Emerging PCR-Based Techniques to Study HIV-1 Reservoir Persistence

 , and
, and
Abstract
1. Introduction
2. The Landscape of PCR-Based Methods to Asses HIV-1 Proviruses
3. Subgenomic Methods
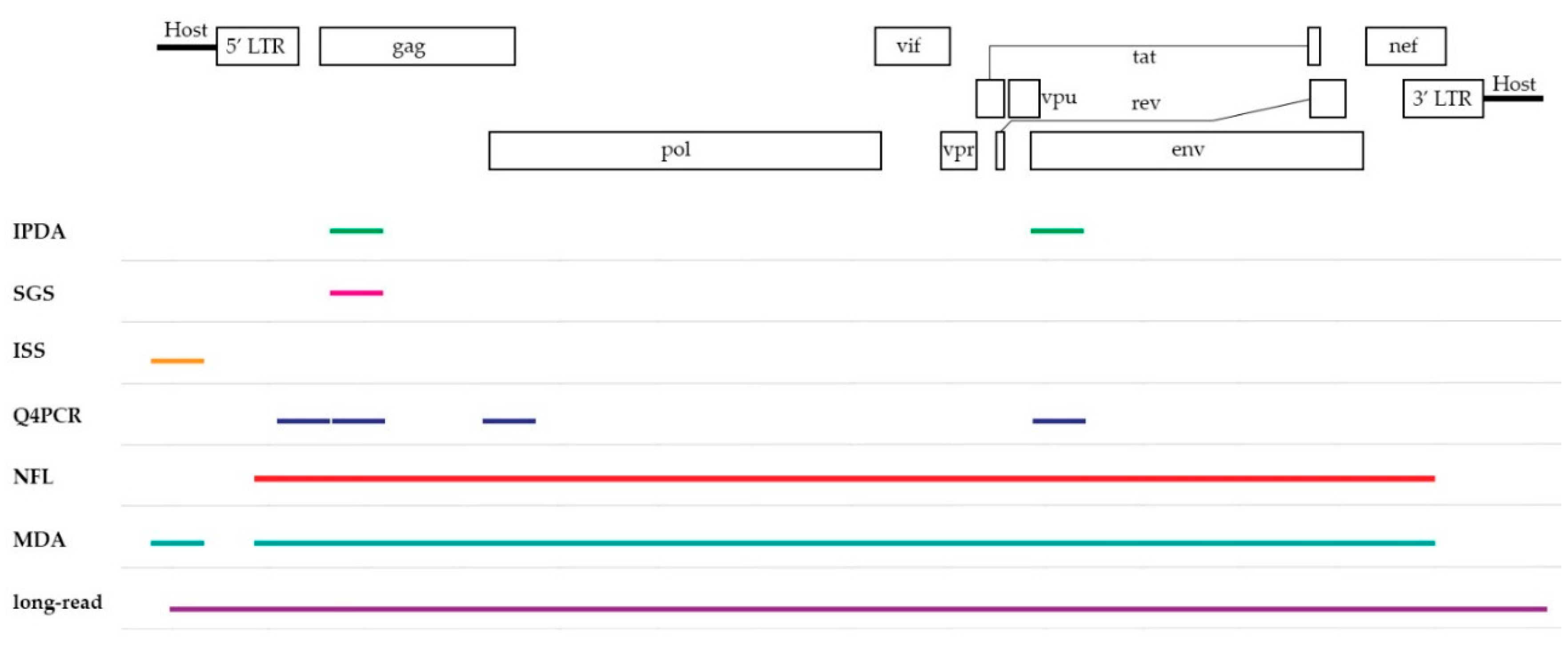
3.1. Intact Proviral DNA Assay (IPDA)
3.2. Single-Genome/Proviral Sequencing (SGS)
3.3. Integration Site Sequencing (ISS)
4. Full-Length Methods
4.1. Near Full-Length (NFL) Sequencing
4.2. Quadruplex qPCR (Q4PCR)
4.3. Multiple-Displacement Amplification (MDA)-Based Techniques
5. Discussion and Future Perspectives
5.1. Shift from Partial to Complete Sequence Information
5.2. Pitfalls of PCR-Based Sequencing
5.3. Need for Standardized Data Analysis
5.4. Promising Technology on the Horizon: Long-Read Sequencing
5.5. Functional Confirmation of Replication-Competent HIV-1 and Beyond
6. Closing Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Barré-Sinoussi, F.; Ross, A.L.; Delfraissy, J.-F. Past, present and future: 30 years of HIV research. Nat. Rev. Microbiol. 2013, 11, 877–883. [Google Scholar] [CrossRef] [PubMed]
- Kulpa, D.A.; Chomont, N. HIV persistence in the setting of antiretroviral therapy: When, where and how does HIV hide? J. Virus Erad. 2015, 1, 59–66. [Google Scholar]
- Wang, Z.; Simonetti, F.R.; Siliciano, R.F.; Laird, G.M. Measuring replication competent HIV-1: Advances and challenges in defining the latent reservoir. Retrovirology 2018, 15, 21. [Google Scholar] [CrossRef]
- Baxter, A.E.; O’Doherty, U.; Kaufmann, D.E. Beyond the replication-competent HIV reservoir: Transcription and translation-competent reservoirs. Retrovirology 2018, 15, 18. [Google Scholar] [CrossRef] [PubMed]
- Siliciano, J.D.; Siliciano, R.F. Enhanced culture assay for detection and quantitation of latently infected, resting CD4+ T-cells carrying replication-competent virus in HIV-1-infected individuals. Methods Mol. Biol. 2005, 304, 3–15. [Google Scholar] [PubMed]
- Procopio, F.A.; Fromentin, R.; Kulpa, D.A.; Brehm, J.H.; Bebin, A.-G.; Strain, M.C.; Richman, D.D.; O’Doherty, U.; Palmer, S.; Hecht, F.M.; et al. A Novel Assay to Measure the Magnitude of the Inducible Viral Reservoir in HIV-infected Individuals. EBioMedicine 2015, 2, 874–883. [Google Scholar] [CrossRef]
- Pardons, M.; Baxter, A.E.; Massanella, M.; Pagliuzza, A.; Fromentin, R.; Dufour, C.; Leyre, L.; Routy, J.-P.; Kaufmann, D.E.; Chomont, N. Single-cell characterization and quantification of translation-competent viral reservoirs in treated and untreated HIV infection. PLoS Pathog. 2019, 15, e1007619. [Google Scholar] [CrossRef]
- Eriksson, S.; Graf, E.H.; Dahl, V.; Strain, M.C.; Yukl, S.A.; Lysenko, E.S.; Bosch, R.J.; Lai, J.; Chioma, S.; Emad, F.; et al. Comparative Analysis of Measures of Viral Reservoirs in HIV-1 Eradication Studies. PLoS Pathog. 2013, 9, e1003174. [Google Scholar] [CrossRef]
- Bruner, K.M.; Wang, Z.; Simonetti, F.R.; Bender, A.M.; Kwon, K.J.; Sengupta, S.; Fray, E.J.; Beg, S.A.; Antar, A.A.R.; Jenike, K.M.; et al. A quantitative approach for measuring the reservoir of latent HIV-1 proviruses. Nature 2019, 1. [Google Scholar] [CrossRef]
