Interplay between Hepatitis D Virus and the Interferon Response


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
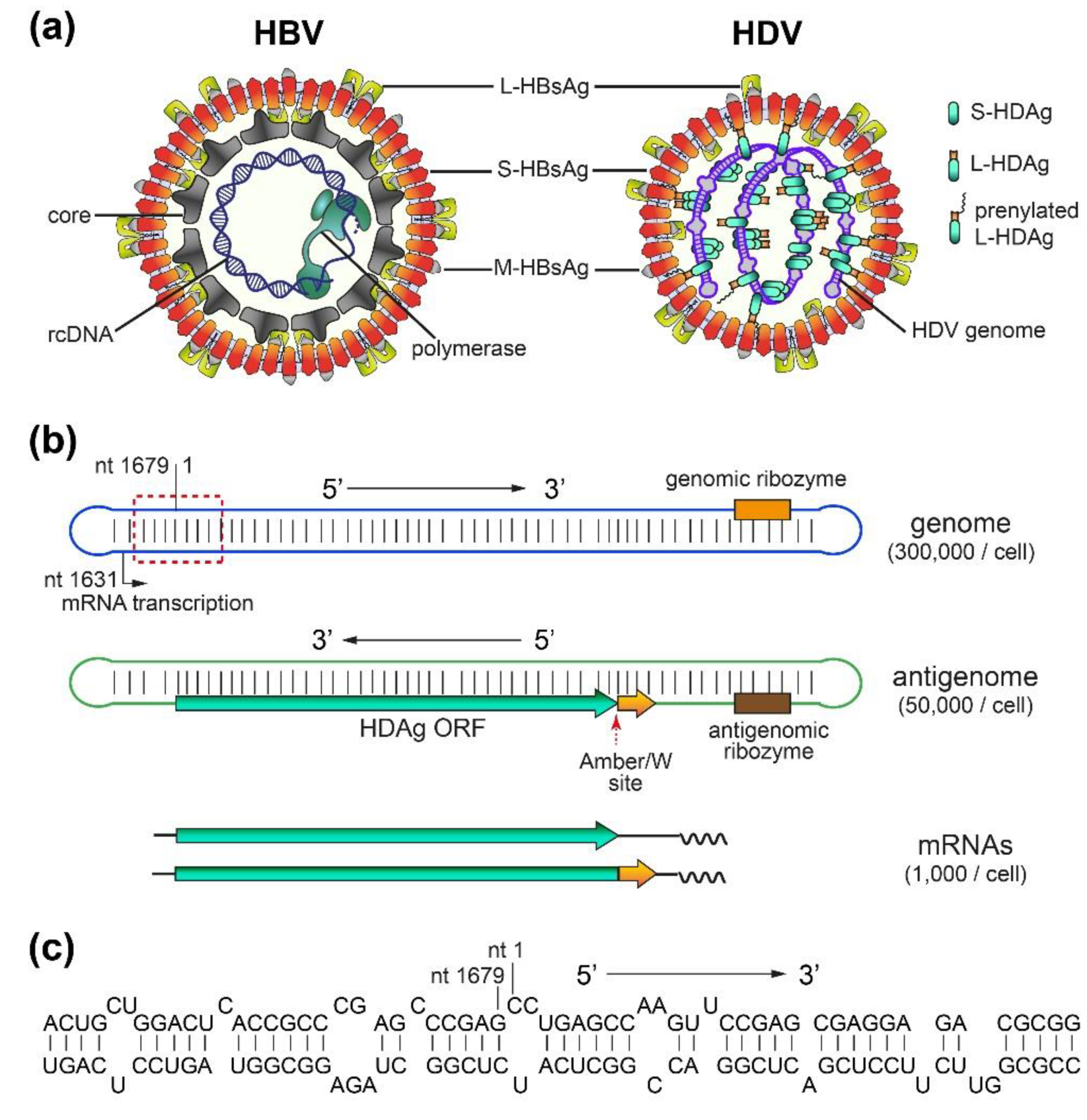
2. HDV Structure, Replication, and Persistence
2.1. HBV/HDV Virions and HDV RNAs
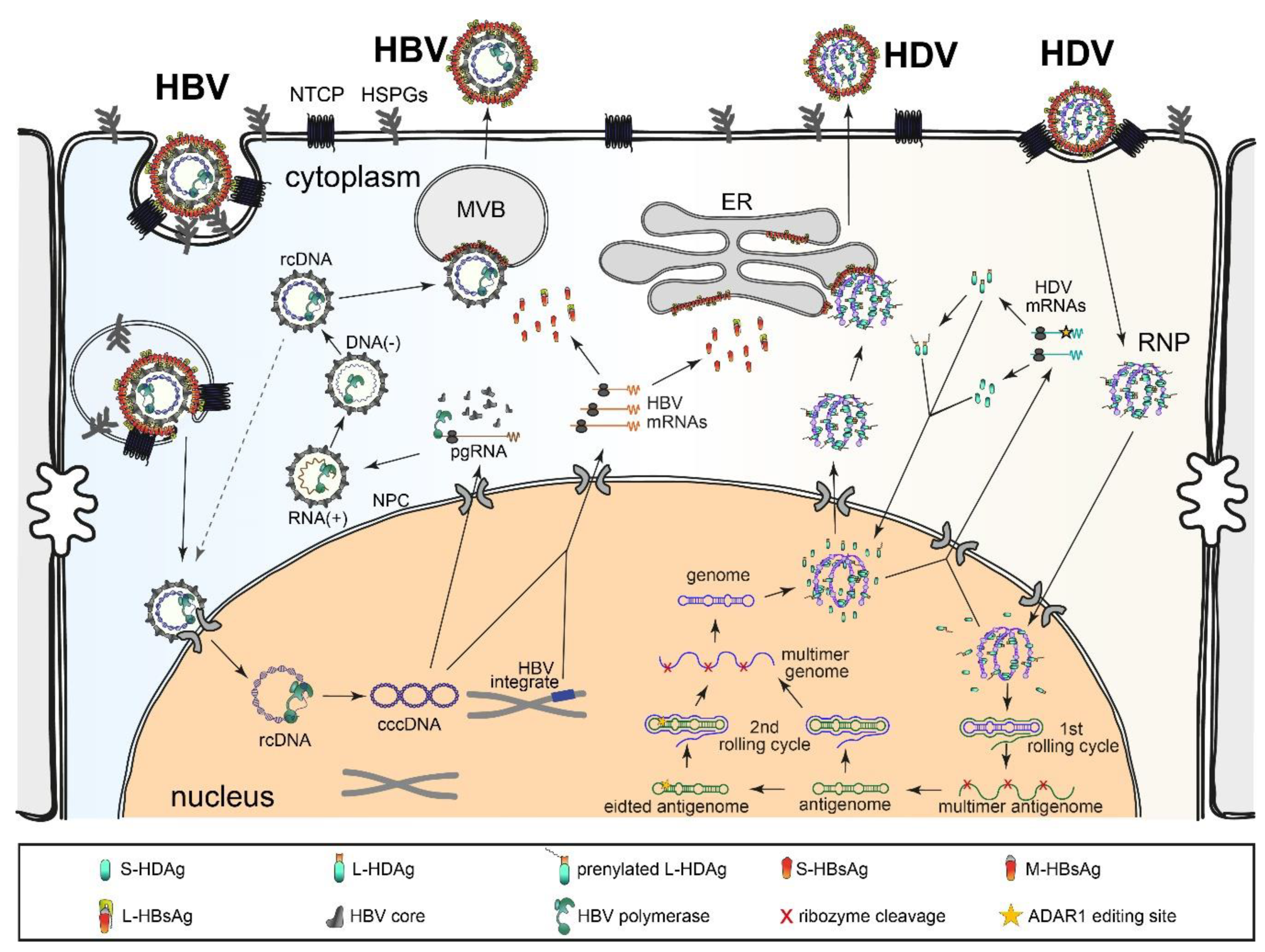
2.2. HDV Replication Cycle
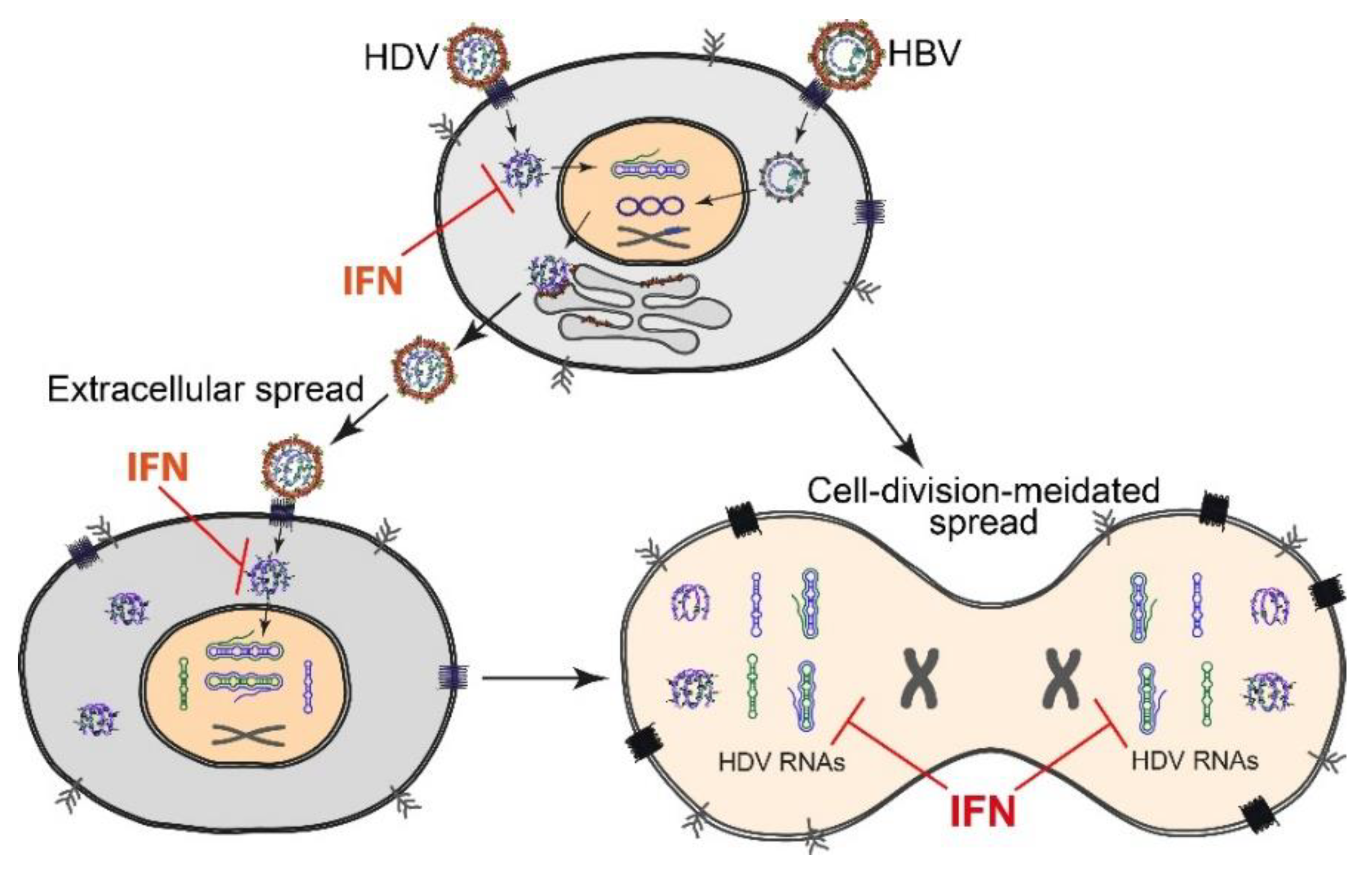
2.3. HDV Spread and Persistence
3. IFN Signaling during RNA Virus Infection
4. IFN Response during HDV Infection
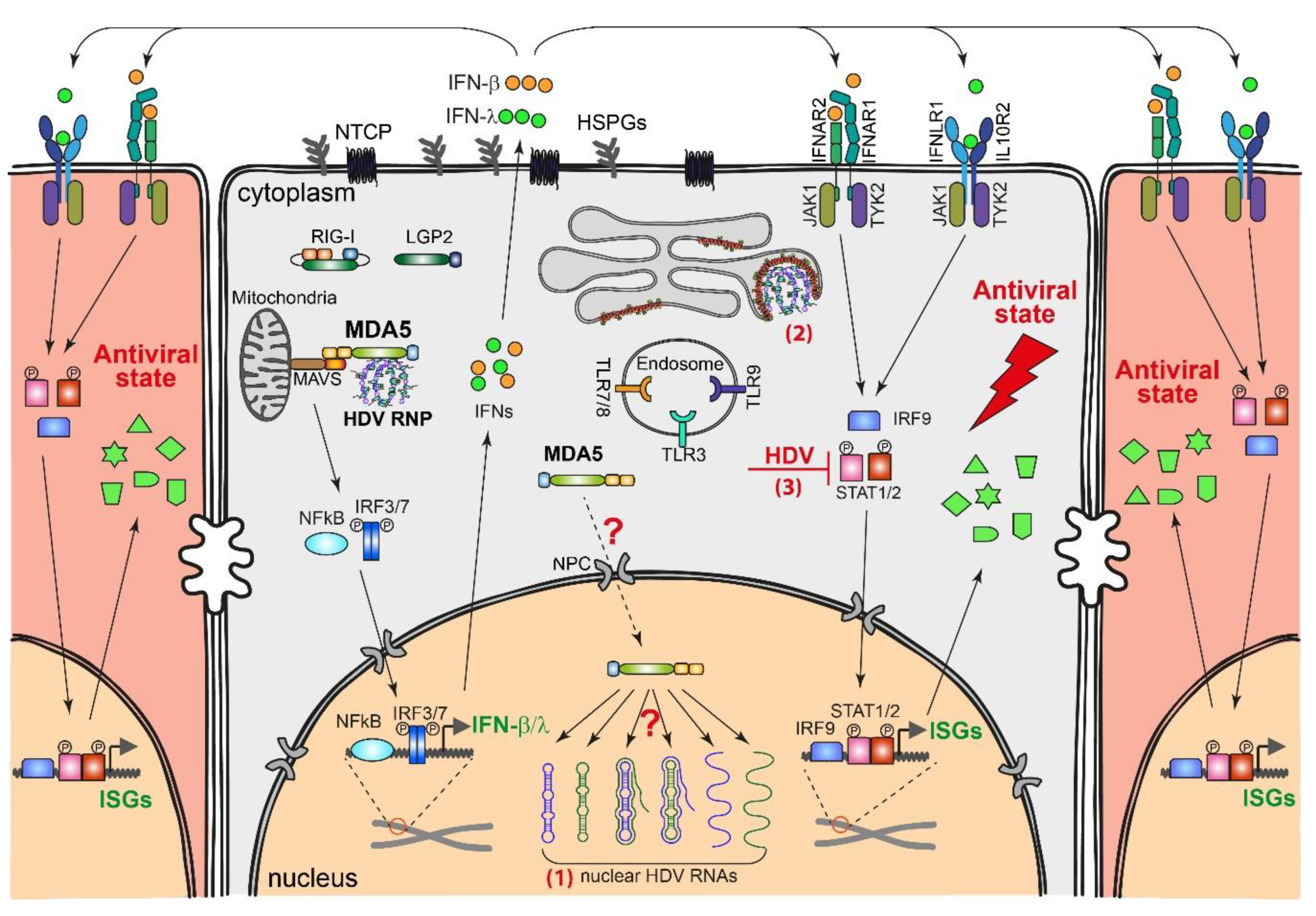
4.1. HDV-Induced Innate Immune Responses
4.2. Innate Immune Sensing of HDV Replication
4.3. Effect of IFN Response on HDV Replication
4.4. Countermeasures by HDV
5. Crosstalk between HDV-Induced IFN Response and HBV
5.1. The Role of HBV in IFN Response Activation during HBV/HDV Co-Infection
5.2. Effect of HDV-Induced IFN Response on HBV Replication
6. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Farci, P.; Niro, G.A. Clinical features of hepatitis D. Semin. Liver Dis. 2012, 32, 228–236. [Google Scholar] [CrossRef] [PubMed]
- Botelho-Souza, L.F.; Vasconcelos, M.P.A.; Dos Santos, A.O.; Salcedo, J.M.V.; Vieira, D.S. Hepatitis delta: Virological and clinical aspects. Virol. J. 2017, 14, 177. [Google Scholar] [CrossRef] [PubMed]
- Negro, F. Hepatitis D virus coinfection and superinfection. Cold Spring Harb. Perspect. Med. 2014, 4, a021550. [Google Scholar] [CrossRef]
- Stockdale, A.J.; Kreuels, B.; Henrion, M.Y.R.; Giorgi, E.; Kyomuhangi, I.; de Martel, C.; Hutin, Y.; Geretti, A.M. The global prevalence of hepatitis D virus infection: Systematic review and meta-analysis. J. Hepatol. 2020, 73, 523–532. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.Y.; Shen, D.T.; Ji, D.Z.; Han, P.C.; Zhang, W.M.; Ma, J.F.; Chen, W.S.; Goyal, H.; Pan, S.; Xu, H.G. Prevalence and burden of hepatitis D virus infection in the global population: A systematic review and meta-analysis. Gut 2019, 68, 512–521. [Google Scholar] [CrossRef] [PubMed]
