
Assessment of Viral Targeted Sequence Capture Using Nanopore Sequencing Directly from Clinical Samples


 and
and
Abstract
1. Introduction
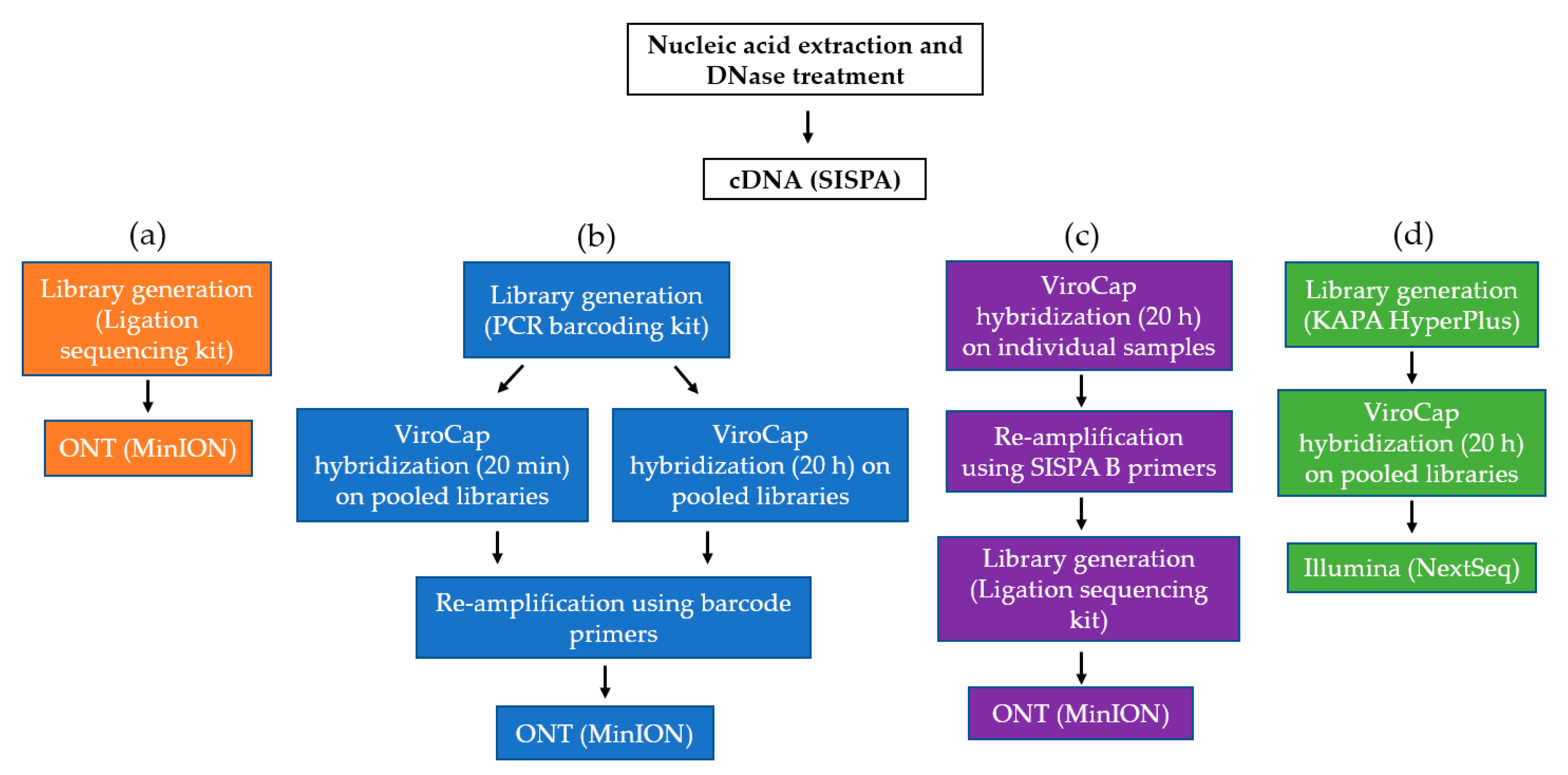
2. Materials and Methods
2.1. Sample Collection
2.2. Nucleic Acid Extraction and cDNA Synthesis
2.3. Oxford Nanopore Technologies SMg and TSC
2.4. Illumina TSC
2.5. Data Analysis
2.6. Ethics Statement
2.7. Data Availability
3. Results
3.1. Hybridization Time and Sample Pooling on the MinION
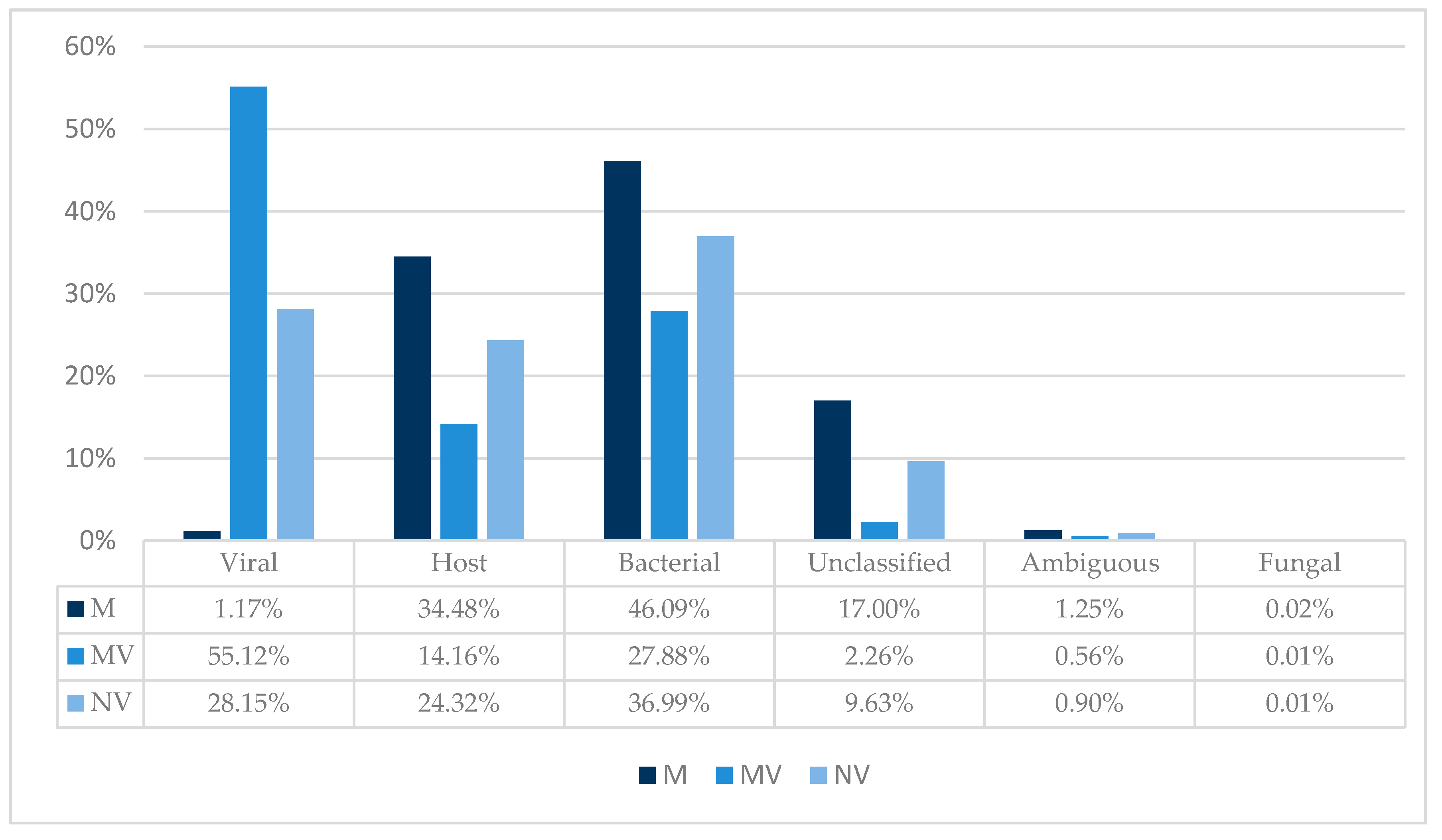
3.2. TSC and Taxonomic Binning
3.3. Viral Genome Coverage and Sequencing Depth
3.4. Viral Detection in Human Samples
3.5. Viral Detection in Animal Samples
3.6. ONT Accuracy
3.7. Coverage Depth of Clinically Relevant Viruses
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Trinh, P.; Zaneveld, J.R.; Safranek, S.; Rabinowitz, P.M. One health relationships between human, animal, and environmental microbiomes: A mini-review. Front. Public Health 2018, 6, 235. [Google Scholar] [CrossRef] [PubMed]
- Wilson, M.R.; Sample, H.A.; Zorn, K.C.; Arevalo, S.; Yu, G.; Neuhaus, J.; Federman, S.; Stryke, D.; Briggs, B.; Langelier, C. Clinical metagenomic sequencing for diagnosis of meningitis and encephalitis. N. Engl. J. Med. 2019, 380, 2327–2340. [Google Scholar] [CrossRef] [PubMed]
- Meredith, L.W.; Hamilton, W.L.; Warne, B.; Houldcroft, C.J.; Hosmillo, M.; Jahun, A.; Curran, M.D.; Parmar, S.; Caller, L.; Caddy, S.L. Rapid implementation of real-time SARS-CoV-2 sequencing to investigate healthcare-associated COVID-19 infections. MedRxiv 2020, 20. [Google Scholar] [CrossRef]
- Smith, G.J.; Vijaykrishna, D.; Bahl, J.; Lycett, S.J.; Worobey, M.; Pybus, O.G.; Ma, S.K.; Cheung, C.L.; Raghwani, J.; Bhatt, S. Origins and evolutionary genomics of the 2009 swine-origin H1N1 influenza A epidemic. Nature 2009, 459, 1122–1125. [Google Scholar] [CrossRef] [PubMed]
