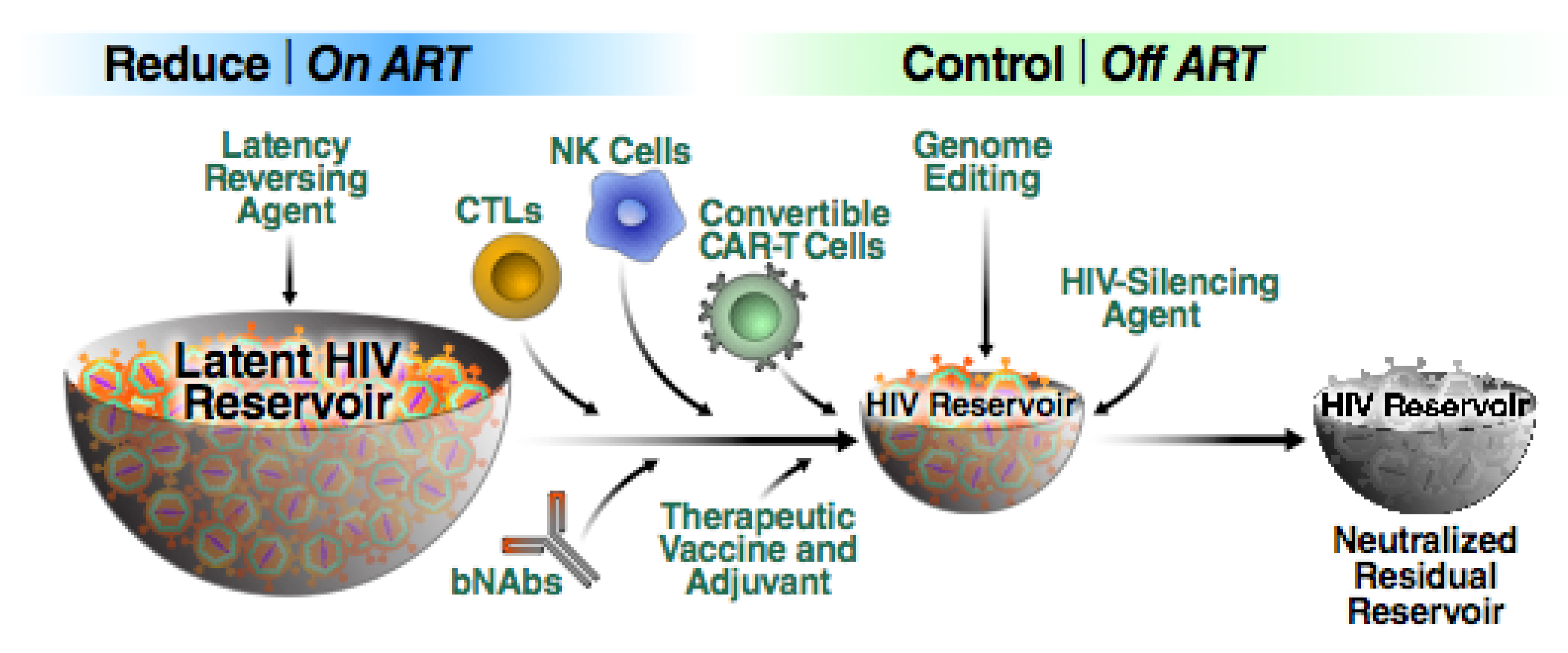
Reduce and Control: A Combinatorial Strategy for Achieving Sustained HIV Remissions in the Absence of Antiretroviral Therapy

Abstract
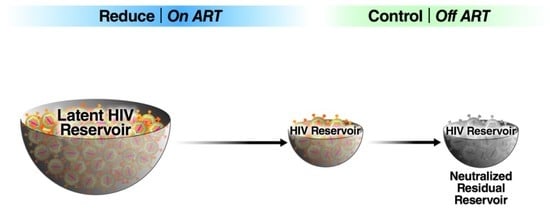
1. Introduction
1.1. HIV Latency and the Latent Reservoir
1.2. Post-Treatment Control: A Blueprint for “Reduce and Control?”
1.3. An Overview of HIV Cure Approaches
1.4. Facilitating Immunological Control of HIV Infections
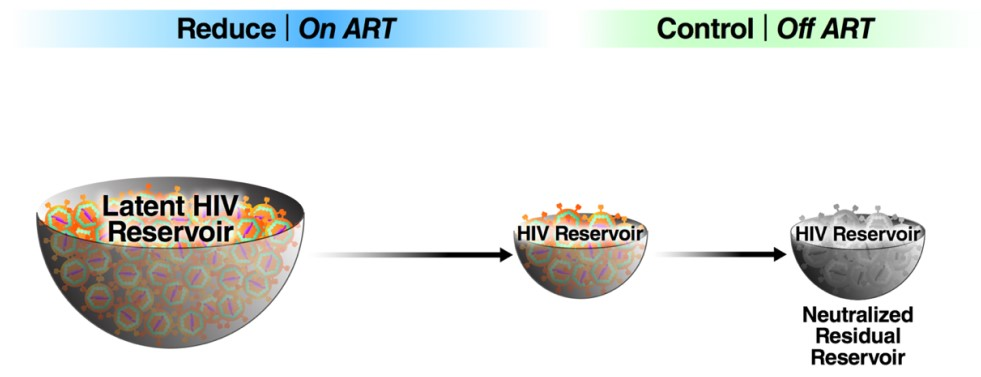
2. In Vitro and In Vivo Reactivation of Latent HIV
2.1. Histone Deacetylase (HDAC) and Histone Methytransferase (HMT) Inhibitors
2.2. BET Inhibitors
2.3. Disulfiram
2.4. PKC Agonists
2.5. Toll-like Receptor (TLR) Agonists
2.6. SMAC Mimetics
2.7. Summary and Conclusions
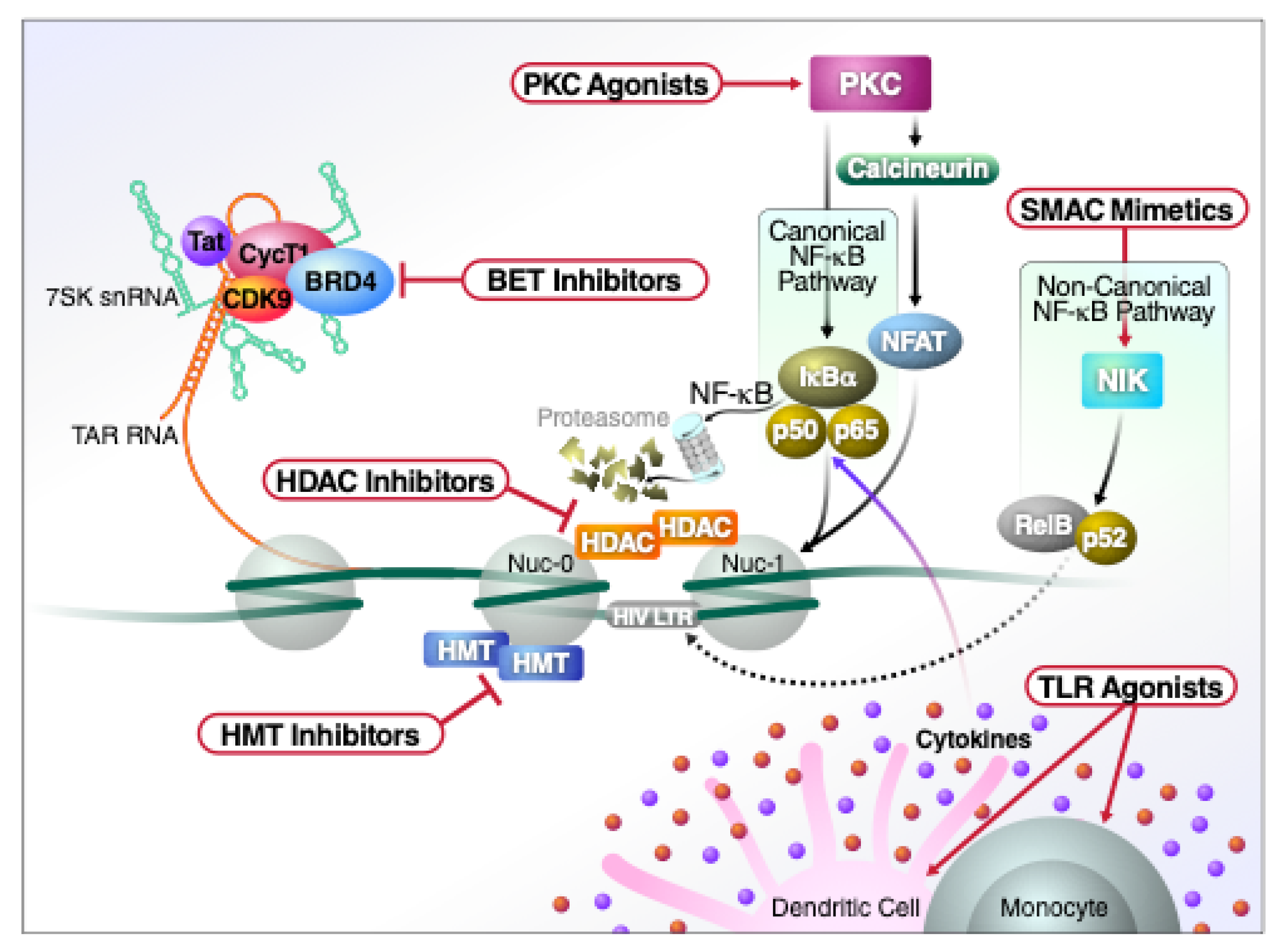
3. “Block and Lock”: An Alternative Strategy for A Functional Cure
3.1. Pharmacologic Inhibition of Host Factors
3.2. Small Molecule Inhibition of HIV Tat
3.3. Targeting HIV-1 mRNAs
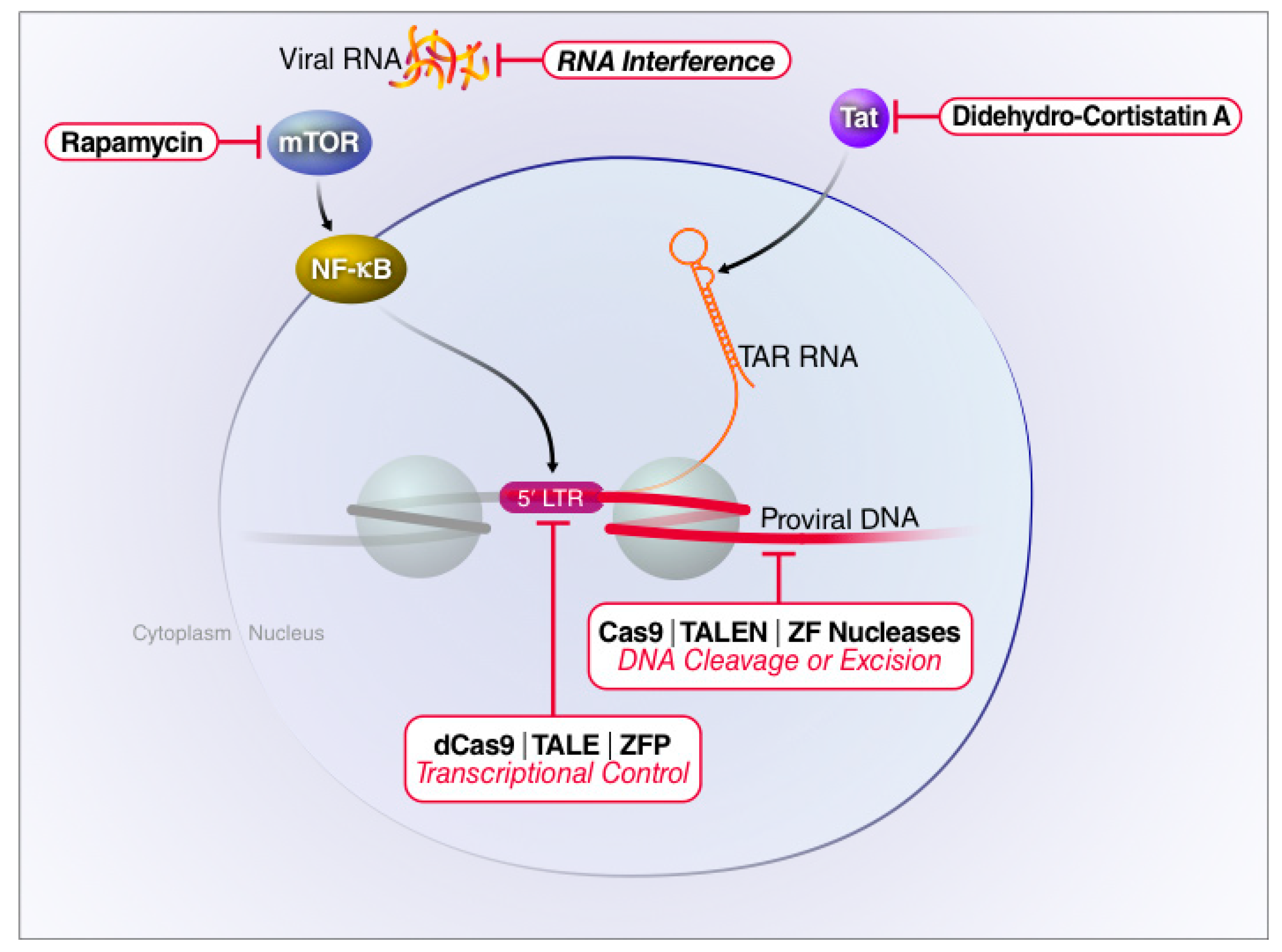
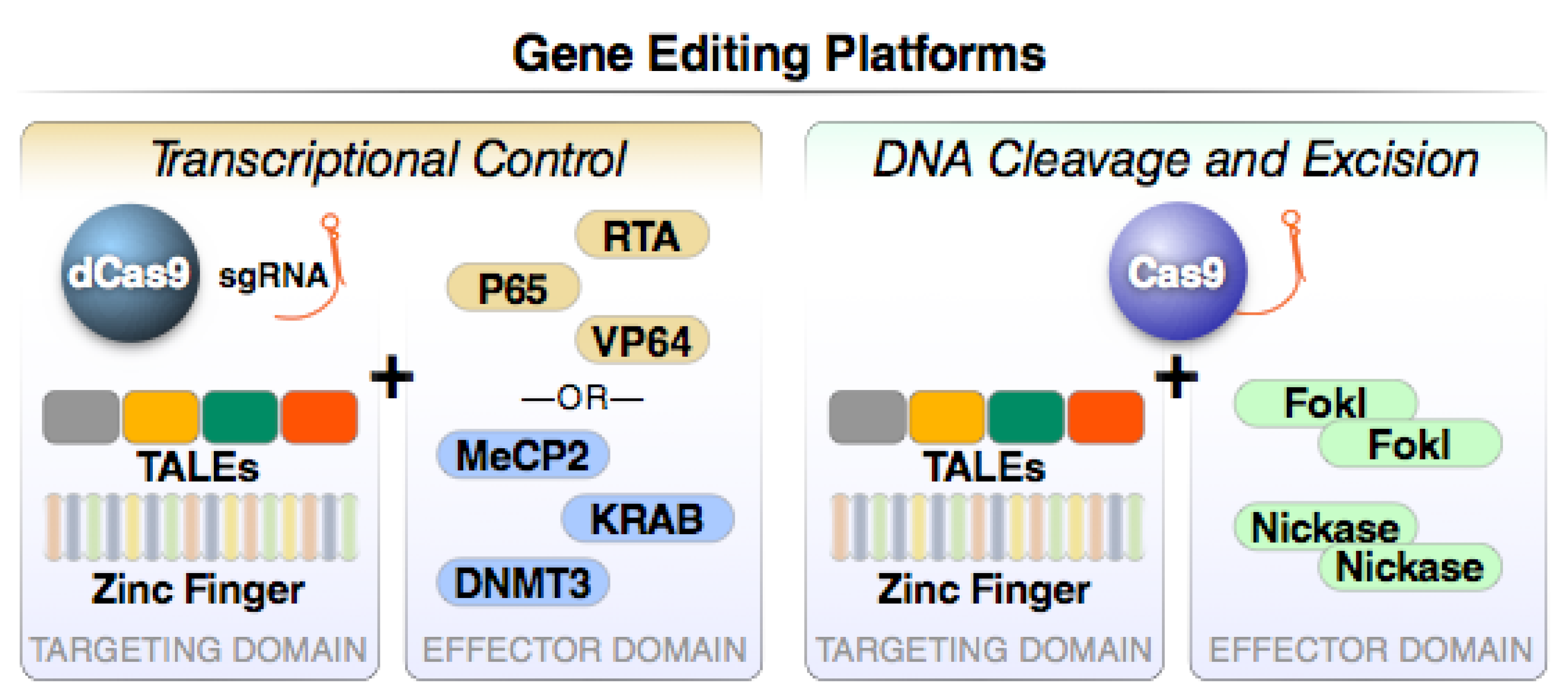
4. Gene Editing Strategies to Attack Latent HIV Proviruses
4.1. Homing Endonucleases
4.2. Zinc Finger Proteins
4.3. TALEs
4.4. CRISPR
4.5. Summary and Conclusions
4.6. Challenges
5. Concluding Thoughts
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Greene, W.C. A history of AIDS: Looking back to see ahead. Eur. J. Immunol. 2007, 37 (Suppl. 1), S94–S102. [Google Scholar] [CrossRef]
- Barre-Sinoussi, F.; Chermann, J.C.; Rey, F.; Nugeyre, M.T.; Chamaret, S.; Gruest, J.; Dauguet, C.; Axler-Blin, C.; Vezinet-Brun, F.; Rouzioux, C.; et al. Isolation of a T-lymphotropic retrovirus from a patient at risk for acquired immune deficiency syndrome (AIDS). Science 1983, 220, 868–871. [Google Scholar] [CrossRef]
- Gallo, R.C.; Salahuddin, S.Z.; Popovic, M.; Shearer, G.M.; Kaplan, M.; Haynes, B.F.; Palker, T.J.; Redfield, R.; Oleske, J.; Safai, B.; et al. Frequent detection and isolation of cytopathic retroviruses (HTLV-III) from patients with AIDS and at risk for AIDS. Science 1984, 224, 500–503. [Google Scholar] [CrossRef]
- Popovic, M.; Sarngadharan, M.G.; Read, E.; Gallo, R.C. Detection, isolation, and continuous production of cytopathic retroviruses (HTLV-III) from patients with AIDS and pre-AIDS. Science 1984, 224, 497–500. [Google Scholar] [CrossRef] [PubMed]
- Schupbach, J.; Popovic, M.; Gilden, R.V.; Gonda, M.A.; Sarngadharan, M.G.; Gallo, R.C. Serological analysis of a subgroup of human T-lymphotropic retroviruses (HTLV-III) associated with AIDS. Science 1984, 224, 503–505. [Google Scholar] [CrossRef] [PubMed]
- Bruner, K.M.; Murray, A.J.; Pollack, R.A.; Soliman, M.G.; Laskey, S.B.; Capoferri, A.A.; Lai, J.; Strain, M.C.; Lada, S.M.; Hoh, R.; et al. Defective proviruses rapidly accumulate during acute HIV-1 infection. Nat. Med. 2016, 22, 1043–1049. [Google Scholar] [CrossRef] [PubMed]
- Pollack, R.A.; Jones, R.B.; Pertea, M.; Bruner, K.M.; Martin, A.R.; Thomas, A.S.; Capoferri, A.A.; Beg, S.A.; Huang, S.H.; Karandish, S.; et al. Defective HIV-1 Proviruses Are Expressed and Can Be Recognized by Cytotoxic T Lymphocytes, which Shape the Proviral Landscape. Cell Host Microbe 2017, 21, 494–506.e494. [Google Scholar] [CrossRef]
- Ho, Y.C.; Shan, L.; Hosmane, N.N.; Wang, J.; Laskey, S.B.; Rosenbloom, D.I.; Lai, J.; Blankson, J.N.; Siliciano, J.D.; Siliciano, R.F. Replication-competent noninduced proviruses in the latent reservoir increase barrier to HIV-1 cure. Cell 2013, 155, 540–551. [Google Scholar] [CrossRef]
- DeMaster, L.K.; Liu, X.; VanBelzen, D.J.; Trinite, B.; Zheng, L.; Agosto, L.M.; Migueles, S.A.; Connors, M.; Sambucetti, L.; Levy, D.N.; et al. A Subset of CD4/CD8 Double-Negative T Cells Expresses HIV Proteins in Patients on Antiretroviral Therapy. J. Virol. 2015, 90, 2165–2179. [Google Scholar] [CrossRef]
- Imamichi, H.; Dewar, R.L.; Adelsberger, J.W.; Rehm, C.A.; O’Doherty, U.; Paxinos, E.E.; Fauci, A.S.; Lane, H.C. Defective HIV-1 proviruses produce novel protein-coding RNA species in HIV-infected patients on combination antiretroviral therapy. Proc. Natl. Acad. Sci. USA 2016, 113, 8783–8788. [Google Scholar] [CrossRef]
- Deeks, S.G.; Tracy, R.; Douek, D.C. Systemic effects of inflammation on health during chronic HIV infection. Immunity 2013, 39, 633–645. [Google Scholar] [CrossRef]
- Bruel, T.; Schwartz, O. Markers of the HIV-1 reservoir: Facts and controversies. Curr. Opin. HIV AIDS 2018, 13, 383–388. [Google Scholar] [CrossRef] [PubMed]
- Tabler, C.O.; Lucera, M.B.; Haqqani, A.A.; McDonald, D.J.; Migueles, S.A.; Connors, M.; Tilton, J.C. CD4+ memory stem cells are infected by HIV-1 in a manner regulated in part by SAMHD1 expression. J. Virol. 2014, 88, 4976–4986. [Google Scholar] [CrossRef]
- Zaikos, T.D.; Terry, V.H.; Sebastian Kettinger, N.T.; Lubow, J.; Painter, M.M.; Virgilio, M.C.; Neevel, A.; Taschuk, F.; Onafuwa-Nuga, A.; McNamara, L.A.; et al. Hematopoietic Stem and Progenitor Cells Are a Distinct HIV Reservoir that Contributes to Persistent Viremia in Suppressed Patients. Cell Rep. 2018, 25, 3759–3773.e3759. [Google Scholar] [CrossRef] [PubMed]
- Aid, M.; Dupuy, F.P.; Moysi, E.; Moir, S.; Haddad, E.K.; Estes, J.D.; Sekaly, R.P.; Petrovas, C.; Ribeiro, S.P. Follicular CD4 T Helper Cells As a Major HIV Reservoir Compartment: A Molecular Perspective. Front. Immunol. 2018, 9, 895. [Google Scholar] [CrossRef]
- Bronnimann, M.P.; Skinner, P.J.; Connick, E. The B-Cell Follicle in HIV Infection: Barrier to a Cure. Front. Immunol. 2018, 9, 20. [Google Scholar] [CrossRef] [PubMed]
- Fukazawa, Y.; Lum, R.; Okoye, A.A.; Park, H.; Matsuda, K.; Bae, J.Y.; Hagen, S.I.; Shoemaker, R.; Deleage, C.; Lucero, C.; et al. B cell follicle sanctuary permits persistent productive simian immunodeficiency virus infection in elite controllers. Nat. Med. 2015, 21, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Bozzi, G.; Simonetti, F.R.; Watters, S.A.; Anderson, E.M.; Gouzoulis, M.; Kearney, M.F.; Rote, P.; Lange, C.; Shao, W.; Gorelick, R.; et al. No evidence of ongoing HIV replication or compartmentalization in tissues during combination antiretroviral therapy: Implications for HIV eradication. Sci. Adv. 2019, 5, eaav2045. [Google Scholar] [CrossRef]
- Chaillon, A.; Gianella, S.; Dellicour, S.; Rawlings, S.A.; Schlub, T.E.; Faria De Oliveira, M.; Ignacio, C.; Porrachia, M.; Vrancken, B.; Smith, D.M. HIV persists throughout deep tissues with repopulation from multiple anatomical sources. J. Clin. Investig. 2020. [Google Scholar] [CrossRef]
- Prevedel, L.; Ruel, N.; Castellano, P.; Smith, C.; Malik, S.; Villeux, C.; Bomsel, M.; Morgello, S.; Eugenin, E.A. Identification, Localization, and Quantification of HIV Reservoirs Using Microscopy. Curr. Protoc. Cell Biol. 2019, 82, e64. [Google Scholar] [CrossRef]
- Henrich, T.J.; Deeks, S.G.; Pillai, S.K. Measuring the Size of the Latent Human Immunodeficiency Virus Reservoir: The Present and Future of Evaluating Eradication Strategies. J. Infect. Dis. 2017, 215, S134–S141. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Prakash, K.; Gianella, S.; Dube, K.; Taylor, J.; Lee, G.; Smith, D.M. Willingness to participate in HIV research at the end of life (EOL). PLoS ONE 2018, 13, e0199670. [Google Scholar] [CrossRef] [PubMed]
- Kulpa, D.A.; Talla, A.; Brehm, J.H.; Ribeiro, S.P.; Yuan, S.; Bebin-Blackwell, A.G.; Miller, M.; Barnard, R.; Deeks, S.G.; Hazuda, D.; et al. Differentiation into an Effector Memory Phenotype Potentiates HIV-1 Latency Reversal in CD4(+) T Cells. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [PubMed]
- Bisgrove, D.; Lewinski, M.; Bushman, F.; Verdin, E. Molecular mechanisms of HIV-1 proviral latency. Expert. Rev. Anti Infect. Ther. 2005, 3, 805–814. [Google Scholar] [CrossRef] [PubMed]
- Cesana, D.; Santoni de Sio, F.R.; Rudilosso, L.; Gallina, P.; Calabria, A.; Beretta, S.; Merelli, I.; Bruzzesi, E.; Passerini, L.; Nozza, S.; et al. HIV-1-mediated insertional activation of STAT5B and BACH2 trigger viral reservoir in T regulatory cells. Nat. Commun. 2017, 8, 498. [Google Scholar] [CrossRef]
- Cohn, L.B.; Silva, I.T.; Oliveira, T.Y.; Rosales, R.A.; Parrish, E.H.; Learn, G.H.; Hahn, B.H.; Czartoski, J.L.; McElrath, M.J.; Lehmann, C.; et al. HIV-1 integration landscape during latent and active infection. Cell 2015, 160, 420–432. [Google Scholar] [CrossRef]
- Hughes, S.H.; Coffin, J.M. What Integration Sites Tell Us about HIV Persistence. Cell Host Microbe 2016, 19, 588–598. [Google Scholar] [CrossRef]
- Maldarelli, F.; Wu, X.; Su, L.; Simonetti, F.R.; Shao, W.; Hill, S.; Spindler, J.; Ferris, A.L.; Mellors, J.W.; Kearney, M.F.; et al. HIV latency. Specific HIV integration sites are linked to clonal expansion and persistence of infected cells. Science 2014, 345, 179–183. [Google Scholar] [CrossRef]
