. 2012, 7, 445–459. [Google Scholar] [CrossRef] [PubMed]
- Avgousti, D.C.; Herrmann, C.; Kulej, K.; Pancholi, N.J.; Sekulic, N.; Petrescu, J.; Molden, R.C.; Blumenthal, D.; Paris, A.J.; Reyes, E.D.; et al. A core viral protein binds host nucleosomes to sequester immune danger signals. Nature 2016, 535, 173–177. [Google Scholar] [CrossRef] [PubMed]
- You, J. Papillomavirus interaction with cellular chromatin. Biochim. Biophys. Acta 2010, 1799, 192–199. [Google Scholar] [CrossRef]
- Heming, J.D.; Conway, J.F.; Homa, F.L. Herpesvirus capsid assembly and DNA packaging. Adv. Anat. Embryol. Cell Biol. 2017, 223, 119–142. [Google Scholar]
- Silva, L.; Cliffe, A.; Chang, L.; Knipe, D.M. Role for A-type lamins in herpesviral DNA targeting and heterochromatin modulation. PLoS Pathog. 2008, 4, e1000071. [Google Scholar] [CrossRef]
- Komatsu, T.; Nagata, K.; Wodrich, H. An Adenovirus DNA Replication Factor, but Not Incoming Genome Complexes, Targets PML Nuclear Bodies. J. Virol. 2016, 90, 1657–1667. [Google Scholar] [CrossRef]
- Hoeben, R.C.; Uil, T.G. Adenovirus DNA replication. Cold Spring Harb. Perspect. Biol. 2013, 5, a013003. [Google Scholar] [CrossRef]
- Weller, S.K.; Coen, D.M. Herpes Simplex Viruses: Mechanisms of DNA Replication. Cold Spring Harb. Perspect. Biol. 2012, 4, a013011. [Google Scholar] [CrossRef]
- McBride, A.A. Replication and Partitioning of Papillomavirus Genomes. Adv. Virus Res. 2008, 72, 155–205. [Google Scholar] [PubMed]
- Cotmore, S.F.; Tattersall, P. Parvoviruses: Small Does Not Mean Simple. Annu. Rev. Virol. 2014, 1, 517–537. [Google Scholar] [CrossRef] [PubMed]
- Fanning, E.; Zhao, K. SV40 DNA replication: From the A gene to a nanomachine. Virology 2009, 384, 352–359. [Google Scholar] [CrossRef] [PubMed]
- Carrington-Lawrence, S.D.; Weller, S.K. Recruitment of polymerase to herpes simplex virus type 1 replication foci in cells expressing mutant primase (UL52) proteins. J. Virol. 2003, 77, 4237–4247. [Google Scholar] [CrossRef]
- Burkham, J.; Coen, D.M.; Weller, S.K. ND10 protein PML is recruited to herpes simplex virus type 1 prereplicative sites and replication compartments in the presence of viral DNA polymerase. J. Virol. 1998, 72, 10100–10107. [Google Scholar] [CrossRef]
- Burkham, J.; Coen, D.M.; Hwang, C.B.; Weller, S.K. Interactions of herpes simplex virus type 1 with ND10 and recruitment of PML to replication compartments. J. Virol. 2001, 75, 2353–2367. [Google Scholar] [CrossRef]
- Shin, Y.; Brangwynne, C.P. Liquid phase condensation in cell physiology and disease. Science 2017, 357, eaaf4382. [Google Scholar] [CrossRef]
- Hyman, A.A.; Weber, C.A.; Jülicher, F. Liquid-liquid phase separation in biology. Annu. Rev. Cell Dev. Biol. 2014, 30, 39–58. [Google Scholar] [CrossRef]
- Alberti, S.; Gladfelter, A.; Mittag, T. Considerations and Challenges in Studying Liquid-Liquid Phase Separation and Biomolecular Condensates. Cell 2019, 176, 419–434. [Google Scholar] [CrossRef]
- Itakura, A.K.; Futia, R.A.; Jarosz, D.F. It Pays To Be in Phase. Biochemistry 2018, 57, 2520–2529. [Google Scholar] [CrossRef]
- Heinrich, B.S.; Maliga, Z.; Stein, D.A.; Hyman, A.A.; Whelan, S.P.J. Phase Transitions Drive the Formation of Vesicular Stomatitis Virus Replication Compartments. MBio 2018, 9, e02290-17. [Google Scholar] [CrossRef] [PubMed]