- Couto, N.; Schuele, L.; Raangs, E.C.; Machado, M.P.; Mendes, C.I.; Jesus, T.F.; Chlebowicz, M.; Rosema, S.; Ramirez, M.; Carriço, J.A. Critical steps in clinical shotgun metagenomics for the concomitant detection and typing of microbial pathogens. Sci. Rep. 2018, 8, 13767. [Google Scholar] [CrossRef] [PubMed]
- Greninger, A.L. The challenge of diagnostic metagenomics. Expert Rev. Mol. Diagn. 2018, 18, 605–615. [Google Scholar] [CrossRef] [PubMed]
- Quick, J.; Grubaugh, N.D.; Pullan, S.T.; Claro, I.M.; Smith, A.D.; Gangavarapu, K.; Oliveira, G.; Robles-Sikisaka, R.; Rogers, T.F.; Beutler, N.A. Multiplex PCR method for MinION and Illumina sequencing of Zika and other virus genomes directly from clinical samples. Nat. Protoc. 2017, 12, 1261. [Google Scholar] [CrossRef]
- Lewandowski, K.; Xu, Y.; Pullan, S.T.; Lumley, S.F.; Foster, D.; Sanderson, N.; Vaughan, A.; Morgan, M.; Bright, N.; Kavanagh, J. Metagenomic nanopore sequencing of influenza virus direct from clinical respiratory samples. J. Clin. Microbiol. 2019, 58. [Google Scholar] [CrossRef]
- Lizarazo, E.; Couto, N.; Vincenti-Gonzalez, M.; Raangs, E.C.; Velasco, Z.; Bethencourt, S.; Jaenisch, T.; Friedrich, A.W.; Tami, A.; Rossen, J.W. Applied shotgun metagenomics approach for the genetic characterization of dengue viruses. J. Biotechnol. 2019, 2, 100009. [Google Scholar] [CrossRef]
- Briese, T.; Kapoor, A.; Mishra, N.; Jain, K.; Kumar, A.; Jabado, O.J.; Lipkin, W.I. Virome capture sequencing enables sensitive viral diagnosis and comprehensive virome analysis. mBio 2015, 6. [Google Scholar] [CrossRef]
- Wylie, T.N.; Wylie, K.M.; Herter, B.N.; Storch, G.A. Enhanced virome sequencing using targeted sequence capture. Genome Res. 2015, 25, 1910–1920. [Google Scholar] [CrossRef] [PubMed]
