
Closely Related Vibrio alginolyticus Strains Encode an Identical Repertoire of Caudovirales-Like Regions and Filamentous Phages

 , ,
, ,  and
and 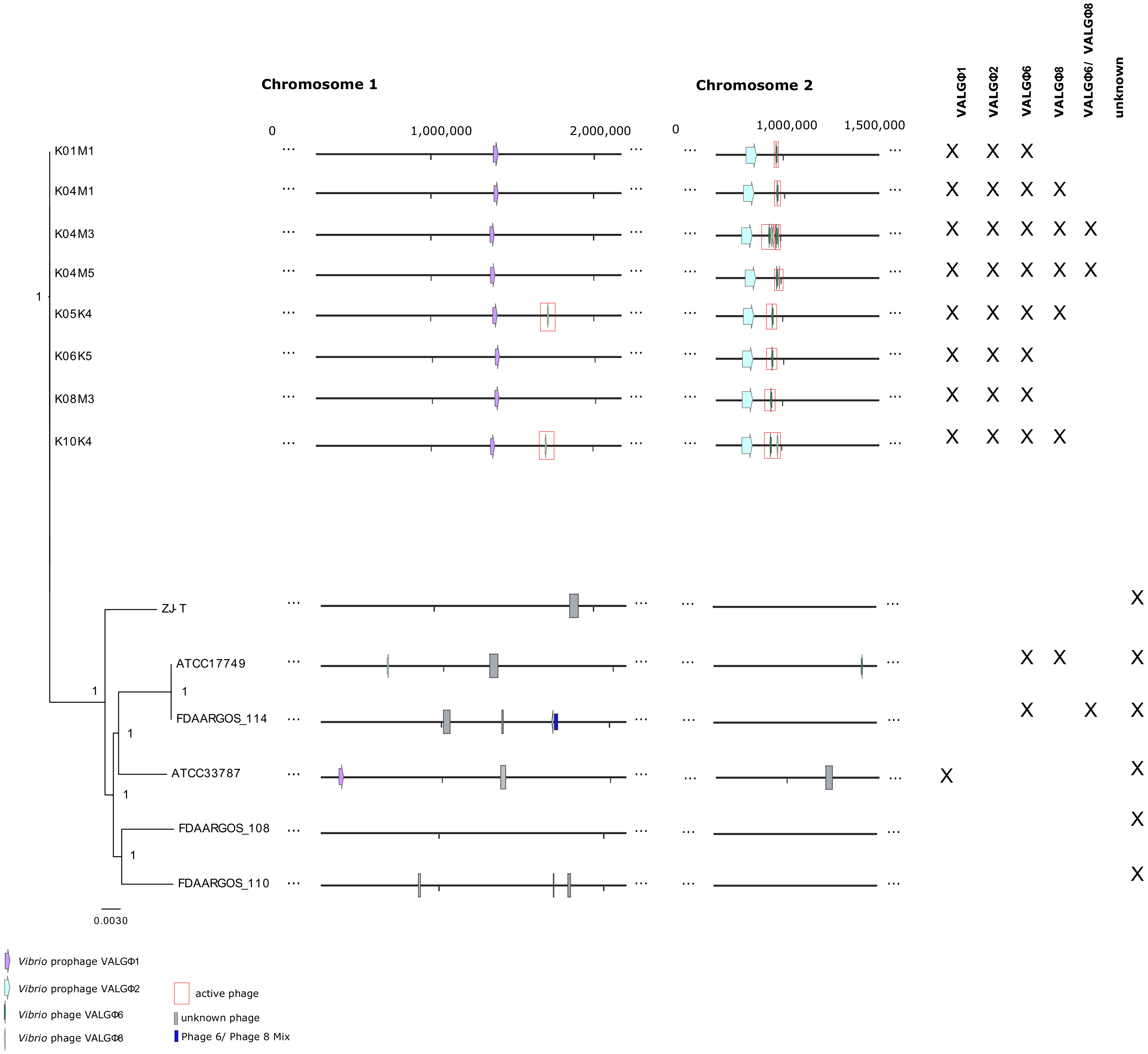{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Methods
2.1. Study Organisms
2.2. Phage Isolation and Sequencing
2.2.1. Prophage Induction
2.2.2. Extraction of Phage DNA
2.2.3. Sequencing of Phage DNA
2.3. Transmission Electron Microscopy
2.4. Genomic Analysis
2.4.1. Prediction of Prophage Regions
2.4.2. Prediction of Active Replicating Phage Regions
2.4.3. Comparative Genomic Analysis
2.4.4. Phage Genomes in Their Host Context
2.4.5. Analysis of Virulence Factors
2.4.6. Data Deposit
2.5. Infection Experiments
3. Results
3.1. General Overview
3.2. Caudovirales
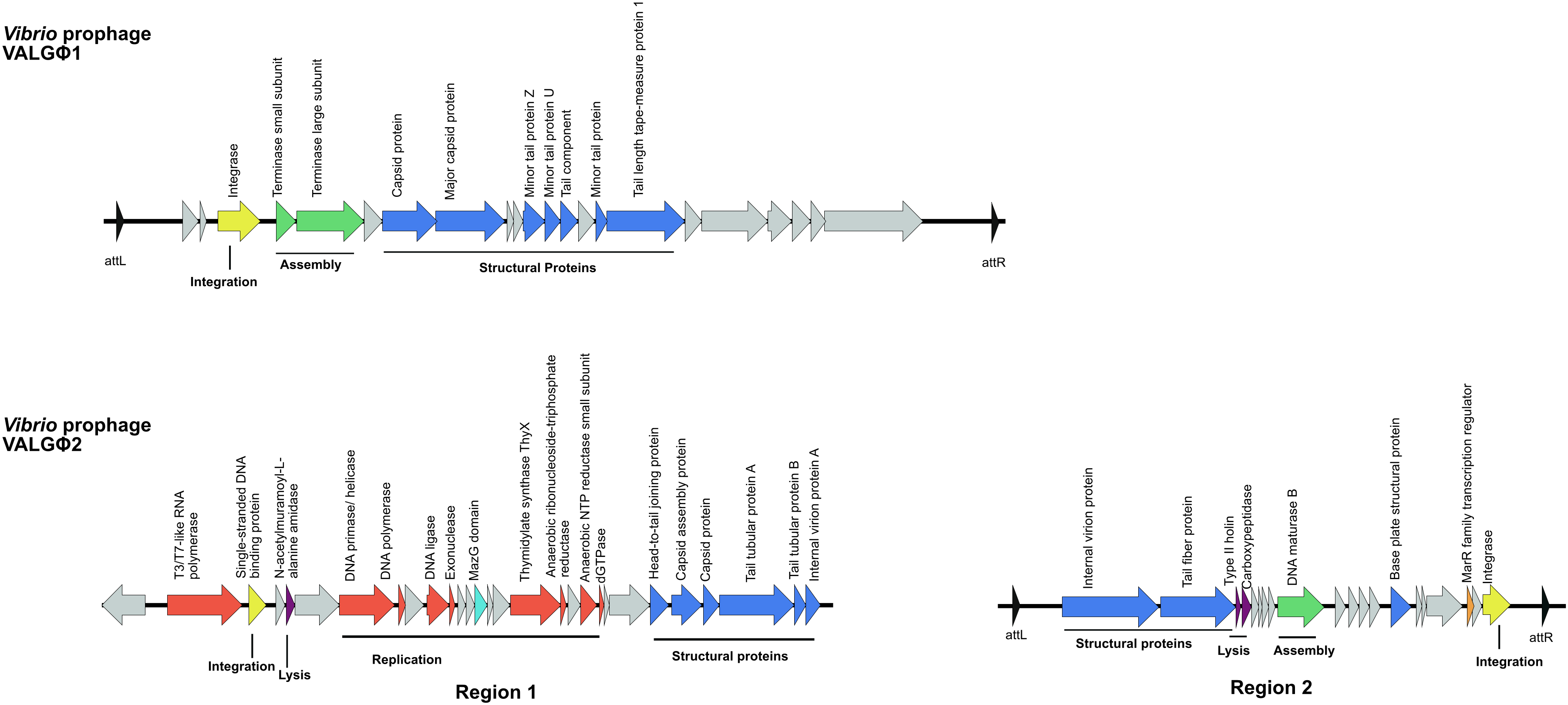
3.2.1. Vibrio Prophage VALGΦ1
3.2.2. Vibrio Prophage VALGΦ2
3.3. Tubulavirales
3.3.1. Phage Morphology
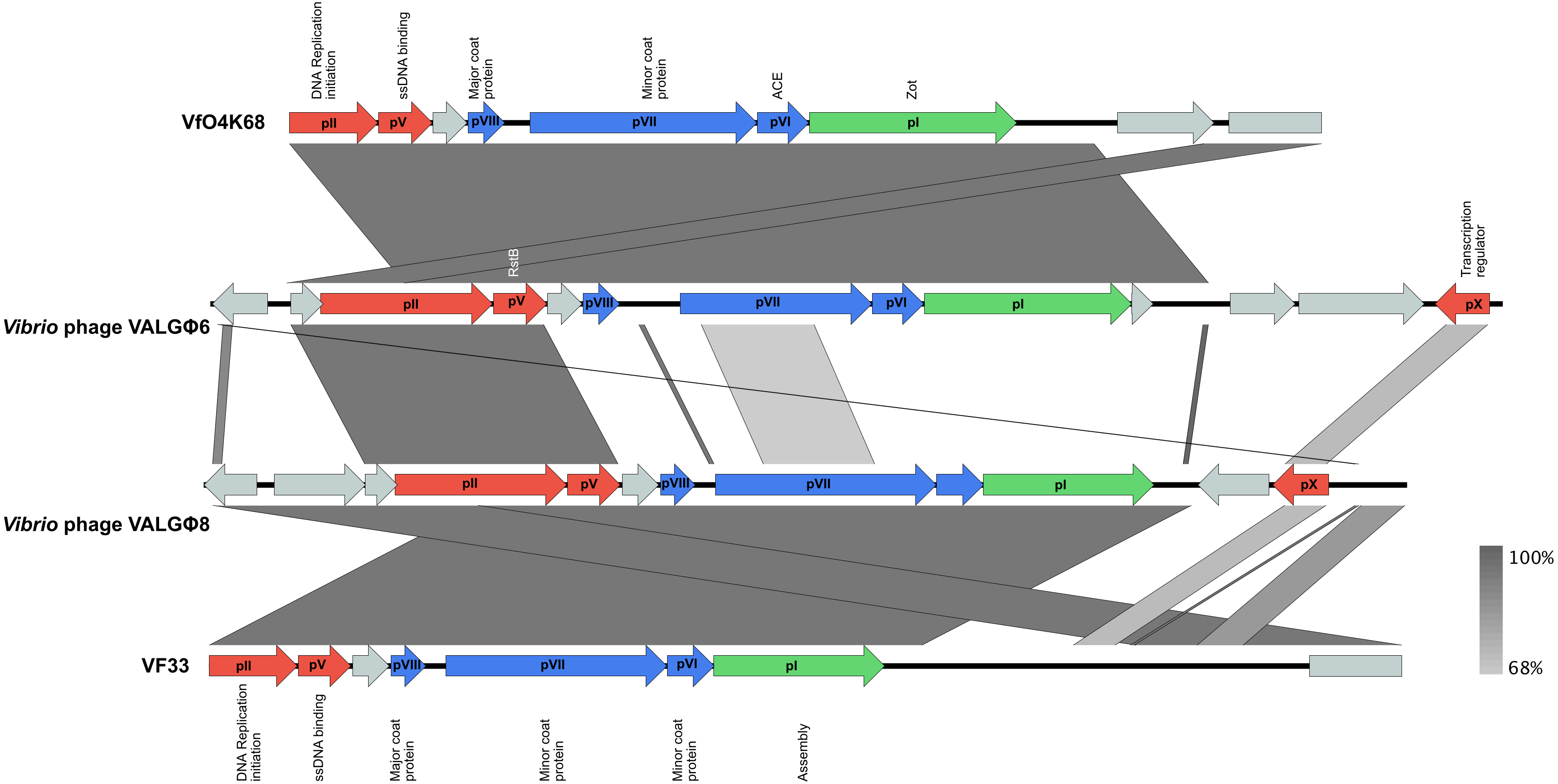
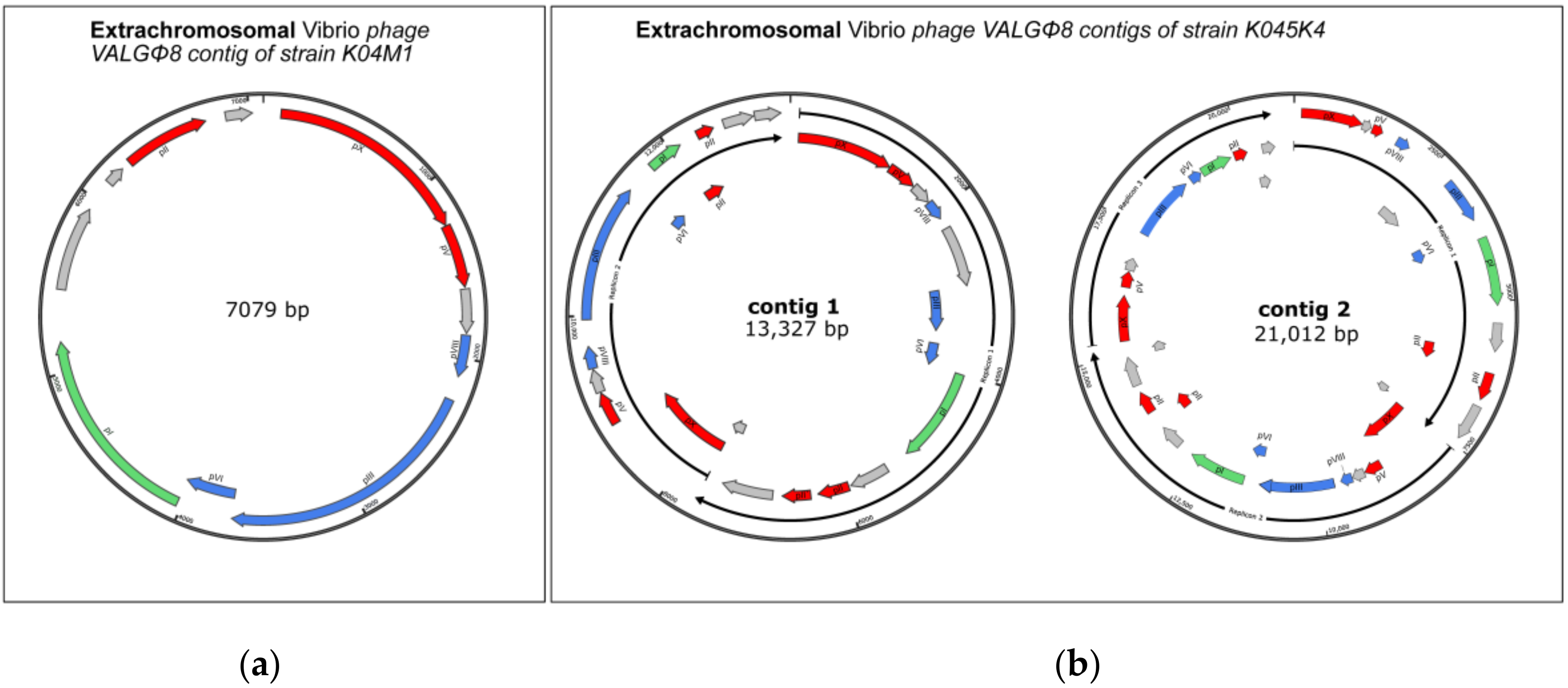
3.3.2. Phage Genomics
3.3.3. Phage Activity and Within-Host Interaction
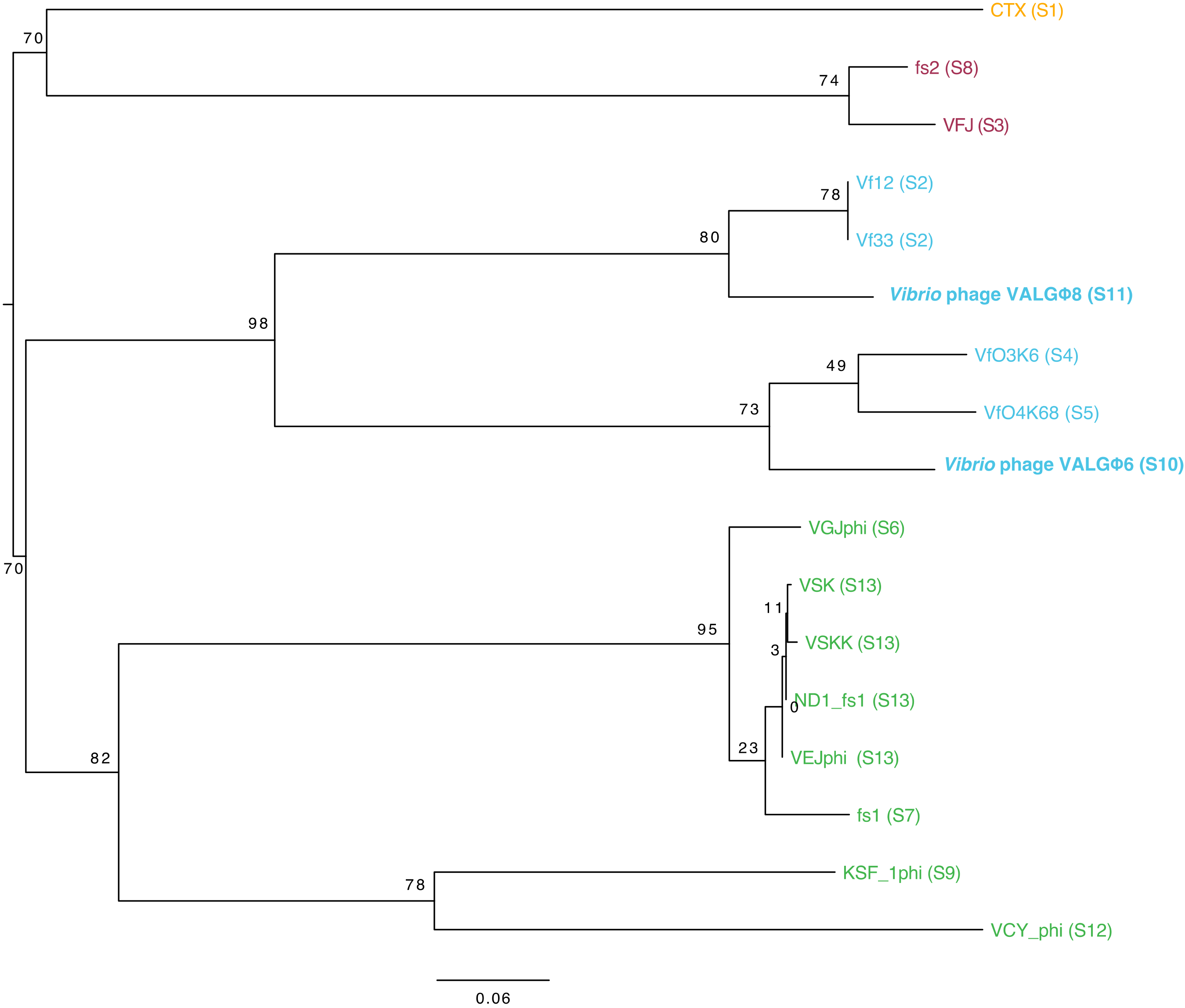
3.3.4. Phylogeny
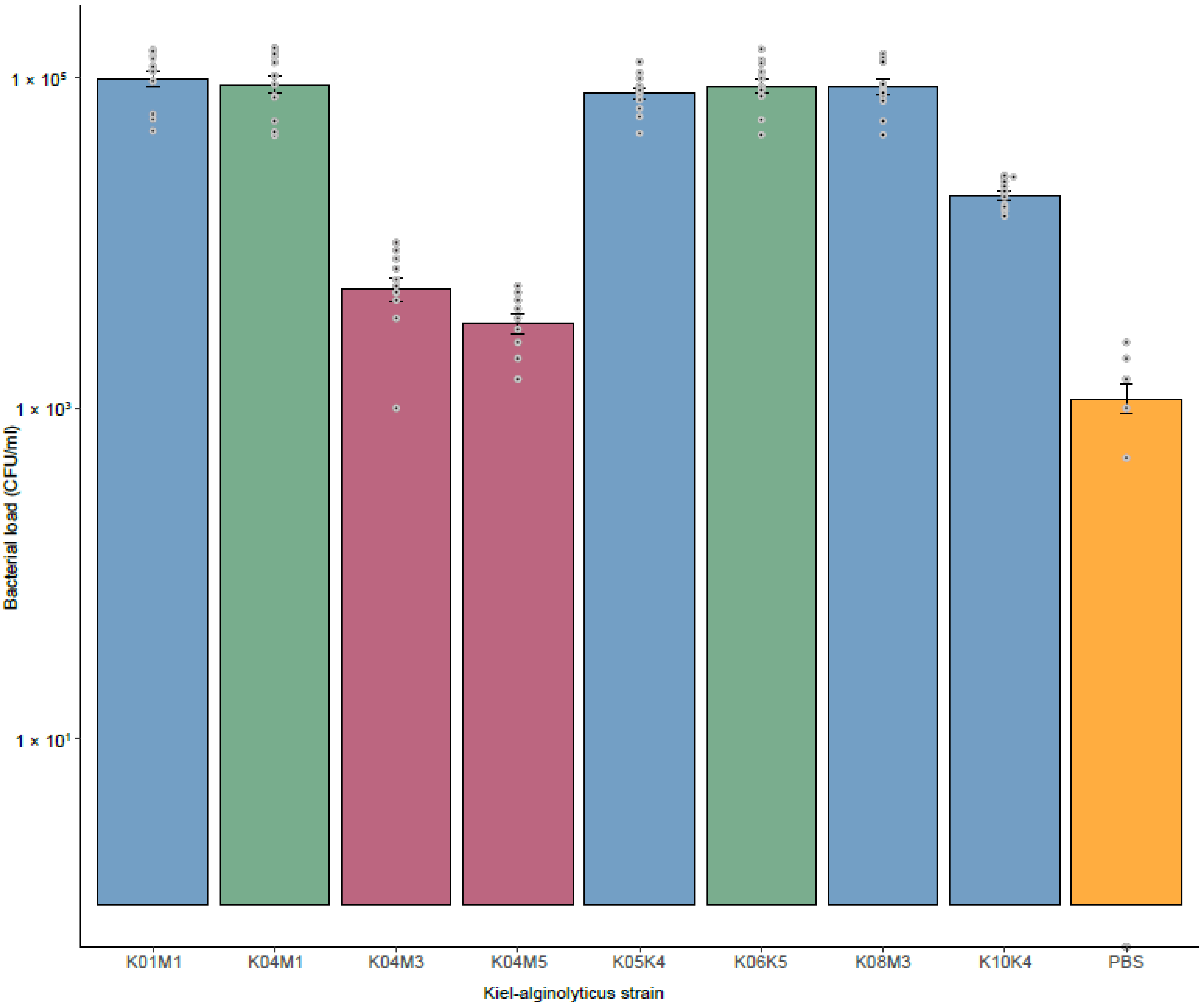
3.4. Virulence of Kiel V. alginolyticus Ecotypes
4. Discussion
4.1. Filamentous Phages Differ in Their Mode of Inheritance
4.2. Within-Host Competition Can Lead to the Reduction of Phage Producing Particles
4.3. HGT of Vibrio Phage VALGΦ6 Containing the Putative Virulence Factors Zot and Ace Might Have Led to the Emergence of the Pathogenic Kiel V. alginolyticus Ecotype
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Ethics Approval
Consent for Publication
Abbreviations
References
- Iguchi, A.; Iyoda, S.; Terajima, J.; Watanabe, H.; Osawa, R. Spontaneous recombination between homologous prophage regions causes large-scale inversions within the Escherichia coli O157:H7 chromosome. Gene 2006, 372, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Wendling, C.C.; Refardt, D.; Hall, A.R. Fitness benefits to bacteria of carrying prophages and prophage-encoded antibiotic-resistance genes peak in different environments. BioRxiv 2020. [Google Scholar] [CrossRef]