- Symons, J.; Cameron, P.U.; Lewin, S.R. HIV integration sites and implications for maintenance of the reservoir. Curr. Opin. HIV AIDS 2018, 13, 152–159. [Google Scholar] [CrossRef]
- Wagner, T.A.; McLaughlin, S.; Garg, K.; Cheung, C.Y.; Larsen, B.B.; Styrchak, S.; Huang, H.C.; Edlefsen, P.T.; Mullins, J.I.; Frenkel, L.M. HIV latency. Proliferation of cells with HIV integrated into cancer genes contributes to persistent infection. Science 2014, 345, 570–573. [Google Scholar] [CrossRef]
- Ruelas, D.S.; Greene, W.C. An integrated overview of HIV-1 latency. Cell 2013, 155, 519–529. [Google Scholar] [CrossRef] [PubMed]
- He, G.; Margolis, D.M. Counterregulation of chromatin deacetylation and histone deacetylase occupancy at the integrated promoter of human immunodeficiency virus type 1 (HIV-1) by the HIV-1 repressor YY1 and HIV-1 activator Tat. Mol. Cell Biol. 2002, 22, 2965–2973. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Iqbal, M.; Tariq, M.; Baig, S.M.; Abbas, W. Epigenetic regulation of HIV-1 latency: Focus on polycomb group (PcG) proteins. Clin. Epigenetics 2018, 10, 14. [Google Scholar] [CrossRef]
- Nguyen, K.; Das, B.; Dobrowolski, C.; Karn, J. Multiple Histone Lysine Methyltransferases Are Required for the Establishment and Maintenance of HIV-1 Latency. MBio 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Tacheny, A.; Michel, S.; Dieu, M.; Payen, L.; Arnould, T.; Renard, P. Unbiased proteomic analysis of proteins interacting with the HIV-1 5’LTR sequence: Role of the transcription factor Meis. Nucleic Acids Res. 2012, 40, e168. [Google Scholar] [CrossRef] [PubMed]
- Dlamini, Z.; Hull, R. Can the HIV-1 splicing machinery be targeted for drug discovery? HIV AIDS (Auckl.) 2017, 9, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Charnay, N.; Ivanyi-Nagy, R.; Soto-Rifo, R.; Ohlmann, T.; Lopez-Lastra, M.; Darlix, J.L. Mechanism of HIV-1 Tat RNA translation and its activation by the Tat protein. Retrovirology 2009, 6, 74. [Google Scholar] [CrossRef]
- Zapata, J.C.; Campilongo, F.; Barclay, R.A.; DeMarino, C.; Iglesias-Ussel, M.D.; Kashanchi, F.; Romerio, F. The Human Immunodeficiency Virus 1 ASP RNA promotes viral latency by recruiting the Polycomb Repressor Complex 2 and promoting nucleosome assembly. Virology 2017, 506, 34–44. [Google Scholar] [CrossRef]
- Maenza, J.; Tapia, K.; Holte, S.; Stekler, J.D.; Stevens, C.E.; Mullins, J.I.; Collier, A.C. How often does treatment of primary HIV lead to post-treatment control? Antivir. Ther. 2015, 20, 855–863. [Google Scholar] [CrossRef]
- Namazi, G.; Fajnzylber, J.M.; Aga, E.; Bosch, R.J.; Acosta, E.P.; Sharaf, R.; Hartogensis, W.; Jacobson, J.M.; Connick, E.; Volberding, P.; et al. The Control of HIV After Antiretroviral Medication Pause (CHAMP) Study: Posttreatment Controllers Identified From 14 Clinical Studies. J. Infect. Dis. 2018, 218, 1954–1963. [Google Scholar] [CrossRef]
- Etemad, B.; Esmaeilzadeh, E.; Li, J.Z. Learning From the Exceptions: HIV Remission in Post-treatment Controllers. Front. Immunol. 2019, 10, 1749. [Google Scholar] [CrossRef] [PubMed]
- Goulder, P.; Deeks, S.G. HIV control: Is getting there the same as staying there? PLoS Pathog. 2018, 14, e1007222. [Google Scholar] [CrossRef] [PubMed]
- Saez-Cirion, A.; Bacchus, C.; Hocqueloux, L.; Avettand-Fenoel, V.; Girault, I.; Lecuroux, C.; Potard, V.; Versmisse, P.; Melard, A.; Prazuck, T.; et al. Post-treatment HIV-1 controllers with a long-term virological remission after the interruption of early initiated antiretroviral therapy ANRS VISCONTI Study. PLoS Pathog. 2013, 9, e1003211. [Google Scholar] [CrossRef] [PubMed]
- Sharaf, R.; Lee, G.Q.; Sun, X.; Etemad, B.; Aboukhater, L.M.; Hu, Z.; Brumme, Z.L.; Aga, E.; Bosch, R.J.; Wen, Y.; et al. HIV-1 proviral landscapes distinguish posttreatment controllers from noncontrollers. J. Clin. Investig. 2018, 128, 4074–4085. [Google Scholar] [CrossRef]
- Wen, Y.; Li, J.Z. Post-treatment HIV controllers or spontaneous controllers in disguise? AIDS 2017, 31, 587–589. [Google Scholar] [CrossRef]
- Pohlmeyer, C.W.; Gonzalez, V.D.; Irrinki, A.; Ramirez, R.N.; Li, L.; Mulato, A.; Murry, J.P.; Arvey, A.; Hoh, R.; Deeks, S.G.; et al. Identification of NK Cell Subpopulations That Differentiate HIV-Infected Subject Cohorts with Diverse Levels of Virus Control. J. Virol. 2019, 93. [Google Scholar] [CrossRef]
- Henrich, T.J.; Hatano, H.; Bacon, O.; Hogan, L.E.; Rutishauser, R.; Hill, A.; Kearney, M.F.; Anderson, E.M.; Buchbinder, S.P.; Cohen, S.E.; et al. HIV-1 persistence following extremely early initiation of antiretroviral therapy (ART) during acute HIV-1 infection: An observational study. PLoS Med. 2017, 14, e1002417. [Google Scholar] [CrossRef]
- Colby, D.J.; Trautmann, L.; Pinyakorn, S.; Leyre, L.; Pagliuzza, A.; Kroon, E.; Rolland, M.; Takata, H.; Buranapraditkun, S.; Intasan, J.; et al. Rapid HIV RNA rebound after antiretroviral treatment interruption in persons durably suppressed in Fiebig I acute HIV infection. Nat. Med. 2018, 24, 923–926. [Google Scholar] [CrossRef]
- Whitney, J.B.; Hill, A.L.; Sanisetty, S.; Penaloza-MacMaster, P.; Liu, J.; Shetty, M.; Parenteau, L.; Cabral, C.; Shields, J.; Blackmore, S.; et al. Rapid seeding of the viral reservoir prior to SIV viraemia in rhesus monkeys. Nature 2014, 512, 74–77. [Google Scholar] [CrossRef]
- Chomont, N.; El-Far, M.; Ancuta, P.; Trautmann, L.; Procopio, F.A.; Yassine-Diab, B.; Boucher, G.; Boulassel, M.R.; Ghattas, G.; Brenchley, J.M.; et al. HIV reservoir size and persistence are driven by T cell survival and homeostatic proliferation. Nat. Med. 2009, 15, 893–900. [Google Scholar] [CrossRef]
- Siliciano, J.D.; Kajdas, J.; Finzi, D.; Quinn, T.C.; Chadwick, K.; Margolick, J.B.; Kovacs, C.; Gange, S.J.; Siliciano, R.F. Long-term follow-up studies confirm the stability of the latent reservoir for HIV-1 in resting CD4+ T cells. Nat. Med. 2003, 9, 727–728. [Google Scholar] [CrossRef] [PubMed]
- Deeks, S.G. HIV: Shock and kill. Nature 2012, 487, 439–440. [Google Scholar] [CrossRef] [PubMed]
- Vargas, B.; Giacobbi, N.S.; Sanyal, A.; Venkatachari, N.J.; Han, F.; Gupta, P.; Sluis-Cremer, N. Inhibitors of Signaling Pathways That Block Reversal of HIV-1 Latency. Antimicrob. Agents Chemother. 2019, 63. [Google Scholar] [CrossRef] [PubMed]
- Stephenson, K.E. Therapeutic vaccination for HIV: Hopes and challenges. Curr. Opin. HIV AIDS 2018, 13, 408–415. [Google Scholar] [CrossRef] [PubMed]
- Fennessey, C.M.; Pinkevych, M.; Immonen, T.T.; Reynaldi, A.; Venturi, V.; Nadella, P.; Reid, C.; Newman, L.; Lipkey, L.; Oswald, K.; et al. Genetically-barcoded SIV facilitates enumeration of rebound variants and estimation of reactivation rates in nonhuman primates following interruption of suppressive antiretroviral therapy. PLoS Pathog. 2017, 13, e1006359. [Google Scholar] [CrossRef] [PubMed]
- Mylvaganam, G.H.; Chea, L.S.; Tharp, G.K.; Hicks, S.; Velu, V.; Iyer, S.S.; Deleage, C.; Estes, J.D.; Bosinger, S.E.; Freeman, G.J.; et al. Combination anti-PD-1 and antiretroviral therapy provides therapeutic benefit against SIV. JCI Insight. 2018, 3. [Google Scholar] [CrossRef]
- Goswami, R.; Nelson, A.N.; Tu, J.J.; Dennis, M.; Feng, L.; Kumar, A.; Mangold, J.; Mangan, R.J.; Mattingly, C.; Curtis, A.D., 2nd; et al. Analytical Treatment Interruption after Short-Term Antiretroviral Therapy in a Postnatally Simian-Human Immunodeficiency Virus-Infected Infant Rhesus Macaque Model. MBio 2019, 10. [Google Scholar] [CrossRef]
- Caskey, M. Delivery of anti-HIV bNAbs by viral vectors. Lancet HIV 2019, 6, e207–e208. [Google Scholar] [CrossRef]
- Blazkova, J.; Refsland, E.W.; Clarridge, K.E.; Shi, V.; Justement, J.S.; Huiting, E.D.; Gittens, K.R.; Chen, X.; Schmidt, S.D.; Liu, C.; et al. Glycan-dependent HIV-specific neutralizing antibodies bind to cells of uninfected individuals. J. Clin. Investig. 2019, 129, 4832–4837. [Google Scholar] [CrossRef]
- Bruel, T.; Guivel-Benhassine, F.; Amraoui, S.; Malbec, M.; Richard, L.; Bourdic, K.; Donahue, D.A.; Lorin, V.; Casartelli, N.; Noel, N.; et al. Elimination of HIV-1-infected cells by broadly neutralizing antibodies. Nat. Commun. 2016, 7, 10844. [Google Scholar] [CrossRef]
- Kim, J.H.; Excler, J.L.; Michael, N.L. Lessons from the RV144 Thai phase III HIV-1 vaccine trial and the search for correlates of protection. Annu. Rev. Med. 2015, 66, 423–437. [Google Scholar] [CrossRef]
- Nishimura, Y.; Gautam, R.; Chun, T.W.; Sadjadpour, R.; Foulds, K.E.; Shingai, M.; Klein, F.; Gazumyan, A.; Golijanin, J.; Donaldson, M.; et al. Early antibody therapy can induce long-lasting immunity to SHIV. Nature 2017, 543, 559–563. [Google Scholar] [CrossRef] [PubMed]
- Bar, K.J.; Sneller, M.C.; Harrison, L.J.; Justement, J.S.; Overton, E.T.; Petrone, M.E.; Salantes, D.B.; Seamon, C.A.; Scheinfeld, B.; Kwan, R.W.; et al. Effect of HIV Antibody VRC01 on Viral Rebound after Treatment Interruption. N. Engl. J. Med. 2016, 375, 2037–2050. [Google Scholar] [CrossRef] [PubMed]