- Wylie, K.M.; Wylie, T.N.; Buller, R.; Herter, B.; Cannella, M.T.; Storch, G.A. Detection of viruses in clinical samples by use of metagenomic sequencing and targeted sequence capture. J. Clin. Microbiol. 2018, 56. [Google Scholar] [CrossRef] [PubMed]
- Deurenberg, R.H.; Bathoorn, E.; Chlebowicz, M.A.; Couto, N.; Ferdous, M.; García-Cobos, S.; Kooistra-Smid, A.M.; Raangs, E.C.; Rosema, S.; Veloo, A.C. Application of next generation sequencing in clinical microbiology and infection prevention. J. Biotechnol. 2017, 243, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Kafetzopoulou, L.E.; Efthymiadis, K.; Lewandowski, K.; Crook, A.; Carter, D.; Osborne, J.; Aarons, E.; Hewson, R.; Hiscox, J.A.; Carroll, M.W. Assessment of metagenomic Nanopore and Illumina sequencing for recovering whole genome sequences of chikungunya and dengue viruses directly from clinical samples. Eurosurveillance 2018, 23, 1800228. [Google Scholar] [CrossRef]
- McNaughton, A.L.; Roberts, H.E.; Bonsall, D.; de Cesare, M.; Mokaya, J.; Lumley, S.F.; Golubchik, T.; Piazza, P.; Martin, J.B.; de Lara, C. Illumina and Nanopore methods for whole genome sequencing of hepatitis B virus (HBV). Sci. Rep. 2019, 9, 7081. [Google Scholar] [CrossRef]
- Eckert, S.E.; Jackie, Z.-M.C.; Houniet, D. Enrichment by hybridisation of long DNA fragments for Nanopore sequencing. Microb. Genom. 2016, 2, e000087. [Google Scholar] [CrossRef]
- Karamitros, T.; Magiorkinis, G. A novel method for the multiplexed target enrichment of MinION next generation sequencing libraries using PCR-generated baits. Nucleic Acids Res. 2015, 43, e152. [Google Scholar] [CrossRef]