- Mai-Prochnow, A.; Hui, J.G.K.; Kjelleberg, S.; Rakonjac, J.; McDougald, D.; Rice, S.A. Big things in small packages: The genetics of filamentous phage and effects on fitness of their host. FEMS Microbiol. Rev. 2015, 39, 465–487. [Google Scholar] [CrossRef] [PubMed]
- Waldor, M.K.; Mekalanos, J.J. Lysogenic Conversion by a Filamentous Phage Encoding Cholera Toxin. Science 1996, 272, 1910–1914. [Google Scholar] [CrossRef] [PubMed]
- Chang, B.; Miyamoto, H.; Taniguchi, H.; Yoshida, S.-I. Isolation and Genetic Characterization of a Novel Filamentous Bacteriophage, a Deleted Form of Phage f237, from a Pandemic Vibrio parahaemolyticus O4:K68 Strain. Microbiol. Immunol. 2002, 46, 565–569. [Google Scholar] [CrossRef] [PubMed]
- Fasano, A.; Baudry, B.; Pumplin, D.W.; Wasserman, S.S.; Tall, B.D.; Ketley, J.M.; Kaper, J.B. Vibrio cholerae produces a second enterotoxin, which affects intestinal tight junctions. Proc. Natl. Acad. Sci. USA 1991, 88, 5242–5246. [Google Scholar] [CrossRef]
- Lan, S.-F.; Huang, C.-H.; Chang, C.-H.; Liao, W.-C.; Lin, I.-H.; Jian, W.-N.; Wu, Y.-G.; Chen, S.-Y.; Wong, H.-C. Characterization of a New Plasmid-Like Prophage in a Pandemic Vibrio parahaemolyticus O3:K6 Strain. Appl. Environ. Microbiol. 2009, 75, 2659–2667. [Google Scholar] [CrossRef]
- Faruque, S.M.; Comstock, L.; Kaper, J.B.; Albert, M.J. Distribution of Zonula occludens toxin (zot) gene among clinical isolates of Vibrio cholerae O1 from Bangladesh and Africa. J. Diarrhoeal Dis. Res. 1994, 12, 222–224. [Google Scholar]
- Kurazono, H.; Pal, A.; Bag, P.K.; Nair, G.B.; Karasawa, T.; Mihara, T.; Takeda, Y. Distribution of genes encoding cholera toxin, zonula occludens toxin, accessory cholera toxin, and El Tor hemolysin Vibrio cholerae of diverse origins. Microb. Pathog. 1995, 18, 231–235. [Google Scholar] [CrossRef]
- Khouadja, S.; Suffredini, E.; Baccouche, B.; Croci, L.; Bakhrouf, A. Occurrence of virulence genes among Vibrio cholerae and Vibrio parahaemolyticus strains from treated wastewaters. Environ. Monit. Assess. 2014, 186, 6935–6945. [Google Scholar] [CrossRef]