- Alenquer, M.; Vale-Costa, S.; Etibor, T.A.; Ferreira, F.; Sousa, A.L.; Amorim, M.J. Influenza A virus ribonucleoproteins form liquid organelles at endoplasmic reticulum exit sites. Nat. Commun. 2019, 10, 1629. [Google Scholar] [CrossRef] [PubMed]
- Salladini, E.; Debarnot, C.; Delauzun, V.; Murrali, M.G.; Sutto-Ortiz, P.; Spinelli, S.; Pierattelli, R.; Bignon, C.; Longhi, S. Phase transition and amyloid formation by a viral protein as an additional molecular mechanism of virus-induced cell toxicity. bioRxiv 2018, 497024. [Google Scholar]
- Zakaryan, H.; Stamminger, T. Nuclear remodelling during viral infections. Cell. Microbiol. 2011, 13, 806–813. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo, P.; Gonzalez, R.A. Formation of adenovirus DNA replication compartments. FEBS Lett. 2019, 593, 3518–3530. [Google Scholar] [CrossRef]
- Tamarozzi, E.R.; Giuliatti, S. Understanding the Role of Intrinsic Disorder of Viral Proteins in the Oncogenicity of Different Types of HPV. Int. J. Mol. Sci. 2018, 19, 198. [Google Scholar] [CrossRef]
- Pelka, P.; Ablack, J.N.G.; Fonseca, G.J.; Yousef, A.F.; Mymryk, J.S. Intrinsic structural disorder in adenovirus E1A: a viral molecular hub linking multiple diverse processes. J. Virol. 2008, 82, 7252–7263. [Google Scholar] [CrossRef]
- Sieber, T.; Scholz, R.; Spoerner, M.; Schumann, F.; Kalbitzer, H.R.; Dobner, T. Intrinsic disorder in the common N-terminus of human adenovirus 5 E1B-55K and its related E1BN proteins indicated by studies on E1B-93R. Virology 2011, 418, 133–143. [Google Scholar] [CrossRef]
- Xue, B.; Blocquel, D.; Habchi, J.; Uversky, A.V.; Kurgan, L.; Uversky, V.N.; Longhi, S. Structural disorder in viral proteins. Chem. Rev. 2014, 114, 6880–6911. [Google Scholar] [CrossRef]
- Xue, B.; Williams, R.W.; Oldfield, C.J.; Goh, G.K.-M.; Dunker, A.K.; Uversky, V.N. Viral disorder or disordered viruses: do viral proteins possess unique features? Protein Pept. Lett. 2010, 17, 932–951. [Google Scholar] [CrossRef]
- Pushker, R.; Mooney, C.; Davey, N.E.; Jacqué, J.-M.; Shields, D.C. Marked Variability in the Extent of Protein Disorder within and between Viral Families. PLOS ONE 2013, 8, e60724. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.; Godinez, W.J.; Kim, I.-H.; Tektonidis, M.; de Lanerolle, P.; Eils, R.; Rohr, K.; Knipe, D.M. Herpesviral replication compartments move and coalesce at nuclear speckles to enhance export of viral late mRNA. Proc. Natl. Acad. Sci. USA 2011, 108, 8539–8540. [Google Scholar] [CrossRef]
- Komatsu, T.; Quentin-Froignant, C.; Carlon-Andres, I.; Lagadec, F.; Rayne, F.; Ragues, J.; Kehlenbach, R.H.; Zhang, W.; Ehrhardt, A.; Bystricky, K.; et al. In Vivo Labelling of Adenovirus DNA Identifies Chromatin Anchoring and Biphasic Genome Replication. J. Virol. 2018, 92, e00795-18. [Google Scholar] [CrossRef]
- Penfold, M.E.; Mocarski, E.S. Formation of cytomegalovirus DNA replication compartments defined by localization of viral proteins and DNA synthesis. Virology 1997, 239, 46–61. [Google Scholar] [CrossRef] [PubMed]