- Poelman, R.; Schölvinck, E.H.; Borger, R.; Niesters, H.G.; van Leer-Buter, C. The emergence of enterovirus D68 in a Dutch University Medical Center and the necessity for routinely screening for respiratory viruses. J. Clin. Virol. 2015, 62, 1–5. [Google Scholar] [CrossRef]
- Chrzastek, K.; Lee, D.-H.; Smith, D.; Sharma, P.; Suarez, D.L.; Pantin-Jackwood, M.; Kapczynski, D.R. Use of Sequence-Independent, Single-Primer-Amplification (SISPA) for rapid detection, identification, and characterization of avian RNA viruses. Virology 2017, 509, 159–166. [Google Scholar] [CrossRef]
- Quick, J. One-pot native barcoding of amplicons. Protoc. Io 2020. [Google Scholar] [CrossRef]
- Flygare, S.; Simmon, K.; Miller, C.; Qiao, Y.; Kennedy, B.; Di Sera, T.; Graf, E.H.; Tardif, K.D.; Kapusta, A.; Rynearson, S. Taxonomer: An interactive metagenomics analysis portal for universal pathogen detection and host mRNA expression profiling. Genome Biol. 2016, 17, 111. [Google Scholar] [CrossRef] [PubMed]
- O’Flaherty, B.M.; Li, Y.; Tao, Y.; Paden, C.R.; Queen, K.; Zhang, J.; Dinwiddie, D.L.; Gross, S.M.; Schroth, G.P.; Tong, S. Comprehensive viral enrichment enables sensitive respiratory virus genomic identification and analysis by next generation sequencing. Genome Res. 2018, 28, 869–877. [Google Scholar] [CrossRef] [PubMed]
- Oba, M.; Tsuchiaka, S.; Omatsu, T.; Katayama, Y.; Otomaru, K.; Hirata, T.; Aoki, H.; Murata, Y.; Makino, S.; Nagai, M. A new comprehensive method for detection of livestock-related pathogenic viruses using a target enrichment system. Biochem. Biophys. Res. Commun. 2018, 495, 1871–1877. [Google Scholar] [CrossRef]
- Leedom-Larson, K.; Barber, E.; Porcine Parainfluenza Virus 1. Swine Health Information Center and Center for Food Security and Pubic Health. 2016. Available online: http://www.cfsph.iastate.edu/pdf/shic-factsheet-porcine-parainfluenza-virus-1 (accessed on 15 May 2020).