- Miao, Z.; Zhang, S.; Ou, X.; Li, S.; Ma, Z.; Wang, W.; Peppelenbosch, M.P.; Liu, J.; Pan, Q. Estimating the Global Prevalence, Disease Progression, and Clinical Outcome of Hepatitis Delta Virus Infection. J. Infect. Dis. 2020, 221, 1677–1687. [Google Scholar] [CrossRef] [PubMed]
- Deny, P. Hepatitis delta virus genetic variability: From genotypes I, II, III to eight major clades? Curr. Top. Microbiol. Immunol. 2006, 307, 151–171. [Google Scholar]
- Le Gal, F.; Brichler, S.; Drugan, T.; Alloui, C.; Roulot, D.; Pawlotsky, J.M.; Deny, P.; Gordien, E. Genetic diversity and worldwide distribution of the deltavirus genus: A study of 2,152 clinical strains. Hepatology 2017, 66, 1826–1841. [Google Scholar] [CrossRef]
- Yan, H.; Zhong, G.; Xu, G.; He, W.; Jing, Z.; Gao, Z.; Huang, Y.; Qi, Y.; Peng, B.; Wang, H.; et al. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. eLife 2012, 1, e00049. [Google Scholar] [CrossRef]
- Ni, Y.; Lempp, F.A.; Mehrle, S.; Nkongolo, S.; Kaufman, C.; Falth, M.; Stindt, J.; Koniger, C.; Nassal, M.; Kubitz, R.; et al. Hepatitis B and D viruses exploit sodium taurocholate co-transporting polypeptide for species-specific entry into hepatocytes. Gastroenterology 2014, 146, 1070–1083. [Google Scholar] [CrossRef]
- Chang, J.; Nie, X.; Chang, H.E.; Han, Z.; Taylor, J. Transcription of hepatitis delta virus RNA by RNA polymerase II. J. Virol. 2008, 82, 1118–1127. [Google Scholar] [CrossRef] [PubMed]
- Macnaughton, T.B.; Shi, S.T.; Modahl, L.E.; Lai, M.M. Rolling circle replication of hepatitis delta virus RNA is carried out by two different cellular RNA polymerases. J. Virol. 2002, 76, 3920–3927. [Google Scholar] [CrossRef] [PubMed]