- Palmer, S.; Kearney, M.; Maldarelli, F.; Halvas, E.K.; Bixby, C.J.; Bazmi, H.; Rock, D.; Falloon, J.; Davey, R.T.; Dewar, R.L.; et al. Multiple, Linked Human Immunodeficiency Virus Type 1 Drug Resistance Mutations in Treatment-Experienced Patients Are Missed by Standard Genotype Analysis. J. Clin. Microbiol. 2005, 43, 406–413. [Google Scholar] [CrossRef]
- Josefsson, L.; Palmer, S.; Faria, N.R.; Lemey, P.; Casazza, J.; Ambrozak, D.; Kearney, M.; Shao, W.; Kottilil, S.; Sneller, M.; et al. Single Cell Analysis of Lymph Node Tissue from HIV-1 Infected Patients Reveals that the Majority of CD4+ T-cells Contain One HIV-1 DNA Molecule. PLoS Pathog. 2013, 9. [Google Scholar] [CrossRef] [PubMed]
- Maldarelli, F.; Wu, X.; Su, L.; Simonetti, F.R.; Shao, W.; Hill, S.; Spindler, J.; Ferris, A.L.; Mellors, J.W.; Kearney, M.F.; et al. Specific HIV integration sites are linked to clonal expansion and persistence of infected cells. Science 2014, 345, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Wagner, T.A.; McLaughlin, S.; Garg, K.; Cheung, C.Y.K.; Larsen, B.B.; Styrchak, S.; Huang, H.C.; Edlefsen, P.T.; Mullins, J.I.; Frenkel, L.M. Proliferation of cells with HIV integrated into cancer genes contributes to persistent infection. Science (New York, N.Y.) 2014, 345, 570–573. [Google Scholar] [CrossRef]
- Ho, Y.-C.; Shan, L.; Hosmane, N.N.; Wang, J.; Laskey, S.B.; Rosenbloom, D.I.S.; Lai, J.; Blankson, J.N.; Siliciano, J.D.; Siliciano, R.F. Replication-competent noninduced proviruses in the latent reservoir increase barrier to HIV-1 cure. Cell 2013, 155, 540–551. [Google Scholar] [CrossRef] [PubMed]
- Hiener, B.; Horsburgh, B.A.; Eden, J.-S.; Barton, K.; Schlub, T.E.; Lee, E.; von Stockenstrom, S.; Odevall, L.; Milush, J.M.; Liegler, T.; et al. Identification of Genetically Intact HIV-1 Proviruses in Specific CD4+ T Cells from Effectively Treated Participants. Cell Rep. 2017, 21, 813–822. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.Q.; Orlova-Fink, N.; Einkauf, K.; Chowdhury, F.Z.; Sun, X.; Harrington, S.; Kuo, H.H.; Hua, S.; Chen, H.R.; Ouyang, Z.; et al. Clonal expansion of genome-intact HIV-1 in functionally polarized Th1 CD4+T cells. J. Clin. Investig. 2017, 127. [Google Scholar] [CrossRef]
- Gaebler, C.; Lorenzi, J.C.C.; Oliveira, T.Y.; Nogueira, L.; Ramos, V.; Lu, C.-L.; Pai, J.A.; Mendoza, P.; Jankovic, M.; Caskey, M.; et al. Combination of quadruplex qPCR and next-generation sequencing for qualitative and quantitative analysis of the HIV-1 latent reservoir. J. Exp. Med. 2019, jem.20190896. [Google Scholar] [CrossRef]
- Einkauf, K.B.; Lee, G.Q.; Gao, C.; Sharaf, R.; Sun, X.; Hua, S.; Chen, S.M.Y.; Jiang, C.; Lian, X.; Chowdhury, F.Z.; et al. Intact HIV-1 proviruses accumulate at distinct chromosomal positions during prolonged antiretroviral therapy. J. Clin. Investig. 2019, 129. [Google Scholar] [CrossRef]