- Lau, S.K.; Woo, P.C.; Wu, Y.; Wong, A.Y.; Wong, B.H.; Lau, C.C.; Fan, R.Y.; Cai, J.-P.; Tsoi, H.-W.; Chan, K.-H. Identification and characterization of a novel paramyxovirus, porcine parainfluenza virus 1, from deceased pigs. J. Gen. Virol. 2013, 94, 2184–2190. [Google Scholar] [CrossRef] [PubMed]
- Kwok, K.T.; Nieuwenhuijse, D.F.; Phan, M.V.; Koopmans, M.P. Virus metagenomics in farm animals: A systematic review. Viruses 2020, 12, 107. [Google Scholar] [CrossRef]
- Gallardo, C.; Fernández-Pinero, J.; Arias, M. African swine fever (ASF) diagnosis, an essential tool in the epidemiological investigation. Virus Res. 2019, 271, 197676. [Google Scholar] [CrossRef]
- Bellehumeur, C.; Boyle, B.; Charette, S.J.; Harel, J.; L’Homme, Y.; Masson, L.; Gagnon, C.A. Propidium monoazide (PMA) and ethidium bromide monoazide (EMA) improve DNA array and high-throughput sequencing of porcine reproductive and respiratory syndrome virus identification. J. Virol. Methods 2015, 222, 182–191. [Google Scholar] [CrossRef]
- Fraaij, P.L.; Wildschut, E.D.; Houmes, R.J.; Swaan, C.M.; Hoebe, C.J.; de Jonge, H.; Tolsma, P.; de Kleer, I.; Pas, S.D.; Munnink, B.B.O. Severe acute respiratory infection caused by swine influenza virus in a child necessitating extracorporeal membrane oxygenation (ECMO), The Netherlands, October 2016. Eurosurveillance 2016, 21, 30416. [Google Scholar] [CrossRef]
- Roux, S.; Adriaenssens, E.M.; Dutilh, B.E.; Koonin, E.V.; Kropinski, A.M.; Krupovic, M.; Kuhn, J.H.; Lavigne, R.; Brister, J.R.; Varsani, A.; et al. Minimum Information about an Uncultivated Virus Genome (MIUViG). Nat. Biotechnol. 2018, 37, 29–37. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Sample ID | Sample Type | qPCR Target | Ct Value | Symptoms | Sampling Date |
|---|---|---|---|---|---|
| H1 | Flocked swab nasopharynx | Enterovirus | 25 | Fever, dyspnea and coughing | 09/2018 |
| H2 | Fecal | Enterovirus | 30 | Chronic diarrhea | 05/2016 |
| H3 | Fecal | Norovirus Enterovirus | 17 29 | Fever, vomiting and abdominal pain | 11/2018 |
| H4 | Fecal | Enterovirus | 21 | Fever, diarrhea and abdominal pain | 10/2016 |
| A1 | Blood plasma | PRSSV 1 | - | None | 12/2017 |
| A2 | Blood plasma | PRSSV 1 | 26 (pool) | None | 10/2018 |
| A3 | Blood plasma | PRSSV 1 | 25 (pool) | Respiratory | 10/2017 |
| A4 | Nasal swab | SIV 2 | 19 | Respiratory (closed enteral system) | 10/2018 |
| Type | Detected Viruses | 20 min (Pool) % | 20 h (Pool) % | 20 h (Individual) 1 % |
|---|---|---|---|---|
| Human samples (n = 4) | Coxsackievirus A22 | 0.001 | 0.004 | 0.192 |
| Enterovirus A71 | 0.00004 | 0.00011 | 0.00028 | |
| Enterovirus D68 | 0.003 | 0.027 | 0.775 | |
| Norovirus GII.4 | 1.42 | 4.77 | 10.56 | |