- Greco-Stewart, V.S.; Miron, P.; Abrahem, A.; Pelchat, M. The human RNA polymerase II interacts with the terminal stem-loop regions of the hepatitis delta virus RNA genome. Virology 2007, 357, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Giersch, K.; Helbig, M.; Volz, T.; Allweiss, L.; Mancke, L.V.; Lohse, A.W.; Polywka, S.; Pollok, J.M.; Petersen, J.; Taylor, J.; et al. Persistent hepatitis D virus mono-infection in humanized mice is efficiently converted by hepatitis B virus to a productive co-infection. J. Hepatol. 2014, 60, 538–544. [Google Scholar] [CrossRef] [PubMed]
- Rizzetto, M.; Macagno, S.; Chiaberge, E.; Verme, G.; Negro, F.; Marinucci, G.; di Giacomo, C.; Alfani, D.; Cortesini, R.; Milazzo, F.; et al. Liver transplantation in hepatitis delta virus disease. Lancet 1987, 2, 469–471. [Google Scholar] [CrossRef]
- Ottobrelli, A.; Marzano, A.; Smedile, A.; Recchia, S.; Salizzoni, M.; Cornu, C.; Lamy, M.E.; Otte, J.B.; De Hemptinne, B.; Geubel, A.; et al. Patterns of hepatitis delta virus reinfection and disease in liver transplantation. Gastroenterology 1991, 101, 1649–1655. [Google Scholar] [CrossRef]
- Samuel, D.; Zignego, A.L.; Reynes, M.; Feray, C.; Arulnaden, J.L.; David, M.F.; Gigou, M.; Bismuth, A.; Mathieu, D.; Gentilini, P.; et al. Long-term clinical and virological outcome after liver transplantation for cirrhosis caused by chronic delta hepatitis. Hepatology 1995, 21, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Mederacke, I.; Filmann, N.; Yurdaydin, C.; Bremer, B.; Puls, F.; Zacher, B.J.; Heidrich, B.; Tillmann, H.L.; Rosenau, J.; Bock, C.T.; et al. Rapid early HDV RNA decline in the peripheral blood but prolonged intrahepatic hepatitis delta antigen persistence after liver transplantation. J. Hepatol. 2012, 56, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Giersch, K.; Bhadra, O.D.; Volz, T.; Allweiss, L.; Riecken, K.; Fehse, B.; Lohse, A.W.; Petersen, J.; Sureau, C.; Urban, S.; et al. Hepatitis delta virus persists during liver regeneration and is amplified through cell division both in vitro and in vivo. Gut 2019, 68, 150–157. [Google Scholar] [CrossRef]