- McSwiggen, D.T.; Hansen, A.S.; Teves, S.S.; Marie-Nelly, H.; Hao, Y.; Heckert, A.B.; Umemoto, K.K.; Dugast-Darzacq, C.; Tjian, R.; Darzacq, X. Evidence for DNA-mediated nuclear compartmentalization distinct from phase separation. Elife 2019, 8, e47098. [Google Scholar] [CrossRef]
- Uchil, P.D.; Satchidanandam, V. Architecture of the flaviviral replication complex. Protease, nuclease, and detergents reveal encasement within double-layered membrane compartments. J. Biol. Chem. 2003, 278, 24388–24398. [Google Scholar] [CrossRef] [PubMed]
- Mazzolini, L.; Bonneville, J.M.; Volovitch, M.; Magazin, M.; Yot, P. Strand-specific viral DNA synthesis in purified viroplasms isolated from turnip leaves infected with cauliflower mosaic virus. Virology 1985, 145, 293–303. [Google Scholar] [CrossRef]
- Paul, D.; Hoppe, S.; Saher, G.; Krijnse-Locker, J.; Bartenschlager, R. Morphological and biochemical characterization of the membranous hepatitis C virus replication compartment. J. Virol. 2013, 87, 10612–10627. [Google Scholar] [CrossRef] [PubMed]
- Green, K.Y.; Mory, A.; Fogg, M.H.; Weisberg, A.; Belliot, G.; Wagner, M.; Mitra, T.; Ehrenfeld, E.; Cameron, C.E.; Sosnovtsev, S.V. Isolation of enzymatically active replication complexes from feline calicivirus-infected cells. J. Virol. 2002, 76, 8582–8595. [Google Scholar] [CrossRef] [PubMed]
- Quinkert, D.; Bartenschlager, R.; Lohmann, V. Quantitative analysis of the hepatitis C virus replication complex. J. Virol. 2005, 79, 13594–13605. [Google Scholar] [CrossRef]
- Van Hemert, M.J.; de Wilde, A.H.; Gorbalenya, A.E.; Snijder, E.J. The in vitro RNA synthesizing activity of the isolated arterivirus replication/transcription complex is dependent on a host factor. J. Biol. Chem. 2008, 283, 16525–16536. [Google Scholar] [CrossRef]
- Van Hemert, M.J.; van den Worm, S.H.E.; Knoops, K.; Mommaas, A.M.; Gorbalenya, A.E.; Snijder, E.J. SARS-coronavirus replication/transcription complexes are membrane-protected and need a host factor for activity in vitro. PLoS Pathog. 2008, 4, e1000054. [Google Scholar] [CrossRef]
- Rincheval, V.; Lelek, M.; Gault, E.; Bouillier, C.; Sitterlin, D.; Blouquit-Laye, S.; Galloux, M.; Zimmer, C.; Eleouet, J.-F.; Rameix-Welti, M.-A. Functional organization of cytoplasmic inclusion bodies in cells infected by respiratory syncytial virus. Nat. Commun. 2017, 8, 563. [Google Scholar] [CrossRef]
- Röder, A.; Koschel, K. Virus-specific proteins associated with the replication complex of poliovirus RNA. J. Gen. Virol. 1975, 28, 85–98. [Google Scholar] [CrossRef]
- Hidalgo, P.; Gonzalez, R.A. Isolation of Viral Replication Compartment-enriched Sub-nuclear Fractions from Adenovirus-infected Normal Human Cells. J. Vis. Exp. 2015, e53296. [Google Scholar] [CrossRef]
- Hidalgo, P.; Anzures, L.; Hernández-Mendoza, A.; Guerrero, A.; Wood, C.D.; Valdés, M.; Dobner, T.; Gonzalez, R.A. Morphological, Biochemical, and Functional Study of Viral Replication Compartments Isolated from Adenovirus-Infected Cells. J. Virol. 2016, 90, 3411–3427. [Google Scholar] [CrossRef]
- Taylor, T.J.; Knipe, D.M. Proteomics of herpes simplex virus replication compartments: association of cellular DNA replication, repair, recombination, and chromatin remodeling proteins with ICP8. J. Virol. 2004, 78, 5856–5866. [Google Scholar] [CrossRef]