- Weynberg, K.D.; Voolstra, C.R.; Neave, M.J.; Buerger, P.; Van Oppen, M.J.H. From cholera to corals: Viruses as drivers of virulence in a major coral bacterial pathogen. Sci. Rep. 2015, 5, 17889. [Google Scholar] [CrossRef]
- Castillo, D.; Alvise, P.D.; Xu, R.; Zhang, F.; Middelboe, M.; Gram, L. Comparative Genome Analyses of Vibrio anguillarum Strains Reveal a Link with Pathogenicity Traits. mSystems 2017, 2, e00001-17. [Google Scholar] [CrossRef] [PubMed]
- Mauritzen, J.J.; Castillo, D.; Tan, D.; Svenningsen, S.L.; Middelboe, M. Beyond Cholera: Characterization of zot-Encoding Filamentous Phages in the Marine Fish Pathogen Vibrio anguillarum. Viruses 2020, 12, 730. [Google Scholar] [CrossRef] [PubMed]
- Castillo, D.; Pérez-Reytor, D.; Plaza, N.; Ramírez-Araya, S.; Blondel, C.J.; Corsini, G.; Bastías, R.; Loyola, D.E.; Jaña, V.; Pavez, L.; et al. Exploring the Genomic Traits of Non-toxigenic Vibrio parahaemolyticus Strains Isolated in Southern Chile. Front. Microbiol. 2018, 9, 161. [Google Scholar] [CrossRef] [PubMed]
- Hada, H.S.; West, P.A.; Lee, J.V.; Stemmler, J.; Colwell, R.R. Vibrio tubiashii sp. nov., a Pathogen of Bivalve Mollusks. Int. J. Syst. Bacteriol. 1984, 34, 1–4. [Google Scholar] [CrossRef]
- Lacoste, A.; Jalabert, F.; Malham, S.; Cueff, A.; Gélébart, F.; Cordevant, C.; Lange, M.; Poulet, S.A. A Vibrio splendidus strain is associated with summer mortality of juvenile oysters Crassostrea gigas in the Bay of Morlaix (North Brittany, France). Dis. Aquat. Org. 2001, 46, 139–145. [Google Scholar] [CrossRef]
- Gómez-León, J.; Villamil, L.; Lemos, M.L.; Novoa, B.; Figueras, A. Isolation of Vibrio alginolyticus and Vibrio splendidus from Aquacultured Carpet Shell Clam (Ruditapes decussatus) Larvae Associated with Mass Mortalities. Appl. Environ. Microbiol. 2005, 71, 98–104. [Google Scholar] [CrossRef]
- Balcázar, J.L.; Gallo-Bueno, A.; Planas, M.; Valverde, J.P.; Planas, M. Isolation of Vibrio alginolyticus and Vibrio splendidus from captive-bred seahorses with disease symptoms. Antonie Van Leeuwenhoek 2009, 97, 207–210. [Google Scholar] [CrossRef]