- Zhang, Z.; Ni, Y.; Urban, S. Endogenous and exogenous IFN responses suppress HDV persistence during proliferation of hepatocytes in vitro. J. Hepatol. 2019, 70, e718–e719. [Google Scholar] [CrossRef]
- Bogomolov, P.; Alexandrov, A.; Voronkova, N.; Macievich, M.; Kokina, K.; Petrachenkova, M.; Lehr, T.; Lempp, F.A.; Wedemeyer, H.; Haag, M.; et al. Treatment of chronic hepatitis D with the entry inhibitor myrcludex B: First results of a phase Ib/IIa study. J. Hepatol. 2016, 65, 490–498. [Google Scholar] [CrossRef] [PubMed]
- Lempp, F.A.; Urban, S. Hepatitis Delta Virus: Replication Strategy and Upcoming Therapeutic Options for a Neglected Human Pathogen. Viruses 2017, 9, 172. [Google Scholar] [CrossRef] [PubMed]
- Tu, T.; Urban, S. Virus entry and its inhibition to prevent and treat hepatitis B and hepatitis D virus infections. Curr. Opin. Virol. 2018, 30, 68–79. [Google Scholar] [CrossRef] [PubMed]
- Streicher, F.; Jouvenet, N. Stimulation of Innate Immunity by Host and Viral RNAs. Trends Immunol. 2019, 40, 1134–1148. [Google Scholar] [CrossRef] [PubMed]
- Chan, Y.K.; Gack, M.U. Viral evasion of intracellular DNA and RNA sensing. Nat. Rev. Microbiol. 2016, 14, 360–373. [Google Scholar] [CrossRef]
- Chen, P.J.; Kalpana, G.; Goldberg, J.; Mason, W.; Werner, B.; Gerin, J.; Taylor, J. Structure and replication of the genome of the hepatitis delta virus. Proc. Natl. Acad. Sci. USA 1986, 83, 8774–8778. [Google Scholar] [CrossRef]
- Kos, A.; Dijkema, R.; Arnberg, A.C.; van der Meide, P.H.; Schellekens, H. The hepatitis delta (delta) virus possesses a circular RNA. Nature 1986, 323, 558–560. [Google Scholar] [CrossRef]
- Hsu, C.W.; Juang, H.H.; Kuo, C.Y.; Li, H.P.; Iang, S.B.; Lin, S.H.; Yeh, C.T.; Chao, M. Structural Pattern Differences in Unbranched Rod-like RNA of Hepatitis Delta Virus affect RNA Editing. Viruses 2019, 11, 934. [Google Scholar] [CrossRef]