- Patro, S.C.; Brandt, L.D.; Bale, M.J.; Halvas, E.K.; Joseph, K.W.; Shao, W.; Wu, X.; Guo, S.; Murrell, B.; Wiegand, A.; et al. Combined HIV-1 sequence and integration site analysis informs viral dynamics and allows reconstruction of replicating viral ancestors. Proc. Natl. Acad. Sci. USA 2019, 116, 25891–25899. [Google Scholar] [CrossRef]
- Artesi, M.; Hahaut, V.; Cole, B.; Lambrechts, L.; Ashrafi, F.; Marçais, A.; Hermine, O.; Griebel, P.; Arsic, N.; van der Meer, F.; et al. Pooled CRISPR Inverse PCR sequencing (PCIP-seq): Simultaneous sequencing of retroviral insertion points and the integrated provirus with long reads. bioRxiv 2019, 558130. [Google Scholar]
- Bender, A.M.; Simonetti, F.R.; Kumar, M.R.; Fray, E.J.; Bruner, K.M.; Timmons, A.E.; Tai, K.Y.; Jenike, K.M.; Antar, A.A.R.; Liu, P.-T.; et al. The Landscape of Persistent Viral Genomes in ART-Treated SIV, SHIV, and HIV-2 Infections. Cell Host Microbe 2019, 26, 73–85.e4. [Google Scholar] [CrossRef] [PubMed]
- De Scheerder, M.-A.; Vrancken, B.; Dellicour, S.; Schlub, T.; Lee, E.; Shao, W.; Rutsaert, S.; Verhofstede, C.; Kerre, T.; Malfait, T.; et al. HIV Rebound Is Predominantly Fueled by Genetically Identical Viral Expansions from Diverse Reservoirs. Cell Host Microbe 2019, 26, 347–358.e7. [Google Scholar] [CrossRef] [PubMed]
- Josefsson, L.; von Stockenstrom, S.; Faria, N.R.; Sinclair, E.; Bacchetti, P.; Killian, M.; Epling, L.; Tan, A.; Ho, T.; Lemey, P.; et al. The HIV-1 reservoir in eight patients on long-term suppressive antiretroviral therapy is stable with few genetic changes over time. Proc. Natl. Acad. Sci. USA 2013, 110, E4987–E4996. [Google Scholar] [CrossRef] [PubMed]
- Laskey, S.B.; Pohlmeyer, C.W.; Bruner, K.M.; Siliciano, R.F. Evaluating Clonal Expansion of HIV-Infected Cells: Optimization of PCR Strategies to Predict Clonality. PLoS Pathog. 2016, 12, e1005689. [Google Scholar] [CrossRef]
- Cesana, D.; Santoni de Sio, F.R.; Rudilosso, L.; Gallina, P.; Calabria, A.; Beretta, S.; Merelli, I.; Bruzzesi, E.; Passerini, L.; Nozza, S.; et al. HIV-1-mediated insertional activation of STAT5B and BACH2 trigger viral reservoir in T regulatory cells. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef]
- Pinzone, M.R.; VanBelzen, D.J.; Weissman, S.; Bertuccio, M.P.; Cannon, L.; Venanzi-Rullo, E.; Migueles, S.; Jones, R.B.; Mota, T.; Joseph, S.B.; et al. Longitudinal HIV sequencing reveals reservoir expression leading to decay which is obscured by clonal expansion. Nat. Commun. 2019, 10, 728. [Google Scholar] [CrossRef]
- Bruner, K.M.; Murray, A.J.; Pollack, R.A.; Soliman, M.G.; Laskey, S.B.; Capoferri, A.A.; Lai, J.; Strain, M.C.; Lada, S.M.; Hoh, R.; et al. Defective proviruses rapidly accumulate during acute HIV-1 infection. Nat. Med. 2016, 22, 1043–1049. [Google Scholar] [CrossRef]
- Lu, C.-L.; Pai, J.A.; Nogueira, L.; Mendoza, P.; Gruell, H.; Oliveira, T.Y.; Barton, J.; Lorenzi, J.C.C.; Cohen, Y.Z.; Cohn, L.B.; et al. Relationship between intact HIV-1 proviruses in circulating CD4+ T cells and rebound viruses emerging during treatment interruption. Proc. Natl. Acad. Sci. USA 2018, 115, E11341–E11348. [Google Scholar] [CrossRef]