- Wendling, C.C.; Wegner, K.M. Relative contribution of reproductive investment, thermal stress and Vibrio infection to summer mortality phenomena in Pacific oysters. Aquaculture 2013, 412–413, 88–96. [Google Scholar] [CrossRef]
- Castillo, D.; Kauffman, K.; Hussain, F.; Kalatzis, P.; Rørbo, N.; Polz, M.F.; Middelboe, M. Widespread distribution of prophage-encoded virulence factors in marine Vibrio communities. Sci. Rep. 2018, 8, 1–9. [Google Scholar] [CrossRef]
- Campos, J.; Martínez, E.; Izquierdo, Y.; Fando, R. VEJφ, a novel filamentous phage of Vibrio cholerae able to transduce the cholera toxin genes. Microbiology 2010, 156, 108–115. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Campos, J.; Martínez, E.; Marrero, K.; Silva, Y.; Rodríguez, B.L.; Suzarte, E.; Ledon, T.; Fando, R. Novel type of specialized transduction for CTX phi or its satellite phage RS1 mediated by filamentous phage VGJ phi in Vibrio cholerae. J. Bacteriol. 2003, 185, 7231–7240. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Munro, J.; Oakey, J.; Bromage, E.; Owens, L. Experimental bacteriophage-mediated virulence in strains of Vibrio harveyi. Dis. Aquat. Org. 2003, 54, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Wendling, C.C.; Goehlich, H.; Roth, O. The structure of temperate phage–bacteria infection networks changes with the phylogenetic distance of the host bacteria. Biol. Lett. 2018, 14, 20180320. [Google Scholar] [CrossRef]
- Goehlich, H.; Roth, O.; Wendling, C.C. Filamentous phages reduce bacterial growth in low salinities. R. Soc. Open Sci. 2019, 6, 191669. [Google Scholar] [CrossRef]
- Wendling, C.C.; Piecyk, A.; Refardt, D.; Chibani, C.; Hertel, R.; Liesegang, H.; Bunk, B.; Overmann, J.; Roth, O. Tripartite species interaction: Eukaryotic hosts suffer more from phage susceptible than from phage resistant bacteria. BMC Evol. Biol. 2017, 17, 1–12. [Google Scholar] [CrossRef]
- Lee, K.-K.; Yu, S.-R.; Yang, T.-I.; Liu, P.-C.; Chen, F.-R. Isolation and characterization of Vibrio alginolyticus isolated from diseased kuruma prawn, Penaeus japonicus. Lett. Appl. Microbiol. 1996, 22, 111–114. [Google Scholar] [CrossRef]