- Abeywickrama-Samarakoon, N.; Cortay, J.C.; Sureau, C.; Muller, S.; Alfaiate, D.; Guerrieri, F.; Chaikuad, A.; Schroder, M.; Merle, P.; Levrero, M.; et al. Hepatitis Delta Virus histone mimicry drives the recruitment of chromatin remodelers for viral RNA replication. Nat. Commun. 2020, 11, 419. [Google Scholar] [CrossRef]
- Seitz, S.; Iancu, C.; Volz, T.; Mier, W.; Dandri, M.; Urban, S.; Bartenschlager, R. A Slow Maturation Process Renders Hepatitis B Virus Infectious. Cell Host Microbe 2016, 20, 25–35. [Google Scholar] [CrossRef]
- Sureau, C. The role of the HBV envelope proteins in the HDV replication cycle. Curr. Top. Microbiol. Immunol. 2006, 307, 113–131. [Google Scholar] [PubMed]
- Sureau, C.; Guerra, B.; Lanford, R.E. Role of the large hepatitis B virus envelope protein in infectivity of the hepatitis delta virion. J. Virol. 1993, 67, 366–372. [Google Scholar] [CrossRef] [PubMed]
- Ganem, D.; Prince, A.M. Hepatitis B virus infection—Natural history and clinical consequences. N. Engl. J. Med. 2004, 350, 1118–1129. [Google Scholar] [CrossRef] [PubMed]
- Chai, N.; Chang, H.E.; Nicolas, E.; Han, Z.; Jarnik, M.; Taylor, J. Properties of subviral particles of hepatitis B virus. J. Virol. 2008, 82, 7812–7817. [Google Scholar] [CrossRef] [PubMed]
- Glenn, J.S.; Watson, J.A.; Havel, C.M.; White, J.M. Identification of a prenylation site in delta virus large antigen. Science 1992, 256, 1331–1333. [Google Scholar] [CrossRef]
- Schulze, A.; Gripon, P.; Urban, S. Hepatitis B virus infection initiates with a large surface protein-dependent binding to heparan sulfate proteoglycans. Hepatology 2007, 46, 1759–1768. [Google Scholar] [CrossRef] [PubMed]
- Leistner, C.M.; Gruen-Bernhard, S.; Glebe, D. Role of glycosaminoglycans for binding and infection of hepatitis B virus. Cell. Microbiol. 2008, 10, 122–133. [Google Scholar] [CrossRef]