- Lee, G.Q.; Reddy, K.; Einkauf, K.B.; Gounder, K.; Chevalier, J.M.; Dong, K.L.; Walker, B.D.; Yu, X.G.; Ndung’u, T.; Lichterfeld, M. HIV-1 DNA sequence diversity and evolution during acute subtype C infection. Nat. Commun. 2019, 10, 2737. [Google Scholar] [CrossRef]
- Rutsaert, S.; Bosman, K.; Trypsteen, W.; Nijhuis, M.; Vandekerckhove, L. Digital PCR as a tool to measure HIV persistence. Retrovirology 2018, 15, 16. [Google Scholar] [CrossRef]
- Kinloch, N.; Ren, Y.; Conce Alberto, W.; Dong, W.; Huang, S.H.; Wilson, A.; Mota, T.; Kikby, D.; Del Rio Estrada, P.M.; Brumme, C.J.; et al. Intra- and inter-individual HIV diversity limits the application of the intact proviral detection assay (IPDA). J. Virus Erad. 2019, 5 (Suppl. S3). [Google Scholar]
- Global HIV & AIDS Statistics — 2018 Fact Sheet | UNAIDS. Available online: https://www.unaids.org/en/resources/fact-sheet (accessed on 19 December 2019).
- Howison, M.; Coetzer, M.; Kantor, R. Measurement error and variant-calling in deep Illumina sequencing of HIV. Bioinformatics 2019, 35, 2029–2035. [Google Scholar] [CrossRef]
- Wymant, C.; Blanquart, F.; Golubchik, T.; Gall, A.; Bakker, M.; Bezemer, D.; Croucher, N.J.; Hall, M.; Hillebregt, M.; Ong, S.H.; et al. Easy and accurate reconstruction of whole HIV genomes from short-read sequence data with shiver. Virus Evol. 2018, 4, vey007. [Google Scholar] [CrossRef] [PubMed]
- Bonsall, D.; Golubchik, T.; de Cesare, M.; Limbada, M.; Kosloff, B.; MacIntyre-Cockett, G.; Hall, M.; Wymant, C.; Ansari, A.; Abeler-Dorner, L.; et al. A comprehensive genomics solution for HIV surveillance and clinical monitoring in a global health setting. BioRxiv 2018, 397083. [Google Scholar] [CrossRef]
- McNaughton, A.L.; Roberts, H.E.; Bonsall, D.; de Cesare, M.; Mokaya, J.; Lumley, S.F.; Golubchik, T.; Piazza, P.; Martin, J.B.; de Lara, C.; et al. Illumina and Nanopore methods for whole genome sequencing of hepatitis B virus (HBV). Sci. Rep. 2019, 9, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Hakre, S.; Chavez, L.; Shirakawa, K.; Verdin, E. Epigenetic regulation of HIV latency. Curr. Opin. HIV AIDS 2011, 6. [Google Scholar] [CrossRef]
- Van Lint, C.; Bouchat, S.; Marcello, A. HIV-1 transcription and latency: An update. Retrovirology 2013, 10, 67. [Google Scholar] [CrossRef]
- Weber, S.; Weiser, B.; Kemal, K.S.; Burger, H.; Ramirez, C.M.; Korn, K.; Anastos, K.; Kaul, R.; Kovacs, C.; Doerfler, W. Epigenetic analysis of HIV-1 proviral genomes from infected individuals: Predominance of unmethylated CpG’s. Virology 2014, 449, 181–189. [Google Scholar] [CrossRef][Green Version]
- Trejbalová, K.; Kovářová, D.; Blažková, J.; Machala, L.; Jilich, D.; Weber, J.; Kučerová, D.; Vencálek, O.; Hirsch, I.; Hejnar, J. Development of 5′ LTR DNA methylation of latent HIV-1 provirus in cell line models and in long-term-infected individuals. Clin. Epigenetics 2016, 8, 19. [Google Scholar] [CrossRef]