- Dembowski, J.A.; DeLuca, N.A. Selective Recruitment of Nuclear Factors to Productively Replicating Herpes Simplex Virus Genomes. PLOS Pathog. 2015, 11, e1004939. [Google Scholar] [CrossRef]
- Dembowski, J.A.; Deluca, N.A. Purification of Viral DNA for the Identification of Associated Viral and Cellular Proteins. J Vis Exp 2017, e56374. [Google Scholar] [CrossRef]
- Majumder, K.; Etingov, I.; Pintel, D.J. Protoparvovirus Interactions with the Cellular DNA Damage Response. Viruses 2017, 9, 323. [Google Scholar] [CrossRef]
- Weitzman, M.D.; Fisher, K.J.; Wilson, J.M. Recruitment of wild-type and recombinant adeno-associated virus into adenovirus replication centers. J. Virol. 1996, 70, 1845–1854. [Google Scholar] [CrossRef]
- Gautam, D.; Bridge, E. The Kinase Activity of Ataxia-Telangiectasia Mutated Interferes with Adenovirus E4 Mutant DNA Replication. J. Virol. 2013, 87, 8687–8696. [Google Scholar] [CrossRef] [PubMed]
- Evans, J.D.; Hearing, P. Relocalization of the Mre11-Rad50-Nbs1 complex by the adenovirus E4 ORF3 protein is required for viral replication. J. Virol. 2005, 79, 6207–6215. [Google Scholar] [CrossRef] [PubMed]
- Spriggs, C.C.; Laimins, L.A. Human Papillomavirus and the DNA Damage Response: Exploiting Host Repair Pathways for Viral Replication. Viruses 2017, 9, 232. [Google Scholar] [CrossRef] [PubMed]
- Justice, J.L.; Verhalen, B.; Jiang, M. Polyomavirus interaction with the DNA damage response. Virol. Sin. 2015, 30, 122–129. [Google Scholar] [CrossRef]
- Chen, R.; Wold, M.S. Replication Protein A: Single-stranded DNA’s first responder: Dynamic DNA-interactions allow Replication Protein A to direct single-strand DNA intermediates into different pathways for synthesis or repair. Bioessays 2014, 36, 1156–1161. [Google Scholar] [CrossRef]
- Ward, P.; Dean, F.B.; O’Donnell, M.E.; Berns, K.I. Role of the Adenovirus DNA-Binding Protein in In Vitro Adeno-Associated Virus DNA Replication. J. Virol. 1998, 72, 420–427. [Google Scholar] [CrossRef]
- Ni, T.H.; McDonald, W.F.; Zolotukhin, I.; Melendy, T.; Waga, S.; Stillman, B.; Muzyczka, N. Cellular proteins required for adeno-associated virus DNA replication in the absence of adenovirus coinfection. J. Virol. 1998, 72, 2777–2787. [Google Scholar] [CrossRef]
- Stracker, T.H.; Cassell, G.D.; Ward, P.; Loo, Y.-M.; van Breukelen, B.; Carrington-Lawrence, S.D.; Hamatake, R.K.; van der Vliet, P.C.; Weller, S.K.; Melendy, T.; et al. The Rep protein of adeno-associated virus type 2 interacts with single-stranded DNA-binding proteins that enhance viral replication. J. Virol. 2004, 78, 441–453. [Google Scholar] [CrossRef]
- Quinlan, M.P.; Chen, L.B.; Knipe, D.M. The intranuclear location of a herpes simplex virus DNA-binding protein is determined by the status of viral DNA replication. Cell 1984, 36, 857–868. [Google Scholar] [CrossRef]
- Puvion-Dutilleul, F.; Puvion, E. Replicating single-stranded adenovirus type 5 DNA molecules accumulate within well-delimited intranuclear areas of lytically infected HeLa cells. Eur. J. Cell Biol. 1990, 52, 379–388. [Google Scholar] [PubMed]