- Verrier, E.R.; Colpitts, C.C.; Bach, C.; Heydmann, L.; Weiss, A.; Renaud, M.; Durand, S.C.; Habersetzer, F.; Durantel, D.; Abou-Jaoude, G.; et al. A targeted functional RNA interference screen uncovers glypican 5 as an entry factor for hepatitis B and D viruses. Hepatology 2016, 63, 35–48. [Google Scholar] [CrossRef]
- Lamas Longarela, O.; Schmidt, T.T.; Schoneweis, K.; Romeo, R.; Wedemeyer, H.; Urban, S.; Schulze, A. Proteoglycans act as cellular hepatitis delta virus attachment receptors. PLoS ONE 2013, 8, e58340. [Google Scholar] [CrossRef]
- Blanchet, M.; Sureau, C. Infectivity determinants of the hepatitis B virus pre-S domain are confined to the N-terminal 75 amino acid residues. J. Virol. 2007, 81, 5841–5849. [Google Scholar] [CrossRef]
- Iwamoto, M.; Saso, W.; Sugiyama, R.; Ishii, K.; Ohki, M.; Nagamori, S.; Suzuki, R.; Aizaki, H.; Ryo, A.; Yun, J.H.; et al. Epidermal growth factor receptor is a host-entry cofactor triggering hepatitis B virus internalization. Proc. Natl. Acad. Sci. USA 2019, 116, 8487–8492. [Google Scholar] [CrossRef] [PubMed]
- Iwamoto, M.; Saso, W.; Nishioka, K.; Ohashi, H.; Sugiyama, R.; Ryo, A.; Ohki, M.; Yun, J.H.; Park, S.Y.; Ohshima, T.; et al. The machinery for endocytosis of epidermal growth factor receptor coordinates the transport of incoming hepatitis B virus to the endosomal network. J. Biol. Chem. 2020, 295, 800–807. [Google Scholar] [CrossRef] [PubMed]
- Chou, H.C.; Hsieh, T.Y.; Sheu, G.T.; Lai, M.M. Hepatitis delta antigen mediates the nuclear import of hepatitis delta virus RNA. J. Virol. 1998, 72, 3684–3690. [Google Scholar] [CrossRef] [PubMed]
- Tavanez, J.P.; Cunha, C.; Silva, M.C.; David, E.; Monjardino, J.; Carmo-Fonseca, M. Hepatitis delta virus ribonucleoproteins shuttle between the nucleus and the cytoplasm. RNA 2002, 8, 637–646. [Google Scholar] [CrossRef]