- Jackson, H.J.; Rafiq, S.; Brentjens, R.J. Driving CAR T-cells forward. Nat. Rev. Clin. Oncol. 2016, 13, 370–383. [Google Scholar] [CrossRef] [PubMed]
- Wagner, T.A. Quarter Century of Anti-HIV CAR T Cells. Curr. HIV/AIDS Rep. 2018, 15, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.B.; Hege, K.; Riley, J.L. CAR Talk: How Cancer-Specific CAR T Cells Can Instruct How to Build CAR T Cells to Cure HIV. Front. Immunol. 2019, 10, 2310. [Google Scholar] [CrossRef]
- Herzig, E.; Kim, K.C.; Packard, T.A.; Vardi, N.; Schwarzer, R.; Gramatica, A.; Deeks, S.G.; Williams, S.R.; Landgraf, K.; Killeen, N.; et al. Attacking Latent HIV with convertibleCAR-T Cells, a Highly Adaptable Killing Platform. Cell 2019, 179, 880–894.e810. [Google Scholar] [CrossRef]
- Liu, B.; Zhang, W.; Zhang, H. Development of CAR-T cells for long-term eradication and surveillance of HIV-1 reservoir. Curr. Opin. Virol. 2019, 38, 21–30. [Google Scholar] [CrossRef]
- Abner, E.; Jordan, A. HIV “shock and kill” therapy: In need of revision. Antiviral Res. 2019, 166, 19–34. [Google Scholar] [CrossRef]
- Cillo, A.R.; Sobolewski, M.D.; Bosch, R.J.; Fyne, E.; Piatak, M., Jr.; Coffin, J.M.; Mellors, J.W. Quantification of HIV-1 latency reversal in resting CD4+ T cells from patients on suppressive antiretroviral therapy. Proc. Natl. Acad. Sci. USA 2014, 111, 7078–7083. [Google Scholar] [CrossRef]
- Archin, N.M.; Espeseth, A.; Parker, D.; Cheema, M.; Hazuda, D.; Margolis, D.M. Expression of latent HIV induced by the potent HDAC inhibitor suberoylanilide hydroxamic acid. AIDS Res. Hum. Retroviruses 2009, 25, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Archin, N.M.; Liberty, A.L.; Kashuba, A.D.; Choudhary, S.K.; Kuruc, J.D.; Crooks, A.M.; Parker, D.C.; Anderson, E.M.; Kearney, M.F.; Strain, M.C.; et al. Administration of vorinostat disrupts HIV-1 latency in patients on antiretroviral therapy. Nature 2012, 487, 482–485. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Swanson, M.; Talla, A.; Graham, D.; Strizki, J.; Gorman, D.; Barnard, R.J.; Blair, W.; Sogaard, O.S.; Tolstrup, M.; et al. HDAC inhibition induces HIV-1 protein and enables immune-based clearance following latency reversal. JCI Insight. 2017, 2. [Google Scholar] [CrossRef] [PubMed]
- Archin, N.M.; Kirchherr, J.L.; Sung, J.A.; Clutton, G.; Sholtis, K.; Xu, Y.; Allard, B.; Stuelke, E.; Kashuba, A.D.; Kuruc, J.D.; et al. Interval dosing with the HDAC inhibitor vorinostat effectively reverses HIV latency. J. Clin. Investig. 2017, 127, 3126–3135. [Google Scholar] [CrossRef]
- Jones, R.B.; O’Connor, R.; Mueller, S.; Foley, M.; Szeto, G.L.; Karel, D.; Lichterfeld, M.; Kovacs, C.; Ostrowski, M.A.; Trocha, A.; et al. Histone deacetylase inhibitors impair the elimination of HIV-infected cells by cytotoxic T-lymphocytes. PLoS Pathog. 2014, 10, e1004287. [Google Scholar] [CrossRef]
- Subramanian, S.; Bates, S.E.; Wright, J.J.; Espinoza-Delgado, I.; Piekarz, R.L. Clinical Toxicities of Histone Deacetylase Inhibitors. Pharmaceuticals (Basel) 2010, 3, 2751–2767. [Google Scholar] [CrossRef]
- Sung, J.A.; Lam, S.; Garrido, C.; Archin, N.; Rooney, C.M.; Bollard, C.M.; Margolis, D.M. Expanded cytotoxic T-cell lymphocytes target the latent HIV reservoir. J. Infect. Dis. 2015, 212, 258–263. [Google Scholar] [CrossRef]
- Imai, K.; Togami, H.; Okamoto, T. Involvement of histone H3 lysine 9 (H3K9) methyltransferase G9a in the maintenance of HIV-1 latency and its reactivation by BIX01294. J. Biol. Chem. 2010, 285, 16538–16545. [Google Scholar] [CrossRef]
- Boehm, D.; Jeng, M.; Camus, G.; Gramatica, A.; Schwarzer, R.; Johnson, J.R.; Hull, P.A.; Montano, M.; Sakane, N.; Pagans, S.; et al. SMYD2-Mediated Histone Methylation Contributes to HIV-1 Latency. Cell Host Microbe 2017, 21, 569–579.e566. [Google Scholar] [CrossRef]
- Friedman, J.; Cho, W.K.; Chu, C.K.; Keedy, K.S.; Archin, N.M.; Margolis, D.M.; Karn, J. Epigenetic silencing of HIV-1 by the histone H3 lysine 27 methyltransferase enhancer of Zeste 2. J. Virol. 2011, 85, 9078–9089. [Google Scholar] [CrossRef]
- Cary, D.C.; Fujinaga, K.; Peterlin, B.M. Molecular mechanisms of HIV latency. J. Clin. Investig. 2016, 126, 448–454. [Google Scholar] [CrossRef] [PubMed]
- Conrad, R.J.; Fozouni, P.; Thomas, S.; Sy, H.; Zhang, Q.; Zhou, M.M.; Ott, M. The Short Isoform of BRD4 Promotes HIV-1 Latency by Engaging Repressive SWI/SNF Chromatin-Remodeling Complexes. Mol. Cell 2017, 67, 1001–1012.e1006. [Google Scholar] [CrossRef] [PubMed]
- Laird, G.M.; Bullen, C.K.; Rosenbloom, D.I.; Martin, A.R.; Hill, A.L.; Durand, C.M.; Siliciano, J.D.; Siliciano, R.F. Ex vivo analysis identifies effective HIV-1 latency-reversing drug combinations. J. Clin. Investig. 2015, 125, 1901–1912. [Google Scholar] [CrossRef] [PubMed]
- Knights, H.D.J. A Critical Review of the Evidence Concerning the HIV Latency Reversing Effect of Disulfiram, the Possible Explanations for Its Inability to Reduce the Size of the Latent Reservoir In Vivo, and the Caveats Associated with Its Use in Practice. AIDS Res. Treat 2017, 2017, 8239428. [Google Scholar] [CrossRef] [PubMed]
- Doyon, G.; Zerbato, J.; Mellors, J.W.; Sluis-Cremer, N. Disulfiram reactivates latent HIV-1 expression through depletion of the phosphatase and tensin homolog. AIDS 2013, 27, F7–F11. [Google Scholar] [CrossRef]
- Elliott, J.H.; McMahon, J.H.; Chang, C.C.; Lee, S.A.; Hartogensis, W.; Bumpus, N.; Savic, R.; Roney, J.; Hoh, R.; Solomon, A.; et al. Short-term administration of disulfiram for reversal of latent HIV infection: A phase 2 dose-escalation study. Lancet HIV 2015, 2, e520–e529. [Google Scholar] [CrossRef]
- Spivak, A.M.; Andrade, A.; Eisele, E.; Hoh, R.; Bacchetti, P.; Bumpus, N.N.; Emad, F.; Buckheit, R., 3rd; McCance-Katz, E.F.; Lai, J.; et al. A pilot study assessing the safety and latency-reversing activity of disulfiram in HIV-1-infected adults on antiretroviral therapy. Clin. Infect. Dis. 2014, 58, 883–890. [Google Scholar] [CrossRef]
- Bullen, C.K.; Laird, G.M.; Durand, C.M.; Siliciano, J.D.; Siliciano, R.F. New ex vivo approaches distinguish effective and ineffective single agents for reversing HIV-1 latency in vivo. Nat. Med. 2014, 20, 425–429. [Google Scholar] [CrossRef]
- Gutierrez, C.; Serrano-Villar, S.; Madrid-Elena, N.; Perez-Elias, M.J.; Martin, M.E.; Barbas, C.; Ruiperez, J.; Munoz, E.; Munoz-Fernandez, M.A.; Castor, T.; et al. Bryostatin-1 for latent virus reactivation in HIV-infected patients on antiretroviral therapy. AIDS 2016, 30, 1385–1392. [Google Scholar] [CrossRef]
- Martinez-Bonet, M.; Clemente, M.I.; Serramia, M.J.; Munoz, E.; Moreno, S.; Munoz-Fernandez, M.A. Synergistic Activation of Latent HIV-1 Expression by Novel Histone Deacetylase Inhibitors and Bryostatin-1. Sci. Rep. 2015, 5, 16445. [Google Scholar] [CrossRef]
- Kupchan, S.M.; Uchida, I.; Branfman, A.R.; Dailey, R.G., Jr.; Fei, B.Y. Antileukemic principles isolated from euphorbiaceae plants. Science 1976, 191, 571–572. [Google Scholar] [CrossRef] [PubMed]
- Pandelo Jose, D.; Bartholomeeusen, K.; da Cunha, R.D.; Abreu, C.M.; Glinski, J.; da Costa, T.B.; Bacchi Rabay, A.F.; Pianowski Filho, L.F.; Dudycz, L.W.; Ranga, U.; et al. Reactivation of latent HIV-1 by new semi-synthetic ingenol esters. Virology 2014, 462–463, 328–339. [Google Scholar] [CrossRef]
- Warrilow, D.; Gardner, J.; Darnell, G.A.; Suhrbier, A.; Harrich, D. HIV type 1 inhibition by protein kinase C modulatory compounds. AIDS Res. Hum. Retroviruses 2006, 22, 854–864. [Google Scholar] [CrossRef] [PubMed]
- Jiang, G.; Mendes, E.A.; Kaiser, P.; Wong, D.P.; Tang, Y.; Cai, I.; Fenton, A.; Melcher, G.P.; Hildreth, J.E.; Thompson, G.R.; et al. Synergistic Reactivation of Latent HIV Expression by Ingenol-3-Angelate, PEP005, Targeted NF-kB Signaling in Combination with JQ1 Induced p-TEFb Activation. PLoS Pathog. 2015, 11, e1005066. [Google Scholar] [CrossRef] [PubMed]
- Gama, L.; Abreu, C.M.; Shirk, E.N.; Price, S.L.; Li, M.; Laird, G.M.; Pate, K.A.; Wietgrefe, S.W.; O’Connor, S.L.; Pianowski, L.; et al. Reactivation of simian immunodeficiency virus reservoirs in the brain of virally suppressed macaques. AIDS 2017, 31, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Dental, C.; Proust, A.; Ouellet, M.; Barat, C.; Tremblay, M.J. HIV-1 Latency-Reversing Agents Prostratin and Bryostatin-1 Induce Blood-Brain Barrier Disruption/Inflammation and Modulate Leukocyte Adhesion/Transmigration. J. Immunol. 2017, 198, 1229–1241. [Google Scholar] [CrossRef]
- Brogdon, J.; Ziani, W.; Wang, X.; Veazey, R.S.; Xu, H. In vitro effects of the small-molecule protein kinase C agonists on HIV latency reactivation. Sci. Rep. 2016, 6, 39032. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef]