- Zhang, D.L.; Manos, J.; Ma, X.R.; Belas, R.; Karaolis, D.K.R. Transcriptional analysis and operon structure of the tagA-orf2-orf3-mop-tagD region on the Vibrio pathogenicity island in epidemic V-cholerae. FEMS Microbiol. Lett. 2004, 235, 199–207. [Google Scholar] [CrossRef]
- Gonzalez-Escalona, N.; Blackstone, G.M.; DePaola, A. Characterization of a Vibrio alginolyticus Strain, Isolated from Alaskan Oysters, Carrying a Hemolysin Gene Similar to the Thermostable Direct Hemolysin-Related Hemolysin Gene (trh) of Vibrio parahaemolyticus. Appl. Environ. Microbiol. 2006, 72, 7925–7929. [Google Scholar] [CrossRef]
- Hörmansdorfer, S.; Wentges, H.; Neugebaur-Büchler, K.; Bauer, J. Isolation of Vibrio alginolyticus from seawater aquaria. Int. J. Hyg. Environ. Health 2000, 203, 169–175. [Google Scholar] [CrossRef]
- Roth, O.; Keller, I.; Landis, S.H.; Salzburger, W.; Reusch, T.B. Hosts are ahead in a marine host-parasite coevolutionary arms race: Innate immune system adaptation in pipefish Syngnathus typhle against Vibrio phylotypes. Evolution 2012, 66, 2528–2539. [Google Scholar] [CrossRef] [PubMed]
- Chibani, C.M.; Roth, O.; Liesegang, H.; Wendling, C.C. Genomic variation among closely related Vibrio alginolyticus strains is located on mobile genetic elements. BMC Genom. 2020, 21, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Chin, C.-S.; Alexander, D.H.; Marks, P.; Klammer, A.A.; Drake, J.P.; Heiner, C.; Clum, A.; Copeland, A.; Huddleston, J.; Eichler, E.E.; et al. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat. Methods 2013, 10, 563–569. [Google Scholar] [CrossRef] [PubMed]
- Hyatt, D.; Chen, G.-L.; Locascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef]
- Willms, I.M.; Hoppert, M.; Hertel, R. Characterization of Bacillus subtilis Viruses vB_BsuM-Goe2 and vB_BsuM-Goe. Viruses 2017, 9, 146. [Google Scholar] [CrossRef]
- Lopes, A.; Tavares, P.; Petit, M.-A.; Guerois, R.; Zinn-Justin, S. Automated classification of tailed bacteriophages according to their neck organization. BMC Genom. 2014, 15, 1027. [Google Scholar] [CrossRef]