- Gudima, S.; Chang, J.; Moraleda, G.; Azvolinsky, A.; Taylor, J. Parameters of human hepatitis delta virus genome replication: The quantity, quality, and intracellular distribution of viral proteins and RNA. J. Virol. 2002, 76, 3709–3719. [Google Scholar] [CrossRef]
- Wong, S.K.; Lazinski, D.W. Replicating hepatitis delta virus RNA is edited in the nucleus by the small form of ADAR1. Proc. Natl. Acad. Sci. USA 2002, 99, 15118–15123. [Google Scholar] [CrossRef]
- Chakraborty, A.; Ko, C.; Henning, C.; Lucko, A.; Harris, J.M.; Chen, F.; Zhuang, X.; Wettengel, J.M.; Roessler, S.; Protzer, U.; et al. Synchronized infection identifies early rate-limiting steps in the hepatitis B virus life cycle. Cell. Microbiol. 2020. [Google Scholar] [CrossRef]
- Watanabe, T.; Sorensen, E.M.; Naito, A.; Schott, M.; Kim, S.; Ahlquist, P. Involvement of host cellular multivesicular body functions in hepatitis B virus budding. Proc. Natl. Acad. Sci. USA 2007, 104, 10205–10210. [Google Scholar] [CrossRef]
- Tu, T.; Budzinska, M.A.; Shackel, N.A.; Urban, S. HBV DNA Integration: Molecular Mechanisms and Clinical Implications. Viruses 2017, 9, 75. [Google Scholar] [CrossRef]
- Tu, T.; Budzinska, M.A.; Vondran, F.W.R.; Shackel, N.A.; Urban, S. Hepatitis B Virus DNA Integration Occurs Early in the Viral Life Cycle in an In Vitro Infection Model via Sodium Taurocholate Cotransporting Polypeptide-Dependent Uptake of Enveloped Virus Particles. J. Virol. 2018, 92, e02007-17. [Google Scholar] [CrossRef]
- Freitas, N.; Cunha, C.; Menne, S.; Gudima, S.O. Envelope proteins derived from naturally integrated hepatitis B virus DNA support assembly and release of infectious hepatitis delta virus particles. J. Virol. 2014, 88, 5742–5754. [Google Scholar] [CrossRef] [PubMed]
- Lempp, F.A.; Schlund, F.; Rieble, L.; Nussbaum, L.; Link, C.; Zhang, Z.; Ni, Y.; Urban, S. Recapitulation of HDV infection in a fully permissive hepatoma cell line allows efficient drug evaluation. Nat. Commun. 2019, 10, 2265. [Google Scholar] [CrossRef] [PubMed]