- Macedo, A.B.; Novis, C.L.; Bosque, A. Targeting Cellular and Tissue HIV Reservoirs With Toll-Like Receptor Agonists. Front. Immunol. 2019, 10, 2450. [Google Scholar] [CrossRef]
- Vibholm, L.; Schleimann, M.H.; Hojen, J.F.; Benfield, T.; Offersen, R.; Rasmussen, K.; Olesen, R.; Dige, A.; Agnholt, J.; Grau, J.; et al. Short-Course Toll-Like Receptor 9 Agonist Treatment Impacts Innate Immunity and Plasma Viremia in Individuals With Human Immunodeficiency Virus Infection. Clin. Infect. Dis. 2017, 64, 1686–1695. [Google Scholar] [CrossRef]
- Macedo, A.B.; Novis, C.L.; De Assis, C.M.; Sorensen, E.S.; Moszczynski, P.; Huang, S.H.; Ren, Y.; Spivak, A.M.; Jones, R.B.; Planelles, V.; et al. Dual TLR2 and TLR7 agonists as HIV latency-reversing agents. JCI Insight. 2018, 3. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.Y.; Osuna, C.E.; Hraber, P.T.; Hesselgesser, J.; Gerold, J.M.; Barnes, T.L.; Sanisetty, S.; Seaman, M.S.; Lewis, M.G.; Geleziunas, R.; et al. TLR7 agonists induce transient viremia and reduce the viral reservoir in SIV-infected rhesus macaques on antiretroviral therapy. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Del Prete, G.Q.; Alvord, W.G.; Li, Y.; Deleage, C.; Nag, M.; Oswald, K.; Thomas, J.A.; Pyle, C.; Bosche, W.J.; Coalter, V.; et al. TLR7 agonist administration to SIV-infected macaques receiving early initiated cART does not induce plasma viremia. JCI Insight. 2019, 4. [Google Scholar] [CrossRef] [PubMed]
- Borducchi, E.N.; Cabral, C.; Stephenson, K.E.; Liu, J.; Abbink, P.; Ng’ang’a, D.; Nkolola, J.P.; Brinkman, A.L.; Peter, L.; Lee, B.C.; et al. Ad26/MVA therapeutic vaccination with TLR7 stimulation in SIV-infected rhesus monkeys. Nature 2016, 540, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Borducchi, E.N.; Liu, J.; Nkolola, J.P.; Cadena, A.M.; Yu, W.H.; Fischinger, S.; Broge, T.; Abbink, P.; Mercado, N.B.; Chandrashekar, A.; et al. Antibody and TLR7 agonist delay viral rebound in SHIV-infected monkeys. Nature 2018, 563, 360–364. [Google Scholar] [CrossRef]
- Bai, L.; Smith, D.C.; Wang, S. Small-molecule SMAC mimetics as new cancer therapeutics. Pharmacol Ther. 2014, 144, 82–95. [Google Scholar] [CrossRef]
- Sun, S.C. The noncanonical NF-kappaB pathway. Immunol. Rev. 2012, 246, 125–140. [Google Scholar] [CrossRef]
- Hattori, S.I.; Matsuda, K.; Tsuchiya, K.; Gatanaga, H.; Oka, S.; Yoshimura, K.; Mitsuya, H.; Maeda, K. Combination of a Latency-Reversing Agent With a Smac Mimetic Minimizes Secondary HIV-1 Infection in vitro. Front Microbiol. 2018, 9, 2022. [Google Scholar] [CrossRef]
- Struzik, J.; Szulc-Dabrowska, L. Manipulation of Non-canonical NF-kappaB Signaling by Non-oncogenic Viruses. Arch. Immunol. Ther. Exp. (Warsz.) 2019, 67, 41–48. [Google Scholar] [CrossRef]
- Kuo, H.H.; Ahmad, R.; Lee, G.Q.; Gao, C.; Chen, H.R.; Ouyang, Z.; Szucs, M.J.; Kim, D.; Tsibris, A.; Chun, T.W.; et al. Anti-apoptotic Protein BIRC5 Maintains Survival of HIV-1-Infected CD4(+) T Cells. Immunity 2018, 48, 1183–1194.e1185. [Google Scholar] [CrossRef]
- Pache, L.; Dutra, M.S.; Spivak, A.M.; Marlett, J.M.; Murry, J.P.; Hwang, Y.; Maestre, A.M.; Manganaro, L.; Vamos, M.; Teriete, P.; et al. BIRC2/cIAP1 Is a Negative Regulator of HIV-1 Transcription and Can Be Targeted by Smac Mimetics to Promote Reversal of Viral Latency. Cell Host Microbe 2015, 18, 345–353. [Google Scholar] [CrossRef] [PubMed]
- Bobardt, M.; Kuo, J.; Chatterji, U.; Chanda, S.; Little, S.J.; Wiedemann, N.; Vuagniaux, G.; Gallay, P.A. The inhibitor apoptosis protein antagonist Debio 1143 Is an attractive HIV-1 latency reversal candidate. PLoS ONE 2019, 14, e0211746. [Google Scholar] [CrossRef] [PubMed]
- Campbell, G.R.; Bruckman, R.S.; Chu, Y.L.; Trout, R.N.; Spector, S.A. SMAC Mimetics Induce Autophagy-Dependent Apoptosis of HIV-1-Infected Resting Memory CD4+ T Cells. Cell Host Microbe 2018, 24, 689–702.e687. [Google Scholar] [CrossRef] [PubMed]
- Cummins, N.W.; Sainski, A.M.; Dai, H.; Natesampillai, S.; Pang, Y.P.; Bren, G.D.; de Araujo Correia, M.C.M.; Sampath, R.; Rizza, S.A.; O’Brien, D.; et al. Prime, Shock, and Kill: Priming CD4 T Cells from HIV Patients with a BCL-2 Antagonist before HIV Reactivation Reduces HIV Reservoir Size. J. Virol. 2016, 90, 4032–4048. [Google Scholar] [CrossRef]
- Kim, Y.; Anderson, J.L.; Lewin, S.R. Getting the “Kill” into “Shock and Kill”: Strategies to Eliminate Latent HIV. Cell Host Microbe 2018, 23, 14–26. [Google Scholar] [CrossRef]
- Spivak, A.M.; Planelles, V. Novel Latency Reversal Agents for HIV-1 Cure. Annu. Rev. Med. 2018, 69, 421–436. [Google Scholar] [CrossRef]
- Battivelli, E.; Dahabieh, M.S.; Abdel-Mohsen, M.; Svensson, J.P.; Da Silva, I.T.; Cohn, L.B.; Gramatica, A.; Deeks, S.; Greene, W.C.; Pillai, S.K.; et al. Distinct chromatin functional states correlate with HIV latency reactivation in infected primary CD4(+) T cells. Elife 2018, 7. [Google Scholar] [CrossRef]
- Kaul, M.; Zheng, J.; Okamoto, S.; Gendelman, H.E.; Lipton, S.A. HIV-1 infection and AIDS: Consequences for the central nervous system. Cell Death Differ. 2005, 12 (Suppl. 1), 878–892. [Google Scholar] [CrossRef]
- Day, C.L.; Kaufmann, D.E.; Kiepiela, P.; Brown, J.A.; Moodley, E.S.; Reddy, S.; Mackey, E.W.; Miller, J.D.; Leslie, A.J.; DePierres, C.; et al. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature 2006, 443, 350–354. [Google Scholar] [CrossRef]
- Infante, J.R.; Dees, E.C.; Olszanski, A.J.; Dhuria, S.V.; Sen, S.; Cameron, S.; Cohen, R.B. Phase I dose-escalation study of LCL161, an oral inhibitor of apoptosis proteins inhibitor, in patients with advanced solid tumors. J. Clin. Oncol. 2014, 32, 3103–3110. [Google Scholar] [CrossRef]
- Bock, M.; Stoye, J.P. Endogenous retroviruses and the human germline. Curr. Opin. Genet. Dev. 2000, 10, 651–655. [Google Scholar] [CrossRef]
- Groh, S.; Schotta, G. Silencing of endogenous retroviruses by heterochromatin. Cell Mol. Life Sci. 2017, 74, 2055–2065. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, C.V.; Staskus, K.; Wietgrefe, S.W.; Rothenberger, M.; Reilly, C.; Chipman, J.G.; Beilman, G.J.; Khoruts, A.; Thorkelson, A.; Schmidt, T.E.; et al. Persistent HIV-1 replication is associated with lower antiretroviral drug concentrations in lymphatic tissues. Proc. Natl. Acad. Sci. USA 2014, 111, 2307–2312. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Picado, J.; Deeks, S.G. Persistent HIV-1 replication during antiretroviral therapy. Curr. Opin. HIV AIDS 2016, 11, 417–423. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.; Jiang, Y. Key factors in mTOR regulation. Cell Mol. Life Sci. 2010, 67, 239–253. [Google Scholar] [CrossRef]
- Heredia, A.; Amoroso, A.; Davis, C.; Le, N.; Reardon, E.; Dominique, J.K.; Klingebiel, E.; Gallo, R.C.; Redfield, R.R. Rapamycin causes down-regulation of CCR5 and accumulation of anti-HIV beta-chemokines: An approach to suppress R5 strains of HIV-1. Proc. Natl. Acad. Sci. USA 2003, 100, 10411–10416. [Google Scholar] [CrossRef]
- Heredia, A.; Latinovic, O.; Gallo, R.C.; Melikyan, G.; Reitz, M.; Le, N.; Redfield, R.R. Reduction of CCR5 with low-dose rapamycin enhances the antiviral activity of vicriviroc against both sensitive and drug-resistant HIV-1. Proc. Natl. Acad. Sci. USA 2008, 105, 20476–20481. [Google Scholar] [CrossRef]
- Roy, J.; Paquette, J.S.; Fortin, J.F.; Tremblay, M.J. The immunosuppressant rapamycin represses human immunodeficiency virus type 1 replication. Antimicrob. Agents Chemother. 2002, 46, 3447–3455. [Google Scholar] [CrossRef]
- Besnard, E.; Hakre, S.; Kampmann, M.; Lim, H.W.; Hosmane, N.N.; Martin, A.; Bassik, M.C.; Verschueren, E.; Battivelli, E.; Chan, J.; et al. The mTOR Complex Controls HIV Latency. Cell Host Microbe 2016, 20, 785–797. [Google Scholar] [CrossRef]
- Stock, P.G.; Barin, B.; Hatano, H.; Rogers, R.L.; Roland, M.E.; Lee, T.H.; Busch, M.; Deeks, S.G.; for Solid Organ Transplantation in, H.I.V.S.I. Reduction of HIV persistence following transplantation in HIV-infected kidney transplant recipients. Am. J. Transpl. 2014, 14, 1136–1141. [Google Scholar] [CrossRef]
- Martin, A.R.; Pollack, R.A.; Capoferri, A.; Ambinder, R.F.; Durand, C.M.; Siliciano, R.F. Rapamycin-mediated mTOR inhibition uncouples HIV-1 latency reversal from cytokine-associated toxicity. J. Clin. Investig. 2017, 127, 651–656. [Google Scholar] [CrossRef] [PubMed]
- Kao, S.Y.; Calman, A.F.; Luciw, P.A.; Peterlin, B.M. Anti-termination of transcription within the long terminal repeat of HIV-1 by tat gene product. Nature 1987, 330, 489–493. [Google Scholar] [CrossRef] [PubMed]
- Salerno, D.; Hasham, M.G.; Marshall, R.; Garriga, J.; Tsygankov, A.Y.; Grana, X. Direct inhibition of CDK9 blocks HIV-1 replication without preventing T-cell activation in primary human peripheral blood lymphocytes. Gene 2007, 405, 65–78. [Google Scholar] [CrossRef] [PubMed]