- Arndt, D.; Grant, J.R.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. PHASTER: A better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016, 44, W16–W21. [Google Scholar] [CrossRef]
- Nayfach, S.; Camargo, A.P.; Eloe-Fadrosh, E.; Roux, S.; Kyrpides, N. CheckV: Assessing the quality of metagenome-assembled viral genomes. BioRxiv 2020. [Google Scholar] [CrossRef]
- Sullivan, M.J.; Petty, N.K.; Beatson, S.A. Easyfig: A genome comparison visualizer. Bioinformatics 2011, 27, 1009–1010. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Dietrich, S.; Wiegand, S.; Liesegang, H. TraV: A Genome Context Sensitive Transcriptome Browser. PLoS ONE 2014, 9, e93677. [Google Scholar] [CrossRef] [PubMed]
- Ramírez, F.; Dündar, F.; Diehl, S.; Grüning, B.A.; Manke, T. deepTools: A flexible platform for exploring deep-sequencing data. Nucleic Acids Res. 2014, 42, W187–W191. [Google Scholar] [CrossRef]
- Larsson, A. AliView: A fast and lightweight alignment viewer and editor for large datasets. Bioinformatics 2014, 30, 3276–3278. [Google Scholar] [CrossRef]
- Meier-Kolthoff, J.P.; Göker, M. VICTOR: Genome-based phylogeny and classification of prokaryotic viruses. Bioinformatics 2017, 33, 3396–3404. [Google Scholar] [CrossRef]
- Meier-Kolthoff, J.P.; Auch, A.F.; Klenk, H.-P.; Göker, M. Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinform. 2013, 14, 60. [Google Scholar] [CrossRef]
- Lefort, V.; Desper, R.; Gascuel, O. FastME 2.0: A Comprehensive, Accurate, and Fast Distance-Based Phylogeny Inference Program: Table. Mol. Biol. Evol. 2015, 32, 2798–2800. [Google Scholar] [CrossRef]
- Farris, J.S. Estimating Phylogenetic Trees from Distance Matrices. Am. Nat. 1972, 106, 645–668. [Google Scholar] [CrossRef]
- Göker, M.; Garcia-Blazquez, G.; Voglmayr, H.; Tellería, M.T.; Martín, M.P. Molecular Taxonomy of Phytopathogenic Fungi: A Case Study in Peronospora. PLoS ONE 2009, 4, e6319. [Google Scholar] [CrossRef]