- Pombo, A.; Ferreira, J.; Bridge, E.; Carmo-Fonseca, M. Adenovirus replication and transcription sites are spatially separated in the nucleus of infected cells. EMBO J. 1994, 13, 5075–5085. [Google Scholar] [CrossRef]
- Murti, K.G.; Davis, D.S.; Kitchingman, G.R. Localization of adenovirus-encoded DNA replication proteins in the nucleus by immunogold electron microscopy. J. Gen. Virol. 1990, 71, 2847–2857. [Google Scholar] [CrossRef]
- Bosher, J.; Dawson, A.; Hay, R.T. Nuclear factor I is specifically targeted to discrete subnuclear sites in adenovirus type 2-infected cells. J. Virol. 1992, 66, 3140–3150. [Google Scholar] [CrossRef] [PubMed]
- Glauser, D.L.; Strasser, R.; Laimbacher, A.S.; Saydam, O.; Clément, N.; Linden, R.M.; Ackermann, M.; Fraefel, C. Live covisualization of competing adeno-associated virus and herpes simplex virus type 1 DNA replication: molecular mechanisms of interaction. J. Virol. 2007, 81, 4732–4743. [Google Scholar] [CrossRef]
- McBride, A.A. The papillomavirus E2 proteins. Virology 2013, 445, 57–79. [Google Scholar] [CrossRef] [PubMed]
- Heino, P.; Zhou, J.; Lambert, P.F. Interaction of the papillomavirus transcription/replication factor, E2, and the viral capsid protein, L2. Virology 2000, 276, 304–314. [Google Scholar] [CrossRef] [PubMed]
- Kadaja, M.; Isok-Paas, H.; Laos, T.; Ustav, E.; Ustav, M. Mechanism of genomic instability in cells infected with the high-risk human papillomaviruses. PLoS Pathog. 2009, 5, e1000397. [Google Scholar] [CrossRef] [PubMed]
- Sakakibara, N.; Chen, D.; Jang, M.K.; Kang, D.W.; Luecke, H.F.; Wu, S.-Y.; Chiang, C.-M.; McBride, A.A. Brd4 is displaced from HPV replication factories as they expand and amplify viral DNA. PLoS Pathog. 2013, 9, e1003777. [Google Scholar] [CrossRef]
- An, P.; Sáenz Robles, M.T.; Pipas, J.M. Large T antigens of polyomaviruses: amazing molecular machines. Annu. Rev. Microbiol. 2012, 66, 213–236. [Google Scholar] [CrossRef]
- De Bruyn Kops, A.; Knipe, D.M. Formation of DNA replication structures in herpes virus-infected cells requires a viral DNA binding protein. Cell 1988, 55, 857–868. [Google Scholar] [CrossRef]
- Wang, I.-H.; Suomalainen, M.; Andriasyan, V.; Kilcher, S.; Mercer, J.; Neef, A.; Luedtke, N.W.; Greber, U.F. Tracking viral genomes in host cells at single-molecule resolution. Cell Host Microbe 2013, 14, 468–480. [Google Scholar] [CrossRef] [PubMed]
- Anacker, D.C.; Gautam, D.; Gillespie, K.A.; Chappell, W.H.; Moody, C.A. Productive replication of human papillomavirus 31 requires DNA repair factor Nbs1. J. Virol. 2014, 88, 8528–8544. [Google Scholar] [CrossRef] [PubMed]
- Gillespie, K.A.; Mehta, K.P.; Laimins, L.A.; Moody, C.A. Human papillomaviruses recruit cellular DNA repair and homologous recombination factors to viral replication centers. J. Virol. 2012, 86, 9520–9526. [Google Scholar] [CrossRef]
- Komatsu, T.; Robinson, D.R.; Hisaoka, M.; Ueshima, S.; Okuwaki, M.; Nagata, K.; Wodrich, H. Tracking adenovirus genomes identifies morphologically distinct late DNA replication compartments. Traffic 2016, 17, 1168–1180. [Google Scholar] [CrossRef]
- Jul-Larsen, A.; Visted, T.; Karlsen, B.O.; Rinaldo, C.H.; Bjerkvig, R.; Lønning, P.E.; Bøe, S.O. PML-nuclear bodies accumulate DNA in response to polyomavirus BK and simian virus 40 replication. Exp. Cell Res. 2004, 298, 58–73. [Google Scholar] [CrossRef]