- Sobhian, B.; Laguette, N.; Yatim, A.; Nakamura, M.; Levy, Y.; Kiernan, R.; Benkirane, M. HIV-1 Tat assembles a multifunctional transcription elongation complex and stably associates with the 7SK snRNP. Mol. Cell 2010, 38, 439–451. [Google Scholar] [CrossRef] [PubMed]
- Nemeth, G.; Varga, Z.; Greff, Z.; Bencze, G.; Sipos, A.; Szantai-Kis, C.; Baska, F.; Gyuris, A.; Kelemenics, K.; Szathmary, Z.; et al. Novel, selective CDK9 inhibitors for the treatment of HIV infection. Curr. Med. Chem. 2011, 18, 342–358. [Google Scholar] [CrossRef] [PubMed]
- Van Duyne, R.; Guendel, I.; Jaworski, E.; Sampey, G.; Klase, Z.; Chen, H.; Zeng, C.; Kovalskyy, D.; El Kouni, M.H.; Lepene, B.; et al. Effect of mimetic CDK9 inhibitors on HIV-1-activated transcription. J. Mol. Biol. 2013, 425, 812–829. [Google Scholar] [CrossRef]
- Okamoto, M.; Hidaka, A.; Toyama, M.; Hosoya, T.; Yamamoto, M.; Hagiwara, M.; Baba, M. Selective inhibition of HIV-1 replication by the CDK9 inhibitor FIT-039. Antiviral Res. 2015, 123, 1–4. [Google Scholar] [CrossRef]
- Medina-Moreno, S.; Dowling, T.C.; Zapata, J.C.; Le, N.M.; Sausville, E.; Bryant, J.; Redfield, R.R.; Heredia, A. Targeting of CDK9 with indirubin 3′-monoxime safely and durably reduces HIV viremia in chronically infected humanized mice. PLoS ONE 2017, 12, e0183425. [Google Scholar] [CrossRef]
- Steens, J.M.; Scherrer, D.; Gineste, P.; Barrett, P.N.; Khuanchai, S.; Winai, R.; Ruxrungtham, K.; Tazi, J.; Murphy, R.; Ehrlich, H. Safety, Pharmacokinetics, and Antiviral Activity of a Novel HIV Antiviral, ABX464, in Treatment-Naive HIV-Infected Subjects in a Phase 2 Randomized, Controlled Study. Antimicrob. Agents Chemother. 2017, 61. [Google Scholar] [CrossRef]
- Hayashi, T.; Jean, M.; Huang, H.; Simpson, S.; Santoso, N.G.; Zhu, J. Screening of an FDA-approved compound library identifies levosimendan as a novel anti-HIV-1 agent that inhibits viral transcription. Antiviral Res. 2017, 146, 76–85. [Google Scholar] [CrossRef]
- Campos, N.; Myburgh, R.; Garcel, A.; Vautrin, A.; Lapasset, L.; Nadal, E.S.; Mahuteau-Betzer, F.; Najman, R.; Fornarelli, P.; Tantale, K.; et al. Long lasting control of viral rebound with a new drug ABX464 targeting Rev - mediated viral RNA biogenesis. Retrovirology 2015, 12, 30. [Google Scholar] [CrossRef]
- Rutsaert, S.; Steens, J.M.; Gineste, P.; Cole, B.; Kint, S.; Barrett, P.N.; Tazi, J.; Scherrer, D.; Ehrlich, H.J.; Vandekerckhove, L. Safety, tolerability and impact on viral reservoirs of the addition to antiretroviral therapy of ABX464, an investigational antiviral drug, in individuals living with HIV-1: A Phase IIa randomised controlled study. J. Virus Erad. 2019, 5, 10–22. [Google Scholar] [PubMed]
- Llano, M.; Saenz, D.T.; Meehan, A.; Wongthida, P.; Peretz, M.; Walker, W.H.; Teo, W.; Poeschla, E.M. An essential role for LEDGF/p75 in HIV integration. Science 2006, 314, 461–464. [Google Scholar] [CrossRef] [PubMed]
- Vandekerckhove, L.; Christ, F.; Van Maele, B.; De Rijck, J.; Gijsbers, R.; Van den Haute, C.; Witvrouw, M.; Debyser, Z. Transient and stable knockdown of the integrase cofactor LEDGF/p75 reveals its role in the replication cycle of human immunodeficiency virus. J. Virol. 2006, 80, 1886–1896. [Google Scholar] [CrossRef] [PubMed]
- Christ, F.; Voet, A.; Marchand, A.; Nicolet, S.; Desimmie, B.A.; Marchand, D.; Bardiot, D.; Van der Veken, N.J.; Van Remoortel, B.; Strelkov, S.V.; et al. Rational design of small-molecule inhibitors of the LEDGF/p75-integrase interaction and HIV replication. Nat. Chem. Biol. 2010, 6, 442–448. [Google Scholar] [CrossRef] [PubMed]
- Vranckx, L.S.; Demeulemeester, J.; Saleh, S.; Boll, A.; Vansant, G.; Schrijvers, R.; Weydert, C.; Battivelli, E.; Verdin, E.; Cereseto, A.; et al. LEDGIN-mediated Inhibition of Integrase-LEDGF/p75 Interaction Reduces Reactivation of Residual Latent HIV. EBioMedicine 2016, 8, 248–264. [Google Scholar] [CrossRef]
- Debyser, Z.; Vansant, G.; Bruggemans, A.; Janssens, J.; Christ, F. Insight in HIV Integration Site Selection Provides a Block-and-Lock Strategy for a Functional Cure of HIV Infection. Viruses 2018, 11, 12. [Google Scholar] [CrossRef]
- Sodroski, J.; Rosen, C.; Wong-Staal, F.; Salahuddin, S.Z.; Popovic, M.; Arya, S.; Gallo, R.C.; Haseltine, W.A. Trans-acting transcriptional regulation of human T-cell leukemia virus type III long terminal repeat. Science 1985, 227, 171–173. [Google Scholar] [CrossRef]
- Burnett, J.C.; Miller-Jensen, K.; Shah, P.S.; Arkin, A.P.; Schaffer, D.V. Control of stochastic gene expression by host factors at the HIV promoter. PLoS Pathog. 2009, 5, e1000260. [Google Scholar] [CrossRef]
- Razooky, B.S.; Pai, A.; Aull, K.; Rouzine, I.M.; Weinberger, L.S. A hardwired HIV latency program. Cell 2015, 160, 990–1001. [Google Scholar] [CrossRef]
- Weinberger, L.S.; Burnett, J.C.; Toettcher, J.E.; Arkin, A.P.; Schaffer, D.V. Stochastic gene expression in a lentiviral positive-feedback loop: HIV-1 Tat fluctuations drive phenotypic diversity. Cell 2005, 122, 169–182. [Google Scholar] [CrossRef] [PubMed]
- Massari, S.; Sabatini, S.; Tabarrini, O. Blocking HIV-1 replication by targeting the Tat-hijacked transcriptional machinery. Curr. Pharm. Des. 2013, 19, 1860–1879. [Google Scholar] [CrossRef] [PubMed]
- Mousseau, G.; Valente, S. Strategies to Block HIV Transcription: Focus on Small Molecule Tat Inhibitors. Biology (Basel) 2012, 1, 668–697. [Google Scholar] [CrossRef] [PubMed]
- Mousseau, G.; Clementz, M.A.; Bakeman, W.N.; Nagarsheth, N.; Cameron, M.; Shi, J.; Baran, P.; Fromentin, R.; Chomont, N.; Valente, S.T. An analog of the natural steroidal alkaloid cortistatin A potently suppresses Tat-dependent HIV transcription. Cell Host Microbe 2012, 12, 97–108. [Google Scholar] [CrossRef]
- Li, C.; Mousseau, G.; Valente, S.T. Tat inhibition by didehydro-Cortistatin A promotes heterochromatin formation at the HIV-1 long terminal repeat. Epigenetics Chromatin 2019, 12, 23. [Google Scholar] [CrossRef]
- Mousseau, G.; Kessing, C.F.; Fromentin, R.; Trautmann, L.; Chomont, N.; Valente, S.T. The Tat Inhibitor Didehydro-Cortistatin A Prevents HIV-1 Reactivation from Latency. MBio 2015, 6, e00465. [Google Scholar] [CrossRef]
- Kessing, C.F.; Nixon, C.C.; Li, C.; Tsai, P.; Takata, H.; Mousseau, G.; Ho, P.T.; Honeycutt, J.B.; Fallahi, M.; Trautmann, L.; et al. In Vivo Suppression of HIV Rebound by Didehydro-Cortistatin A, a “Block-and-Lock” Strategy for HIV-1 Treatment. Cell Rep. 2017, 21, 600–611. [Google Scholar] [CrossRef]
- Mousseau, G.; Aneja, R.; Clementz, M.A.; Mediouni, S.; Lima, N.S.; Haregot, A.; Kessing, C.F.; Jablonski, J.A.; Thenin-Houssier, S.; Nagarsheth, N.; et al. Resistance to the Tat Inhibitor Didehydro-Cortistatin A Is Mediated by Heightened Basal HIV-1 Transcription. MBio 2019, 10. [Google Scholar] [CrossRef]
- Sen, G.L.; Blau, H.M. A brief history of RNAi: The silence of the genes. FASEB J. 2006, 20, 1293–1299. [Google Scholar] [CrossRef]
- Setten, R.L.; Rossi, J.J.; Han, S.P. The current state and future directions of RNAi-based therapeutics. Nat. Rev. Drug Discov. 2019, 18, 421–446. [Google Scholar] [CrossRef]
- Stevenson, M. Dissecting HIV-1 through RNA interference. Nat. Rev. Immunol. 2003, 3, 851–858. [Google Scholar] [CrossRef] [PubMed]
- Boden, D.; Pusch, O.; Lee, F.; Tucker, L.; Ramratnam, B. Human immunodeficiency virus type 1 escape from RNA interference. J. Virol. 2003, 77, 11531–11535. [Google Scholar] [CrossRef] [PubMed]
- Das, A.T.; Brummelkamp, T.R.; Westerhout, E.M.; Vink, M.; Madiredjo, M.; Bernards, R.; Berkhout, B. Human immunodeficiency virus type 1 escapes from RNA interference-mediated inhibition. J. Virol. 2004, 78, 2601–2605. [Google Scholar] [CrossRef] [PubMed]
- Westerhout, E.M.; Ooms, M.; Vink, M.; Das, A.T.; Berkhout, B. HIV-1 can escape from RNA interference by evolving an alternative structure in its RNA genome. Nucleic Acids Res. 2005, 33, 796–804. [Google Scholar] [CrossRef]
- Bobbin, M.L.; Burnett, J.C.; Rossi, J.J. RNA interference approaches for treatment of HIV-1 infection. Genome Med. 2015, 7, 50. [Google Scholar] [CrossRef]
- Kretova, O.V.; Fedoseeva, D.M.; Gorbacheva, M.A.; Gashnikova, N.M.; Gashnikova, M.P.; Melnikova, N.V.; Chechetkin, V.R.; Kravatsky, Y.V.; Tchurikov, N.A. Six Highly Conserved Targets of RNAi Revealed in HIV-1-Infected Patients from Russia Are Also Present in Many HIV-1 Strains Worldwide. Mol. Ther. Nucleic Acids 2017, 8, 330–344. [Google Scholar] [CrossRef]
- Choi, J.G.; Bharaj, P.; Abraham, S.; Ma, H.; Yi, G.; Ye, C.; Dang, Y.; Manjunath, N.; Wu, H.; Shankar, P. Multiplexing seven miRNA-Based shRNAs to suppress HIV replication. Mol. Ther. 2015, 23, 310–320. [Google Scholar] [CrossRef]
- Singh, S.K.; Gaur, R.K. Progress towards therapeutic application of RNA interference for HIV infection. BioDrugs 2009, 23, 269–276. [Google Scholar] [CrossRef]