- Meier-Kolthoff, J.P.; Hahnke, R.L.; Petersen, J.; Scheuner, C.; Michael, V.; Fiebig, A.; Rohde, C.; Rohde, M.; Fartmann, B.A.; Goodwin, L.; et al. Complete genome sequence of DSM 30083T, the type strain (U5/41T) of Escherichia coli, and a proposal for delineating subspecies in microbial taxonomy. Stand. Genom. Sci. 2014, 9, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Huelsenbeck, J.P.; Ronquist, F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 2001, 17, 754–755. [Google Scholar] [CrossRef] [PubMed]
- Hertel, R.; Rodríguez, D.P.; Hollensteiner, J.; Dietrich, S.; Leimbach, A.; Hoppert, M.; Liesegang, H.; Volland, S. Genome-Based Identification of Active Prophage Regions by Next Generation Sequencing in Bacillus licheniformis DSM. PLoS ONE 2015, 10, e0120759. [Google Scholar] [CrossRef] [PubMed]
- Chibani, C.; Farr, A.; Klama, S.; Dietrich, S.; Liesegang, H. Classifying the Unclassified: A Phage Classification Method. Viruses 2019, 11, 195. [Google Scholar] [CrossRef] [PubMed]
- Askora, A.; Abdel-Haliem, M.E.F.; Yamada, T. Site-specific recombination systems in filamentous phages. Mol. Genet. Genom. 2012, 287, 525–530. [Google Scholar] [CrossRef]
- Refardt, D. Within-host competition determines reproductive success of temperate bacteriophages. ISME J. 2011, 5, 1451–1460. [Google Scholar] [CrossRef]
- Davis, B.M.; Moyer, K.E.; Boyd, E.F.; Waldor, M.K. CTX Prophages in Classical Biotype Vibrio cholerae: Functional Phage Genes but Dysfunctional Phage Genomes. J. Bacteriol. 2000, 182, 6992–6998. [Google Scholar] [CrossRef]
- Hazen, T.H.; Pan, L.; Gu, J.-D.; Sobecky, P.A. The contribution of mobile genetic elements to the evolution and ecology of Vibrios. FEMS Microbiol. Ecol. 2010, 74, 485–499. [Google Scholar] [CrossRef]
- Faruque, S.M.; Rahman, M.M.; Islam, K.M.N.; Mekalanos, J.J. Lysogenic Conversion of Environmental Vibrio mimicus Strains by CTXΦ. Infect. Immun. 1999, 67, 5723–5729. [Google Scholar] [CrossRef]
- Boyd, E.F.; Moyer, K.E.; Shi, L.; Waldor, M.K. Infectious CTXΦ and the Vibrio pathogenicity Island Prophage in Vibrio mimicus: Evidence for Recent Horizontal Transfer between V. mimicus and V. cholerae. Infect. Immun. 2000, 68, 1507–1513. [Google Scholar] [CrossRef]