- Ahn, J.H.; Jang, W.J.; Hayward, G.S. The human cytomegalovirus IE2 and UL112-113 proteins accumulate in viral DNA replication compartments that initiate from the periphery of promyelocytic leukemia protein-associated nuclear bodies (PODs or ND10). J. Virol. 1999, 73, 10458–10471. [Google Scholar] [CrossRef]
- Yamamoto, T.; Suzuki, S.; Radsak, K.; Hirai, K. The UL112/113 gene products of human cytomegalovirus which colocalize with viral DNA in infected cell nuclei are related to efficient viral DNA replication. Virus Res. 1998, 56, 107–114. [Google Scholar] [CrossRef]
- Fox, H.L.; Dembowski, J.A.; DeLuca, N.A. A Herpesviral Immediate Early Protein Promotes Transcription Elongation of Viral Transcripts. MBio 2017, 8, e00745-17. [Google Scholar] [CrossRef]
- Everett, R.D.; Maul, G.G. HSV-1 IE protein Vmw110 causes redistribution of PML. EMBO J. 1994, 13, 5062–5069. [Google Scholar] [CrossRef]
- Everett, R.D.; Sourvinos, G.; Orr, A. Recruitment of herpes simplex virus type 1 transcriptional regulatory protein ICP4 into foci juxtaposed to ND10 in live, infected cells. J. Virol. 2003, 77, 3680–3689. [Google Scholar] [CrossRef] [PubMed]
- Day, P.M.; Roden, R.B.; Lowy, D.R.; Schiller, J.T. The papillomavirus minor capsid protein, L2, induces localization of the major capsid protein, L1, and the viral transcription/replication protein, E2, to PML oncogenic domains. J. Virol. 1998, 72, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Heiser, K.; Nicholas, C.; Garcea, R.L. Activation of DNA damage repair pathways by murine polyomavirus. Virology 2016, 497, 346–356. [Google Scholar] [CrossRef] [PubMed]
- James, N.J.; Howell, G.J.; Walker, J.H.; Blair, G.E. The role of Cajal bodies in the expression of late phase adenovirus proteins. Virology 2010, 399, 299–311. [Google Scholar] [CrossRef]
- Bridge, E.; Xia, D.X.; Carmo-Fonseca, M.; Cardinali, B.; Lamond, A.I.; Pettersson, U. Dynamic organization of splicing factors in adenovirus-infected cells. J. Virol. 1995, 69, 281–290. [Google Scholar] [CrossRef]
- Gama-Carvalho, M.; Condado, I.; Carmo-Fonseca, M. Regulation of adenovirus alternative RNA splicing correlates with a reorganization of splicing factors in the nucleus. Exp. Cell Res. 2003, 289, 77–85. [Google Scholar] [CrossRef]
- Puvion-Dutilleul, F.; Bachellerie, J.P.; Visa, N.; Puvion, E. Rearrangements of intranuclear structures involved in RNA processing in response to adenovirus infection. J. Cell. Sci. 1994, 107 Pt 6, 1457–1468. [Google Scholar]
- Feichtinger, S.; Stamminger, T.; Müller, R.; Graf, L.; Klebl, B.; Eickhoff, J.; Marschall, M. Recruitment of cyclin-dependent kinase 9 to nuclear compartments during cytomegalovirus late replication: importance of an interaction between viral pUL69 and cyclin T1. J. Gen. Virol. 2011, 92, 1519–1531. [Google Scholar] [CrossRef]
- Snaar, S.P.; Vincent, M.; Dirks, R.W. RNA polymerase II localizes at sites of human cytomegalovirus immediate-early RNA synthesis and processing. J. Histochem. Cytochem. 1999, 47, 245–254. [Google Scholar] [CrossRef]
- Jenkins, H.L.; Spencer, C.A. RNA polymerase II holoenzyme modifications accompany transcription reprogramming in herpes simplex virus type 1-infected cells. J. Virol. 2001, 75, 9872–9884. [Google Scholar] [CrossRef]