- Soejitno, A.; Wihandani, D.M.; Kuswardhani, T. The therapeutic potential of RNA interference in controlling HIV-1 replication. Acta Med. Indones. 2009, 41, 215–221. [Google Scholar]
- Swamy, M.N.; Wu, H.; Shankar, P. Recent advances in RNAi-based strategies for therapy and prevention of HIV-1/AIDS. Adv. Drug Deliv. Rev. 2016, 103, 174–186. [Google Scholar] [CrossRef]
- DiGiusto, D.L.; Krishnan, A.; Li, L.; Li, H.; Li, S.; Rao, A.; Mi, S.; Yam, P.; Stinson, S.; Kalos, M.; et al. RNA-based gene therapy for HIV with lentiviral vector-modified CD34(+) cells in patients undergoing transplantation for AIDS-related lymphoma. Sci. Transl. Med. 2010, 2, 36ra43. [Google Scholar] [CrossRef] [PubMed]
- Smith, I.; Greenside, P.G.; Natoli, T.; Lahr, D.L.; Wadden, D.; Tirosh, I.; Narayan, R.; Root, D.E.; Golub, T.R.; Subramanian, A.; et al. Evaluation of RNAi and CRISPR technologies by large-scale gene expression profiling in the Connectivity Map. PLoS Biol. 2017, 15, e2003213. [Google Scholar] [CrossRef] [PubMed]
- Fellmann, C.; Lowe, S.W. Stable RNA interference rules for silencing. Nat. Cell Biol. 2014, 16, 10–18. [Google Scholar] [CrossRef] [PubMed]
- McManus, W.R.; Bale, M.J.; Spindler, J.; Wiegand, A.; Musick, A.; Patro, S.C.; Sobolewski, M.D.; Musick, V.K.; Anderson, E.M.; Cyktor, J.C.; et al. HIV-1 in lymph nodes is maintained by cellular proliferation during antiretroviral therapy. J. Clin. Investig. 2019, 130, 4629–4642. [Google Scholar] [CrossRef]
- Pardons, M.; Baxter, A.E.; Massanella, M.; Pagliuzza, A.; Fromentin, R.; Dufour, C.; Leyre, L.; Routy, J.P.; Kaufmann, D.E.; Chomont, N. Single-cell characterization and quantification of translation-competent viral reservoirs in treated and untreated HIV infection. PLoS Pathog. 2019, 15, e1007619. [Google Scholar] [CrossRef]
- Wiegand, A.; Spindler, J.; Hong, F.F.; Shao, W.; Cyktor, J.C.; Cillo, A.R.; Halvas, E.K.; Coffin, J.M.; Mellors, J.W.; Kearney, M.F. Single-cell analysis of HIV-1 transcriptional activity reveals expression of proviruses in expanded clones during ART. Proc. Natl. Acad. Sci. USA 2017, 114, E3659–E3668. [Google Scholar] [CrossRef]
- Stone, D.; Kiem, H.P.; Jerome, K.R. Targeted gene disruption to cure HIV. Curr. Opin. HIV AIDS 2013, 8, 217–223. [Google Scholar] [CrossRef]
- Maeder, M.L.; Gersbach, C.A. Genome-editing Technologies for Gene and Cell Therapy. Mol. Ther. 2016, 24, 430–446. [Google Scholar] [CrossRef]
- Rogers, G.L.; Cannon, P.M. Gene Therapy Approaches to Human Immunodeficiency Virus and Other Infectious Diseases. Hematol. Oncol. Clin. North Am. 2017, 31, 883–895. [Google Scholar] [CrossRef]
- Belfort, M.; Bonocora, R.P. Homing endonucleases: From genetic anomalies to programmable genomic clippers. Methods Mol. Biol. 2014, 1123, 1–26. [Google Scholar] [CrossRef]
- Dhanasekaran, M.; Negi, S.; Sugiura, Y. Designer zinc finger proteins: Tools for creating artificial DNA-binding functional proteins. Acc. Chem. Res. 2006, 39, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Papworth, M.; Kolasinska, P.; Minczuk, M. Designer zinc-finger proteins and their applications. Gene 2006, 366, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Wright, D.A.; Thibodeau-Beganny, S.; Sander, J.D.; Winfrey, R.J.; Hirsh, A.S.; Eichtinger, M.; Fu, F.; Porteus, M.H.; Dobbs, D.; Voytas, D.F.; et al. Standardized reagents and protocols for engineering zinc finger nucleases by modular assembly. Nat. Protoc. 2006, 1, 1637–1652. [Google Scholar] [CrossRef] [PubMed]
- Shim, G.; Kim, D.; Park, G.T.; Jin, H.; Suh, S.K.; Oh, Y.K. Therapeutic gene editing: Delivery and regulatory perspectives. Acta Pharmacol. Sin. 2017, 38, 738–753. [Google Scholar] [CrossRef] [PubMed]
- Qu, X.; Wang, P.; Ding, D.; Li, L.; Wang, H.; Ma, L.; Zhou, X.; Liu, S.; Lin, S.; Wang, X.; et al. Zinc-finger-nucleases mediate specific and efficient excision of HIV-1 proviral DNA from infected and latently infected human T cells. Nucleic Acids Res. 2013, 41, 7771–7782. [Google Scholar] [CrossRef]
- Wayengera, M. Proviral HIV-genome-wide and pol-gene specific zinc finger nucleases: Usability for targeted HIV gene therapy. Theor. Biol. Med. Model. 2011, 8, 26. [Google Scholar] [CrossRef]
- Ji, H.; Lu, P.; Liu, B.; Qu, X.; Wang, Y.; Jiang, Z.; Yang, X.; Zhong, Y.; Yang, H.; Pan, H.; et al. Zinc-Finger Nucleases Induced by HIV-1 Tat Excise HIV-1 from the Host Genome in Infected and Latently Infected Cells. Mol. Ther. Nucleic Acids 2018, 12, 67–74. [Google Scholar] [CrossRef]
- Eberhardy, S.R.; Goncalves, J.; Coelho, S.; Segal, D.J.; Berkhout, B.; Barbas, C.F., 3rd. Inhibition of human immunodeficiency virus type 1 replication with artificial transcription factors targeting the highly conserved primer-binding site. J. Virol. 2006, 80, 2873–2883. [Google Scholar] [CrossRef]
- Kim, Y.S.; Kim, J.M.; Jung, D.L.; Kang, J.E.; Lee, S.; Kim, J.S.; Seol, W.; Shin, H.C.; Kwon, H.S.; Van Lint, C.; et al. Artificial zinc finger fusions targeting Sp1-binding sites and the trans-activator-responsive element potently repress transcription and replication of HIV-1. J. Biol. Chem. 2005, 280, 21545–21552. [Google Scholar] [CrossRef]
- Segal, D.J.; Goncalves, J.; Eberhardy, S.; Swan, C.H.; Torbett, B.E.; Li, X.; Barbas, C.F., 3rd. Attenuation of HIV-1 replication in primary human cells with a designed zinc finger transcription factor. J. Biol. Chem. 2004, 279, 14509–14519. [Google Scholar] [CrossRef]
- Reynolds, L.; Ullman, C.; Moore, M.; Isalan, M.; West, M.J.; Clapham, P.; Klug, A.; Choo, Y. Repression of the HIV-1 5’ LTR promoter and inhibition of HIV-1 replication by using engineered zinc-finger transcription factors. Proc. Natl. Acad. Sci. USA 2003, 100, 1615–1620. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Qu, X.; Lu, P.; Yang, X.; Zhu, Y.; Ji, H.; Wang, Y.; Jiang, Z.; Li, X.; Zhong, Y.; et al. Specific and Stable Suppression of HIV Provirus Expression In Vitro by Chimeric Zinc Finger DNA Methyltransferase 1. Mol. Ther. Nucleic Acids 2017, 6, 233–242. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Qu, X.; Wang, X.; Zhu, X.; Zeng, H.; Chen, H.; Zhu, H. Specific reactivation of latent HIV-1 with designer zinc-finger transcription factors targeting the HIV-1 5′-LTR promoter. Gene Ther. 2014, 21, 490–495. [Google Scholar] [CrossRef] [PubMed]
- Bogdanove, A.J.; Schornack, S.; Lahaye, T. TAL effectors: Finding plant genes for disease and defense. Curr. Opin. Plant Biol. 2010, 13, 394–401. [Google Scholar] [CrossRef]
- Samson, M.; Libert, F.; Doranz, B.J.; Rucker, J.; Liesnard, C.; Farber, C.M.; Saragosti, S.; Lapoumeroulie, C.; Cognaux, J.; Forceille, C.; et al. Resistance to HIV-1 infection in caucasian individuals bearing mutant alleles of the CCR-5 chemokine receptor gene. Nature 1996, 382, 722–725. [Google Scholar] [CrossRef]
- Holt, N.; Wang, J.; Kim, K.; Friedman, G.; Wang, X.; Taupin, V.; Crooks, G.M.; Kohn, D.B.; Gregory, P.D.; Holmes, M.C.; et al. Human hematopoietic stem/progenitor cells modified by zinc-finger nucleases targeted to CCR5 control HIV-1 in vivo. Nat. Biotechnol. 2010, 28, 839–847. [Google Scholar] [CrossRef]
- Didigu, C.A.; Wilen, C.B.; Wang, J.; Duong, J.; Secreto, A.J.; Danet-Desnoyers, G.A.; Riley, J.L.; Gregory, P.D.; June, C.H.; Holmes, M.C.; et al. Simultaneous zinc-finger nuclease editing of the HIV coreceptors ccr5 and cxcr4 protects CD4+ T cells from HIV-1 infection. Blood 2014, 123, 61–69. [Google Scholar] [CrossRef]
- Tebas, P.; Stein, D.; Tang, W.W.; Frank, I.; Wang, S.Q.; Lee, G.; Spratt, S.K.; Surosky, R.T.; Giedlin, M.A.; Nichol, G.; et al. Gene editing of CCR5 in autologous CD4 T cells of persons infected with HIV. N. Engl. J. Med. 2014, 370, 901–910. [Google Scholar] [CrossRef]
- Allers, K.; Schneider, T. CCR5Delta32 mutation and HIV infection: Basis for curative HIV therapy. Curr. Opin. Virol. 2015, 14, 24–29. [Google Scholar] [CrossRef]
- Joung, J.K.; Sander, J.D. TALENs: A widely applicable technology for targeted genome editing. Nat. Rev. Mol. Cell Biol. 2013, 14, 49–55. [Google Scholar] [CrossRef]
- Ebina, H.; Kanemura, Y.; Misawa, N.; Sakuma, T.; Kobayashi, T.; Yamamoto, T.; Koyanagi, Y. A high excision potential of TALENs for integrated DNA of HIV-based lentiviral vector. PLoS ONE 2015, 10, e0120047. [Google Scholar] [CrossRef] [PubMed]
- Strong, C.L.; Guerra, H.P.; Mathew, K.R.; Roy, N.; Simpson, L.R.; Schiller, M.R. Damaging the Integrated HIV Proviral DNA with TALENs. PLoS ONE 2015, 10, e0125652. [Google Scholar] [CrossRef] [PubMed]
- Mock, U.; Machowicz, R.; Hauber, I.; Horn, S.; Abramowski, P.; Berdien, B.; Hauber, J.; Fehse, B. mRNA transfection of a novel TAL effector nuclease (TALEN) facilitates efficient knockout of HIV co-receptor CCR5. Nucleic Acids Res. 2015, 43, 5560–5571. [Google Scholar] [CrossRef]