| Animal samples (n = 4) | Astrovirus wild boar (n = 2) | 0.002 | 0.010 | 0.001 |
| Bocavirus pig | 0.001 | 0.007 | 0.003 | |
| Influenza A virus * | 55.31 | 51.15 | 9.39 | |
| Mamastrovirus 2 | 0.0004 | 0.0017 | 0.0003 | |
| Pasivirus A1 | 0.002 | 0.010 | 0.053 | |
| PERV 2 (n = 4) | 10.63 | 11.83 | 11.34 | |
| Porcine astrovirus 4 | 0.001 | 0.008 | 0.001 | |
| Porcine bocavirus H18 | 0.009 | 0.029 | 0.016 | |
| Porcine enterovirus B | 0.0002 | 0.0002 | 4.75 × 10−5 | |
| Porcine kobuvirus | 0.49 | 1.35 | 0.28 | |
| Porcine respirovirus 1 3 | 0.019 | 0.064 | 0.033 | |
| Porcine sapelovirus 1 | 0.0001 | 0.0019 | 0.0003 | |
| PRRSV 4 (n = 3) | 9.75 | 6.82 | 15.75 | |
| Rotavirus C * | 0.00004 | 0.00005 | 0.00007 | |
| Ungulate tetraparvovirus 3 | 10.02 | 0.44 | 5.24 | |
| Total viral reads % | 87.66 | 76.53 | 53.64 |
| ID | GenBank | Reference Length | Reference | Genome Coverage (%) | Average Sequencing Depth | ||||
|---|---|---|---|---|---|---|---|---|---|
| M | MV | NV | M | MV | NV | ||||
| H1 | MH341731.1 | 7345 | Enterovirus D68 | 89 | 100 | 96 | 2 | 3342 | 2664 |
| H2 | ─ | ─ | No virus detected | ─ | ─ | ─ | ─ | ─ | ─ |
| H3 | MK073885.1 | 7555 | Norovirus GII.4 | 99 | 100 | 100 | 55 | 44,148 | 374,921 |
| H3 | LR027546.1 | 7410 | Enterovirus A71 | 20 | 39 | 40 | <1 | 1 | 4 |
| H4 | DQ995647.1 | 7401 | Coxsackievirus A22 | 35 | 100 | 85 | <1 | 898 | 152 |
| Average (Human Samples) | 61 | 85 | 80 | 14 | 12097 | 94,435 | |||
| A1 | HM159246 | 8774 | PERV 1 C | 83 | 71 | 64 | 28 | 7536 | 608 |
| A1 | NC_038546 | 5114 | Porcine hokovirus HK7 | 18 | 41 | 74 | 1 | 977 | 6893 |
| A1 | NC_035180 | 5533 | Ungulate tetraparvovirus 3 | 100 | 100 | 96 | 773 | 35518 | 380,898 |
| A2 | NC_038537 | 4786 | Bocavirus pig/SX/China | 43 | 59 | 82 | 5 | 143 | 425 |
| A2 | NC_038538 | 5267 | Porcine bocavirus P18 | 39 | 56 | 61 | 5 | 130 | 479 |
| A2 | HM159246 | 8774 | PERV 1 C | 100 | 100 | 92 | 408 | 37,379 | 22,903 |
| A2 | NC_025402 | 15396 | Porcine respirovirus 1 | 79 | 85 | 98 | 9 | 75 | 980 |
| A2 | GU067771.1 | 15098 | PRRSV 2 (Amervac) | 89 | 100 | 100 | 21 | 21,836 | 11,949 |
| A3 | NC_016896 | 6707 | Astrovirus wild boar | 10 | 17 | 32 | 60 | 1 | 144 |
| A3 | NC_030653 | 10908 | Atypical porcine pestivirus 1 | 100 | 20 | 100 | 69 | <1 | 36 |
| A3 | NC_018226 | 6916 | Pasivirus A1 | 91 | 78 | 63 | 38 | 243 | 478 |
| A3 | HM159246 | 8774 | PERV 1 C | 33 | 81 | 49 | 2 | 1627 | 62 |
| A3 | KT344816.1 | 15095 | PRRSV 2 (GER09-613) | 99 | 100 | 96 | 85 | 17,451 | 8459 |
| A4 | NC_016896 | 6707 | Astrovirus wild boar | 98 | 81 | 76 | 20 | 12 | 128 |
| A4 | NC_027711 | 6327 | Dromedary astrovirus | 83 | 44 | 38 | 5 | 5 | 23 |
| A4 | KY250316-23 | 13200 | Influenza A virus * | 100 | 100 | 100 | 1512 | 24,292 | 197,905 |