- Puvion-Dutilleul, F.; Puvion, E. Sites of transcription of adenovirus type 5 genomes in relation to early viral DNA replication in infected HeLa cells. A high resolution in situ hybridization and autoradiographical study. Biol. Cell 1991, 71, 135–147. [Google Scholar] [CrossRef]
- Shishido-Hara, Y.; Ichinose, S.; Higuchi, K.; Hara, Y.; Yasui, K. Major and minor capsid proteins of human polyomavirus JC cooperatively accumulate to nuclear domain 10 for assembly into virions. J. Virol. 2004, 78, 9890–9903. [Google Scholar] [CrossRef] [PubMed]
- Erickson, K.D.; Garcea, R.L. Viral replication centers and the DNA damage response in JC virus-infected cells. Virology 2019, 528, 198–206. [Google Scholar] [CrossRef] [PubMed]
- Florin, L.; Schäfer, F.; Sotlar, K.; Streeck, R.E.; Sapp, M. Reorganization of nuclear domain 10 induced by papillomavirus capsid protein l2. Virology 2002, 295, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Condezo, G.N.; San Martín, C. Localization of adenovirus morphogenesis players, together with visualization of assembly intermediates and failed products, favor a model where assembly and packaging occur concurrently at the periphery of the replication center. PLoS Pathog. 2017, 13, e1006320. [Google Scholar] [CrossRef]
- Ahi, Y.S.; Mittal, S.K. Components of Adenovirus Genome Packaging. Front. Microbiol. 2016, 7, 1503. [Google Scholar] [CrossRef]
- Hasson, T.B.; Soloway, P.D.; Ornelles, D.A.; Doerfler, W.; Shenk, T. Adenovirus L1 52- and 55-kilodalton proteins are required for assembly of virions. J. Virol. 1989, 63, 3612–3621. [Google Scholar] [CrossRef]
- Ostapchuk, P.; Hearing, P. Control of adenovirus packaging. J. Cell. Biochem. 2005, 96, 25–35. [Google Scholar] [CrossRef]
- Ruhge, L.L.; Huet, A.G.E.; Conway, J.F.; Smith, G.A. The Apical Region of the Herpes Simplex Virus Major Capsid Protein Promotes Capsid Maturation. J. Virol. 2018, 92, e00821-18. [Google Scholar] [CrossRef]
- Lamberti, C.; Weller, S.K. The herpes simplex virus type 1 cleavage/packaging protein, UL32, is involved in efficient localization of capsids to replication compartments. J. Virol. 1998, 72, 2463–2473. [Google Scholar] [CrossRef]
- Albright, B.S.; Kosinski, A.; Szczepaniak, R.; Cook, E.A.; Stow, N.D.; Conway, J.F.; Weller, S.K. The putative herpes simplex virus 1 chaperone protein UL32 modulates disulfide bond formation during infection. J. Virol. 2015, 89, 443–453. [Google Scholar] [CrossRef] [PubMed]
- De Bruyn Kops, A.; Uprichard, S.L.; Chen, M.; Knipe, D.M. Comparison of the intranuclear distributions of herpes simplex virus proteins involved in various viral functions. Virology 1998, 252, 162–178. [Google Scholar] [CrossRef] [PubMed]
- Giesen, K.; Radsak, K.; Bogner, E. Targeting of the gene product encoded by ORF UL56 of human cytomegalovirus into viral replication centers. FEBS Lett. 2000, 471, 215–218. [Google Scholar] [CrossRef]
- Li, Z.; Fang, C.; Su, Y.; Liu, H.; Lang, F.; Li, X.; Chen, G.; Lu, D.; Zhou, J. Visualizing the replicating HSV-1 virus using STED super-resolution microscopy. Virol. J. 2016, 13, 65. [Google Scholar] [CrossRef]