- Liu, J.; Gaj, T.; Patterson, J.T.; Sirk, S.J.; Barbas, C.F., 3rd. Cell-penetrating peptide-mediated delivery of TALEN proteins via bioconjugation for genome engineering. PLoS ONE 2014, 9, e85755. [Google Scholar] [CrossRef]
- Ru, R.; Yao, Y.; Yu, S.; Yin, B.; Xu, W.; Zhao, S.; Qin, L.; Chen, X. Targeted genome engineering in human induced pluripotent stem cells by penetrating TALENs. Cell Regen. (Lond.) 2013, 2, 5. [Google Scholar] [CrossRef] [PubMed]
- Mussolino, C.; Morbitzer, R.; Lutge, F.; Dannemann, N.; Lahaye, T.; Cathomen, T. A novel TALE nuclease scaffold enables high genome editing activity in combination with low toxicity. Nucleic Acids Res. 2011, 39, 9283–9293. [Google Scholar] [CrossRef]
- Shi, B.; Li, J.; Shi, X.; Jia, W.; Wen, Y.; Hu, X.; Zhuang, F.; Xi, J.; Zhang, L. TALEN-Mediated Knockout of CCR5 Confers Protection Against Infection of Human Immunodeficiency Virus. J. Acquir. Immune Defic. Syndr. 2017, 74, 229–241. [Google Scholar] [CrossRef]
- Perdigao, P.; Gaj, T.; Santa-Marta, M.; Barbas, C.F., 3rd; Goncalves, J. Reactivation of Latent HIV-1 Expression by Engineered TALE Transcription Factors. PLoS ONE 2016, 11, e0150037. [Google Scholar] [CrossRef]
- Wang, X.; Wang, P.; Fu, Z.; Ji, H.; Qu, X.; Zeng, H.; Zhu, X.; Deng, J.; Lu, P.; Zha, S.; et al. Designed transcription activator-like effector proteins efficiently induced the expression of latent HIV-1 in latently infected cells. AIDS Res. Hum. Retroviruses 2015, 31, 98–106. [Google Scholar] [CrossRef]
- Geissler, R.; Hauber, I.; Funk, N.; Richter, A.; Behrens, M.; Renner, I.; Chemnitz, J.; Hofmann-Sieber, H.; Baum, H.; van Lunzen, J.; et al. Patient-adapted, specific activation of HIV-1 by customized TAL effectors (TALEs), a proof of principle study. Virology 2015, 486, 248–254. [Google Scholar] [CrossRef][Green Version]
- Zych, A.O.; Bajor, M.; Zagozdzon, R. Application of Genome Editing Techniques in Immunology. Arch. Immunol. Ther. Exp. (Warsz.) 2018, 66, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Lino, C.A.; Harper, J.C.; Carney, J.P.; Timlin, J.A. Delivering CRISPR: A review of the challenges and approaches. Drug Deliv. 2018, 25, 1234–1257. [Google Scholar] [CrossRef] [PubMed]
- Bloom, K.; Mussolino, C.; Arbuthnot, P. Transcription Activator-Like Effector (TALE) Nucleases and Repressor TALEs for Antiviral Gene Therapy. Curr. Stem Cell Rep. 2015, 1, 1–8. [Google Scholar] [CrossRef]
- Ishino, Y.; Shinagawa, H.; Makino, K.; Amemura, M.; Nakata, A. Nucleotide sequence of the iap gene, responsible for alkaline phosphatase isozyme conversion in Escherichia coli, and identification of the gene product. J. Bacteriol. 1987, 169, 5429–5433. [Google Scholar] [CrossRef] [PubMed]
- Sorek, R.; Kunin, V.; Hugenholtz, P. CRISPR--a widespread system that provides acquired resistance against phages in bacteria and archaea. Nat. Rev. Microbiol. 2008, 6, 181–186. [Google Scholar] [CrossRef]
- Xiao, Q.; Guo, D.; Chen, S. Application of CRISPR/Cas9-Based Gene Editing in HIV-1/AIDS Therapy. Front Cell Infect. Microbiol. 2019, 9, 69. [Google Scholar] [CrossRef]
- Gilbert, L.A.; Larson, M.H.; Morsut, L.; Liu, Z.; Brar, G.A.; Torres, S.E.; Stern-Ginossar, N.; Brandman, O.; Whitehead, E.H.; Doudna, J.A.; et al. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell 2013, 154, 442–451. [Google Scholar] [CrossRef]
- Charlesworth, C.T.; Deshpande, P.S.; Dever, D.P.; Camarena, J.; Lemgart, V.T.; Cromer, M.K.; Vakulskas, C.A.; Collingwood, M.A.; Zhang, L.; Bode, N.M.; et al. Identification of preexisting adaptive immunity to Cas9 proteins in humans. Nat. Med. 2019, 25, 249–254. [Google Scholar] [CrossRef]
- Ebina, H.; Misawa, N.; Kanemura, Y.; Koyanagi, Y. Harnessing the CRISPR/Cas9 system to disrupt latent HIV-1 provirus. Sci. Rep. 2013, 3, 2510. [Google Scholar] [CrossRef]
- Khalili, W.H.; Rafal, K.; Fan, Y.; Yonggang, Z.; Laura, C.; Fang, L.; Biao, L.; David, A.-C.; Yoelvis, G.-M.; Jonathan, K.; et al. RNA-directed gene editing specifically eradicates latent and prevents new HIV-1 infection. Proc. Natl. Acad. Sci. USA 2014. [Google Scholar] [CrossRef]
- Belmonte, H.-K.L.; Ying, G.; Arturo, D.; John, M.; Yuta, T.; Mo, L.; Keiichiro, S.; Ruo, X.; Tomoaki, H.; Chan-Jung, C.; et al. Use of the CRISPR/Cas9 system as an intracellular defense against HIV-1 infection in human cells. Nat. Commun. 2015, 6, 6413. [Google Scholar] [CrossRef]
- Wang, Q.; Liu, S.; Liu, Z.; Ke, Z.; Li, C.; Yu, X.; Chen, S.; Guo, D. Genome scale screening identification of SaCas9/gRNAs for targeting HIV-1 provirus and suppression of HIV-1 infection. Virus Res. 2018, 250, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Ophinni, Y.; Inoue, M.; Kotaki, T.; Kameoka, M. CRISPR/Cas9 system targeting regulatory genes of HIV-1 inhibits viral replication in infected T-cell cultures. Sci. Rep. 2018, 8, 7784. [Google Scholar] [CrossRef]
- Lebbink, R.J.; de Jong, D.C.; Wolters, F.; Kruse, E.M.; van Ham, P.M.; Wiertz, E.J.; Nijhuis, M. A combinational CRISPR/Cas9 gene-editing approach can halt HIV replication and prevent viral escape. Sci. Rep. 2017, 7, 41968. [Google Scholar] [CrossRef]
- Kaminski, R.; Chen, Y.; Fischer, T.; Tedaldi, E.; Napoli, A.; Zhang, Y.; Karn, J.; Hu, W.; Khalili, K. Elimination of HIV-1 Genomes from Human T-lymphoid Cells by CRISPR/Cas9 Gene Editing. Sci. Rep. 2016, 6, 22555. [Google Scholar] [CrossRef] [PubMed]
- Bella, R.; Kaminski, R.; Mancuso, P.; Young, W.B.; Chen, C.; Sariyer, R.; Fischer, T.; Amini, S.; Ferrante, P.; Jacobson, J.M.; et al. Removal of HIV DNA by CRISPR from Patient Blood Engrafts in Humanized Mice. Mol. Ther. Nucleic Acids 2018, 12, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Kaminski, R.; Bella, R.; Yin, C.; Otte, J.; Ferrante, P.; Gendelman, H.E.; Li, H.; Booze, R.; Gordon, J.; Hu, W.; et al. Excision of HIV-1 DNA by gene editing: A proof-of-concept in vivo study. Gene Ther. 2016, 23, 690–695. [Google Scholar] [CrossRef]
- Yin, C.; Zhang, T.; Qu, X.; Zhang, Y.; Putatunda, R.; Xiao, X.; Li, F.; Xiao, W.; Zhao, H.; Dai, S.; et al. In Vivo Excision of HIV-1 Provirus by saCas9 and Multiplex Single-Guide RNAs in Animal Models. Mol. Ther. 2017, 25, 1168–1186. [Google Scholar] [CrossRef]
- Dash, P.K.; Kaminski, R.; Bella, R.; Su, H.; Mathews, S.; Ahooyi, T.M.; Chen, C.; Mancuso, P.; Sariyer, R.; Ferrante, P.; et al. Sequential LASER ART and CRISPR Treatments Eliminate HIV-1 in a Subset of Infected Humanized Mice. Nat. Commun. 2019, 10, 2753. [Google Scholar] [CrossRef]
- Yoder, K.E.; Bundschuh, R. Host Double Strand Break Repair Generates HIV-1 Strains Resistant to CRISPR/Cas9. Sci. Rep. 2016, 6, 29530. [Google Scholar] [CrossRef]
- Wang, Z.; Pan, Q.; Gendron, P.; Zhu, W.; Guo, F.; Cen, S.; Wainberg, M.A.; Liang, C. CRISPR/Cas9-Derived Mutations Both Inhibit HIV-1 Replication and Accelerate Viral Escape. Cell Rep. 2016, 15, 481–489. [Google Scholar] [CrossRef]
- Wang, G.; Zhao, N.; Berkhout, B.; Das, A.T. CRISPR-Cas9 Can Inhibit HIV-1 Replication but NHEJ Repair Facilitates Virus Escape. Mol. Ther. 2016, 24, 522–526. [Google Scholar] [CrossRef] [PubMed]
- Ueda, S.; Ebina, H.; Kanemura, Y.; Misawa, N.; Koyanagi, Y. Anti-HIV-1 potency of the CRISPR/Cas9 system insufficient to fully inhibit viral replication. Microbiol. Immunol. 2016, 60, 483–496. [Google Scholar] [CrossRef]
- Wang, G.; Zhao, N.; Berkhout, B.; Das, A.T. A Combinatorial CRISPR-Cas9 Attack on HIV-1 DNA Extinguishes All Infectious Provirus in Infected T Cell Cultures. Cell Rep. 2016, 17, 2819–2826. [Google Scholar] [CrossRef] [PubMed]
- Jiang, F.; Doudna, J.A. CRISPR-Cas9 Structures and Mechanisms. Annu. Rev. Biophys. 2017, 46, 505–529. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.H.; Tee, L.Y.; Wang, X.G.; Huang, Q.S.; Yang, S.H. Off-target Effects in CRISPR/Cas9-mediated Genome Engineering. Mol. Ther. Nucleic Acids 2015, 4, e264. [Google Scholar] [CrossRef]
- Ye, L.; Wang, J.; Beyer, A.I.; Teque, F.; Cradick, T.J.; Qi, Z.; Chang, J.C.; Bao, G.; Muench, M.O.; Yu, J.; et al. Seamless modification of wild-type induced pluripotent stem cells to the natural CCR5Delta32 mutation confers resistance to HIV infection. Proc. Natl. Acad. Sci. USA 2014, 111, 9591–9596. [Google Scholar] [CrossRef]
- Li, C.; Guan, X.; Du, T.; Jin, W.; Wu, B.; Liu, Y.; Wang, P.; Hu, B.; Griffin, G.E.; Shattock, R.J.; et al. Inhibition of HIV-1 infection of primary CD4+ T-cells by gene editing of CCR5 using adenovirus-delivered CRISPR/Cas9. J. Gen. Virol. 2015, 96, 2381–2393. [Google Scholar] [CrossRef]
- Xu, L.; Yang, H.; Gao, Y.; Chen, Z.; Xie, L.; Liu, Y.; Liu, Y.; Wang, X.; Li, H.; Lai, W.; et al. CRISPR/Cas9-Mediated CCR5 Ablation in Human Hematopoietic Stem/Progenitor Cells Confers HIV-1 Resistance In Vivo. Mol. Ther. 2017, 25, 1782–1789. [Google Scholar] [CrossRef]