- Ruby, E.G.; Urbanowski, M.; Campbell, J.; Dunn, A.; Faini, M.; Gunsalus, R.; Lostroh, P.; Lupp, C.; McCann, J.; Millikan, D.; et al. Complete genome sequence of Vibrio fischeri: A symbiotic bacterium with pathogenic congeners. Proc. Natl. Acad. Sci. USA 2005, 102, 3004–3009. [Google Scholar] [CrossRef] [PubMed]
- Trucksis, M.; Galen, J.E.; Michalski, J.; Fasano, A.; Kaper, J.B. Accessory cholera enterotoxin (Ace), the third toxin of a Vibrio cholerae virulence cassette. Proc. Natl. Acad. Sci. USA 1993, 90, 5267–5271. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Reytor, D.; Jaña, V.; Pavez, L.; Navarrete, P.; García, K. Accessory Toxins of Vibrio pathogens and Their Role in Epithelial Disruption during Infection. Front. Microbiol. 2018, 9, 2248. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Cho, Y.J.; Chun, J.; Seok, Y.J.; Lee, J.K.; Kim, K.S.; Choi, S.H. Complete genome sequence of Vibrio vulnificus MO6-24/O. J. Bacteriol 2011, 193, 2062–2063. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chibani, C.M.; Hertel, R.; Hoppert, M.; Liesegang, H.; Wendling, C.C. Closely Related Vibrio alginolyticus Strains Encode an Identical Repertoire of Caudovirales-Like Regions and Filamentous Phages. Viruses 2020, 12, 1359. https://doi.org/10.3390/v12121359
Chibani CM, Hertel R, Hoppert M, Liesegang H, Wendling CC. Closely Related Vibrio alginolyticus Strains Encode an Identical Repertoire of Caudovirales-Like Regions and Filamentous Phages. Viruses. 2020; 12(12):1359. https://doi.org/10.3390/v12121359
Chicago/Turabian StyleChibani, Cynthia Maria, Robert Hertel, Michael Hoppert, Heiko Liesegang, and Carolin Charlotte Wendling. 2020. "Closely Related Vibrio alginolyticus Strains Encode an Identical Repertoire of Caudovirales-Like Regions and Filamentous Phages" Viruses 12, no. 12: 1359. https://doi.org/10.3390/v12121359
APA StyleChibani, C. M., Hertel, R., Hoppert, M., Liesegang, H., & Wendling, C. C. (2020). Closely Related Vibrio alginolyticus Strains Encode an Identical Repertoire of Caudovirales-Like Regions and Filamentous Phages. Viruses, 12(12), 1359. https://doi.org/10.3390/v12121359