- Khan, S.; Iqbal, M.; Tariq, M.; Baig, S.M.; Abbas, W. Epigenetic regulation of HIV-1 latency: Focus on polycomb group (PcG) proteins. Clin. Epigenetics 2018, 10, 14. [Google Scholar] [CrossRef]
- Lucic, B.; Chen, H.-C.; Kuzman, M.; Zorita, E.; Wegner, J.; Minneker, V.; Wang, W.; Fronza, R.; Laufs, S.; Schmidt, M.; et al. Spatially clustered loci with multiple enhancers are frequent targets of HIV-1 integration. Nat. Commun. 2019, 1, 1. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Assay | Assay Overview | Aspect | Advantage | Limitations | Key References | |
|---|---|---|---|---|---|---|
| Subgenomic coverage | IPDA | ddPCR using two assays targeting subgenomic regions | Info on intactness and hypermutation | High-throughput Easy set-up Fast Quantification, better estimation than total HIV-1 DNA | Prone to overestimation No info on (full-length) sequence | [9] |
| SGS | Sequencing at single-genome level | Info on intactness of subgenomic regions | Semi-high-throughput Single genome Easy set-up | Prone to overestimation No info on (full-length) sequence | [10,11] | |
| ISS | Sequencing of flanking host regions | Chromosomal Integration site | Detection of clonality Info on spatial context | No proviral sequence | [12,13] | |
| Full-length coverage | NFL | Nested PCR, followed by Illumina-based sequencing at single-genome level | Full-length sequences | Relative high-throughput Distinction intact vs. defective provirus | More laborious workflow Cost No info on clonality | [14,15,16] |
| Q4PCR | Modified NFL sequencing with addition of qPCR step for intactness filtering | Full-length sequences | See NFL Enrichment of full-length sequences | See NFL Increased bench time (qPCR) | [17] | |
| MDA | MDA at single-genome level, followed by NFL and ISS | Matching full-length and integration site | See NFL and ISS Combined info | Expensive Challenging workflow | [18,19] | |
| Long-read | Acquire single-genome and flanking host sequences via PCIP-seq in bulk | Fragments containing both full-length and integration site | See NFL and ISS Combined info Cost-effective compared to MDA | Lower sensitivity for small and/or non-clonal proviruses | [20] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lambrechts, L.; Cole, B.; Rutsaert, S.; Trypsteen, W.; Vandekerckhove, L. Emerging PCR-Based Techniques to Study HIV-1 Reservoir Persistence. Viruses 2020, 12, 149. https://doi.org/10.3390/v12020149
Lambrechts L, Cole B, Rutsaert S, Trypsteen W, Vandekerckhove L. Emerging PCR-Based Techniques to Study HIV-1 Reservoir Persistence. Viruses. 2020; 12(2):149. https://doi.org/10.3390/v12020149
Chicago/Turabian StyleLambrechts, Laurens, Basiel Cole, Sofie Rutsaert, Wim Trypsteen, and Linos Vandekerckhove. 2020. "Emerging PCR-Based Techniques to Study HIV-1 Reservoir Persistence" Viruses 12, no. 2: 149. https://doi.org/10.3390/v12020149
APA StyleLambrechts, L., Cole, B., Rutsaert, S., Trypsteen, W., & Vandekerckhove, L. (2020). Emerging PCR-Based Techniques to Study HIV-1 Reservoir Persistence. Viruses, 12(2), 149. https://doi.org/10.3390/v12020149