| A4 | NC_034974 | 6347 | Mamastrovirus 2 | 83 | 39 | 51 | 4 | 5 | 22 |
| A4 | NC_023675 | 6639 | Porcine astrovirus 4 | 99 | 82 | 65 | 18 | 12 | 151 |
| A4 | NC_023636 | 6500 | Porcine astrovirus 5 | 46 | 41 | 76 | 1 | 1 | 4 |
| A4 | NC_016647 | 5076 | Porcine bocavirus 5 | 0 | 0 | 12 | 0 | 0 | 4 |
| A4 | HQ540591 | 9182 | PERV 1 A | 73 | 96 | 58 | 1 | 7 | 6 |
| A4 | KY214435 | 8043 | Porcine enterovirus b | 94 | 21 | 85 | 5 | 0.4 | 6 |
| A4 | NC_016769 | 8210 | Porcine kobuvirus | 99 | 100 | 99 | 152 | 1457 | 4056 |
| A4 | NC_003987 | 7491 | Porcine sapelovirus 1 | 98 | 59 | 66 | 3 | 2 | 26 |
| A4 | GU067771.1 | 15098 | PRRSV (Amervac) 2 | 20 | 93 | 100 | <1 | 10 | 66 |
| A4 | MN102366-75 | 18286 | Rotavirus A pig * | 4 | 2 | 10 | <1 | <1 | <1 |
| A4 | NC_003985 | 7117 | Teschovirus A | 23 | 41 | 62 | <1 | 1 | 13 |
| Average (animal samples) | 67 | 63 | 71 | 120 | 5508 | 23,582 | |||
| Detected Virus | Percentage Identity (%) | Average Sequencing Depth (MV) |
|---|---|---|
| Norovirus GII.4 | 99.89 | 44,148 |
| Influenza A virus * | 99.88 | 24,292 |
| Enterovirus D68 | 99.83 | 3342 |
| Ungulate tateraparvovirus 3 | 99.37 | 35,518 |
| PRRSV 1 | 98.95 | 17,451 |
| Porcine kobuvirus | 98.71 | 1457 |
| Average | 99.44 | 21,035 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schuele, L.; Cassidy, H.; Lizarazo, E.; Strutzberg-Minder, K.; Schuetze, S.; Loebert, S.; Lambrecht, C.; Harlizius, J.; Friedrich, A.W.; Peter, S.; et al. Assessment of Viral Targeted Sequence Capture Using Nanopore Sequencing Directly from Clinical Samples. Viruses 2020, 12, 1358. https://doi.org/10.3390/v12121358
Schuele L, Cassidy H, Lizarazo E, Strutzberg-Minder K, Schuetze S, Loebert S, Lambrecht C, Harlizius J, Friedrich AW, Peter S, et al. Assessment of Viral Targeted Sequence Capture Using Nanopore Sequencing Directly from Clinical Samples. Viruses. 2020; 12(12):1358. https://doi.org/10.3390/v12121358
Chicago/Turabian StyleSchuele, Leonard, Hayley Cassidy, Erley Lizarazo, Katrin Strutzberg-Minder, Sabine Schuetze, Sandra Loebert, Claudia Lambrecht, Juergen Harlizius, Alex W. Friedrich, Silke Peter, and et al. 2020. "Assessment of Viral Targeted Sequence Capture Using Nanopore Sequencing Directly from Clinical Samples" Viruses 12, no. 12: 1358. https://doi.org/10.3390/v12121358
APA StyleSchuele, L., Cassidy, H., Lizarazo, E., Strutzberg-Minder, K., Schuetze, S., Loebert, S., Lambrecht, C., Harlizius, J., Friedrich, A. W., Peter, S., Niesters, H. G. M., Rossen, J. W. A., & Couto, N. (2020). Assessment of Viral Targeted Sequence Capture Using Nanopore Sequencing Directly from Clinical Samples. Viruses, 12(12), 1358. https://doi.org/10.3390/v12121358