- Strang, B.L.; Boulant, S.; Chang, L.; Knipe, D.M.; Kirchhausen, T.; Coen, D.M. Human cytomegalovirus UL44 concentrates at the periphery of replication compartments, the site of viral DNA synthesis. J. Virol. 2012, 86, 2089–2095. [Google Scholar] [CrossRef]
- Genoveso, M.J.; Hisaoka, M.; Komatsu, T.; Wodrich, H.; Nagata, K.; Okuwaki, M. Formation of adenovirus DNA replication compartments and viral DNA accumulation sites by host chromatin regulatory proteins including NPM1. FEBS J. 2019, 205–217. [Google Scholar] [CrossRef]
- Mostafa, H.H.; Davido, D.J. Herpes simplex virus 1 ICP22 but not US 1.5 is required for efficient acute replication in mice and VICE domain formation. J. Virol. 2013, 87, 13510–13519. [Google Scholar] [CrossRef]
- Bastian, T.W.; Livingston, C.M.; Weller, S.K.; Rice, S.A. Herpes simplex virus type 1 immediate-early protein ICP22 is required for VICE domain formation during productive viral infection. J. Virol. 2010, 84, 2384–2394. [Google Scholar] [CrossRef]
- Burch, A.D.; Weller, S.K. Nuclear sequestration of cellular chaperone and proteasomal machinery during herpes simplex virus type 1 infection. J. Virol. 2004, 78, 7175–7185. [Google Scholar] [CrossRef]
- Livingston, C.M.; Ifrim, M.F.; Cowan, A.E.; Weller, S.K. Virus-Induced Chaperone-Enriched (VICE) domains function as nuclear protein quality control centers during HSV-1 infection. PLoS Pathog. 2009, 5, e1000619. [Google Scholar] [CrossRef]
- Li, L.; Johnson, L.A.; Dai-Ju, J.Q.; Sandri-Goldin, R.M. Hsc70 focus formation at the periphery of HSV-1 transcription sites requires ICP27. PLoS ONE 2008, 3, e1491. [Google Scholar] [CrossRef]
- Su Hui Teo, C.; Serwa, R.A.; O’Hare, P. Spatial and Temporal Resolution of Global Protein Synthesis during HSV Infection Using Bioorthogonal Precursors and Click Chemistry. PLoS Pathog. 2016, 12, e1005927. [Google Scholar] [CrossRef]
- Tran, K.; Mahr, J.A.; Spector, D.H. Proteasome subunits relocalize during human cytomegalovirus infection, and proteasome activity is necessary for efficient viral gene transcription. J. Virol. 2010, 84, 3079–3093. [Google Scholar] [CrossRef]
- Rosa-Calatrava, M.; Grave, L.; Puvion-Dutilleul, F.; Chatton, B.; Kedinger, C. Functional analysis of adenovirus protein IX identifies domains involved in capsid stability, transcriptional activity, and nuclear reorganization. J. Virol. 2001, 75, 7131–7141. [Google Scholar] [CrossRef]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Charman, M.; Weitzman, M.D. Replication Compartments of DNA Viruses in the Nucleus: Location, Location, Location. Viruses 2020, 12, 151. https://doi.org/10.3390/v12020151
Charman M, Weitzman MD. Replication Compartments of DNA Viruses in the Nucleus: Location, Location, Location. Viruses. 2020; 12(2):151. https://doi.org/10.3390/v12020151
Chicago/Turabian StyleCharman, Matthew, and Matthew D. Weitzman. 2020. "Replication Compartments of DNA Viruses in the Nucleus: Location, Location, Location" Viruses 12, no. 2: 151. https://doi.org/10.3390/v12020151
APA StyleCharman, M., & Weitzman, M. D. (2020). Replication Compartments of DNA Viruses in the Nucleus: Location, Location, Location. Viruses, 12(2), 151. https://doi.org/10.3390/v12020151