- Xiao, Q.; Chen, S.; Wang, Q.; Liu, Z.; Liu, S.; Deng, H.; Hou, W.; Wu, D.; Xiong, Y.; Li, J.; et al. CCR5 editing by Staphylococcus aureus Cas9 in human primary CD4(+) T cells and hematopoietic stem/progenitor cells promotes HIV-1 resistance and CD4(+) T cell enrichment in humanized mice. Retrovirology 2019, 16, 15. [Google Scholar] [CrossRef]
- Hou, P.; Chen, S.; Wang, S.; Yu, X.; Chen, Y.; Jiang, M.; Zhuang, K.; Ho, W.; Hou, W.; Huang, J.; et al. Genome editing of CXCR4 by CRISPR/cas9 confers cells resistant to HIV-1 infection. Sci. Rep. 2015, 5, 15577. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Chen, S.; Xiao, Q.; Liu, Z.; Liu, S.; Hou, P.; Zhou, L.; Hou, W.; Ho, W.; Li, C.; et al. Genome modification of CXCR4 by Staphylococcus aureus Cas9 renders cells resistance to HIV-1 infection. Retrovirology 2017, 14, 51. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Yao, Y.; Xiao, H.; Li, J.; Liu, Q.; Yang, Y.; Adah, D.; Lu, J.; Zhao, S.; Qin, L.; et al. Simultaneous Knockout of CXCR4 and CCR5 Genes in CD4+ T Cells via CRISPR/Cas9 Confers Resistance to Both X4- and R5-Tropic Human Immunodeficiency Virus Type 1 Infection. Hum. Gene Ther. 2018, 29, 51–67. [Google Scholar] [CrossRef] [PubMed]
- Dar, A.; Kollet, O.; Lapidot, T. Mutual, reciprocal SDF-1/CXCR4 interactions between hematopoietic and bone marrow stromal cells regulate human stem cell migration and development in NOD/SCID chimeric mice. Exp. Hematol. 2006, 34, 967–975. [Google Scholar] [CrossRef] [PubMed]
- Kim, V.; Mears, B.M.; Powell, B.H.; Witwer, K.W. Mutant Cas9-transcriptional activator activates HIV-1 in U1 cells in the presence and absence of LTR-specific guide RNAs. Matters (Zur.) 2017, 2017. [Google Scholar] [CrossRef] [PubMed]
- Saayman, S.M.; Lazar, D.C.; Scott, T.A.; Hart, J.R.; Takahashi, M.; Burnett, J.C.; Planelles, V.; Morris, K.V.; Weinberg, M.S. Potent and Targeted Activation of Latent HIV-1 Using the CRISPR/dCas9 Activator Complex. Mol. Ther. 2016, 24, 488–498. [Google Scholar] [CrossRef]
- Limsirichai, P.; Gaj, T.; Schaffer, D.V. CRISPR-mediated Activation of Latent HIV-1 Expression. Mol. Ther. 2016, 24, 499–507. [Google Scholar] [CrossRef]
- Ji, H.; Jiang, Z.; Lu, P.; Ma, L.; Li, C.; Pan, H.; Fu, Z.; Qu, X.; Wang, P.; Deng, J.; et al. Specific Reactivation of Latent HIV-1 by dCas9-SunTag-VP64-mediated Guide RNA Targeting the HIV-1 Promoter. Mol. Ther. 2016, 24, 508–521. [Google Scholar] [CrossRef]
- Bialek, J.K.; Dunay, G.A.; Voges, M.; Schafer, C.; Spohn, M.; Stucka, R.; Hauber, J.; Lange, U.C. Targeted HIV-1 Latency Reversal Using CRISPR/Cas9-Derived Transcriptional Activator Systems. PLoS ONE 2016, 11, e0158294. [Google Scholar] [CrossRef]
- Zhang, Y.; Yin, C.; Zhang, T.; Li, F.; Yang, W.; Kaminski, R.; Fagan, P.R.; Putatunda, R.; Young, W.B.; Khalili, K.; et al. CRISPR/gRNA-directed synergistic activation mediator (SAM) induces specific, persistent and robust reactivation of the HIV-1 latent reservoirs. Sci. Rep. 2015, 5, 16277. [Google Scholar] [CrossRef]
- Qu, D.; Li, C.; Sang, F.; Li, Q.; Jiang, Z.Q.; Xu, L.R.; Guo, H.J.; Zhang, C.; Wang, J.H. The variances of Sp1 and NF-kappaB elements correlate with the greater capacity of Chinese HIV-1 B’-LTR for driving gene expression. Sci. Rep. 2016, 6, 34532. [Google Scholar] [CrossRef] [PubMed]
- Bogerd, H.P.; Kornepati, A.V.; Marshall, J.B.; Kennedy, E.M.; Cullen, B.R. Specific induction of endogenous viral restriction factors using CRISPR/Cas-derived transcriptional activators. Proc. Natl. Acad. Sci. USA 2015, 112, E7249–E7256. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Ozono, S.; Yao, W.; Tobiume, M.; Yamaoka, S.; Kishigami, S.; Fujita, H.; Tokunaga, K. CRISPR-mediated activation of endogenous BST-2/tetherin expression inhibits wild-type HIV-1 production. Sci. Rep. 2019, 9, 3134. [Google Scholar] [CrossRef] [PubMed]
- Guha, T.K.; Wai, A.; Hausner, G. Programmable Genome Editing Tools and their Regulation for Efficient Genome Engineering. Comput. Struct. Biotechnol. J. 2017, 15, 146–160. [Google Scholar] [CrossRef]
- Yu, K.R.; Natanson, H.; Dunbar, C.E. Gene Editing of Human Hematopoietic Stem and Progenitor Cells: Promise and Potential Hurdles. Hum. Gene Ther. 2016, 27, 729–740. [Google Scholar] [CrossRef] [PubMed]
- Knipping, F.; Osborn, M.J.; Petri, K.; Tolar, J.; Glimm, H.; von Kalle, C.; Schmidt, M.; Gabriel, R. Genome-wide Specificity of Highly Efficient TALENs and CRISPR/Cas9 for T Cell Receptor Modification. Mol. Ther. Methods Clin. Dev. 2017, 4, 213–224. [Google Scholar] [CrossRef]
- Guha, T.K.; Edgell, D.R. Applications of Alternative Nucleases in the Age of CRISPR/Cas9. Int J. Mol. Sci. 2017, 18, 2565. [Google Scholar] [CrossRef]
- Straubeta, A.; Lahaye, T. Zinc fingers, TAL effectors, or Cas9-based DNA binding proteins: What’s best for targeting desired genome loci? Mol. Plant 2013, 6, 1384–1387. [Google Scholar] [CrossRef]
- Chevalier, B.S.; Stoddard, B.L. Homing endonucleases: Structural and functional insight into the catalysts of intron/intein mobility. Nucleic Acids Res. 2001, 29, 3757–3774. [Google Scholar] [CrossRef]
- Stoddard, B.L. Homing endonucleases from mobile group I introns: Discovery to genome engineering. Mob DNA 2014, 5, 7. [Google Scholar] [CrossRef]
- Daboussi, F.; Stoddard, T.J.; Zhang, F. Engineering Meganuclease for Precise Plant Genome Modification. In Engineering Meganuclease for Precise Plant Genome Modification. In Advances in New Technology for Targeted Modification of Plant Genomes; Feng Zhang, H.P., Thomson, J.G., Eds.; Springer Science+Business Media: New York, NY, USA, 2015; pp. 21–38. [Google Scholar] [CrossRef]
- Waryah, C.B.; Moses, C.; Arooj, M.; Blancafort, P. Zinc Fingers, TALEs, and CRISPR Systems: A Comparison of Tools for Epigenome Editing. Methods Mol. Biol. 2018, 1767, 19–63. [Google Scholar] [CrossRef] [PubMed]
- Mehta, A.; Haber, J.E. Sources of DNA double-strand breaks and models of recombinational DNA repair. Cold Spring Harb. Perspect. Biol. 2014, 6, a016428. [Google Scholar] [CrossRef] [PubMed]
- Rodgers, K.; McVey, M. Error-Prone Repair of DNA Double-Strand Breaks. J. Cell Physiol. 2016, 231, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Clarridge, K.E.; Blazkova, J.; Einkauf, K.; Petrone, M.; Refsland, E.W.; Justement, J.S.; Shi, V.; Huiting, E.D.; Seamon, C.A.; Lee, G.Q.; et al. Effect of analytical treatment interruption and reinitiation of antiretroviral therapy on HIV reservoirs and immunologic parameters in infected individuals. PLoS Pathog. 2018, 14, e1006792. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HE | ZFP | TALE | CRISPR | |
|---|---|---|---|---|
| Target sequence constraints | High (pre-defined targets) | No constraints (any sequence) | No constraints (any sequence) | Low (PAM required) |
| Target size [213,254] | 12–44 | 18–36 | 24–40 | 17–23 |
| Design and assembly [213] | Difficult | Moderate | Easy | Very easy |
| Toxicity [206,211,255] | Low | High | Low | Unclear, high in some cell lineages |
| Specificity [211,254,256,257] | High | Low to Moderate | Moderate to High | Low to Moderate |
| Multiplexing suitability [254] | Low | Low | Moderate | High |
| Source [254] | Organelles, Bacteria, Phages | Bacteria, Eukaryotes | Bacteria | Bacteria |
| Cost to generate knockout reagent [254] | 4000–5000 USD | 5000–10,000 USD | <1000 USD | <100 USD |
| Immunogenicity [218,254] | Unknown | Low | Unknown | Prevalent pre-existing immunity |
| Size of effector protein in kDa [258,259] | <40 | ~40 | ~105 | ~160 |
| Average length of effector protein [258,260] | 200–300 aa | 120–180 aa | 660–700 aa | 1400 aa |
| Sensitivity to chromatin condensation [261,262] | Sensitive to chromatin compactation and CpG Methylation | Binds condensed and hypermethylated DNA | Potentially decreased binding of condensed DNA | Targeting of hypermethylated CpG islands may be limited |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schwarzer, R.; Gramatica, A.; Greene, W.C. Reduce and Control: A Combinatorial Strategy for Achieving Sustained HIV Remissions in the Absence of Antiretroviral Therapy. Viruses 2020, 12, 188. https://doi.org/10.3390/v12020188
Schwarzer R, Gramatica A, Greene WC. Reduce and Control: A Combinatorial Strategy for Achieving Sustained HIV Remissions in the Absence of Antiretroviral Therapy. Viruses. 2020; 12(2):188. https://doi.org/10.3390/v12020188
Chicago/Turabian StyleSchwarzer, Roland, Andrea Gramatica, and Warner C. Greene. 2020. "Reduce and Control: A Combinatorial Strategy for Achieving Sustained HIV Remissions in the Absence of Antiretroviral Therapy" Viruses 12, no. 2: 188. https://doi.org/10.3390/v12020188
APA StyleSchwarzer, R., Gramatica, A., & Greene, W. C. (2020). Reduce and Control: A Combinatorial Strategy for Achieving Sustained HIV Remissions in the Absence of Antiretroviral Therapy. Viruses, 12(2), 188. https://doi.org/10.3390/v12020188