- Ni, Y.; Zhang, Z.; Engelskircher, L.; Verch, G.; Tu, T.; Lempp, F.A.; Urban, S. Generation and characterization of a stable cell line persistently replicating and secreting the human hepatitis delta virus. Sci. Rep. 2019, 9, 10021. [Google Scholar] [CrossRef] [PubMed]
- Filipovska, J.; Konarska, M.M. Specific HDV RNA-templated transcription by pol II in vitro. RNA 2000, 6, 41–54. [Google Scholar] [CrossRef] [PubMed]
- Modahl, L.E.; Macnaughton, T.B.; Zhu, N.; Johnson, D.L.; Lai, M.M. RNA-Dependent replication and transcription of hepatitis delta virus RNA involve distinct cellular RNA polymerases. Mol. Cell. Biol. 2000, 20, 6030–6039. [Google Scholar] [CrossRef] [PubMed]
- Modahl, L.E.; Lai, M.M. Transcription of hepatitis delta antigen mRNA continues throughout hepatitis delta virus (HDV) replication: A new model of HDV RNA transcription and replication. J. Virol. 1998, 72, 5449–5456. [Google Scholar] [CrossRef]
- Kuo, M.Y.; Chao, M.; Taylor, J. Initiation of replication of the human hepatitis delta virus genome from cloned DNA: Role of delta antigen. J. Virol. 1989, 63, 1945–1950. [Google Scholar] [CrossRef]
- Harichandran, K.; Shen, Y.; Stephenson Tsoris, S.; Lee, S.C.; Casey, J.L. Hepatitis Delta Antigen Regulates mRNA and Antigenome RNA Levels during Hepatitis Delta Virus Replication. J. Virol. 2019, 93. [Google Scholar] [CrossRef]
- Lehmann, E.; Brueckner, F.; Cramer, P. Molecular basis of RNA-dependent RNA polymerase II activity. Nature 2007, 450, 445–449. [Google Scholar] [CrossRef]
- Lai, M.M. RNA replication without RNA-dependent RNA polymerase: Surprises from hepatitis delta virus. J. Virol. 2005, 79, 7951–7958. [Google Scholar] [CrossRef]
- Cao, D.; Haussecker, D.; Huang, Y.; Kay, M.A. Combined proteomic-RNAi screen for host factors involved in human hepatitis delta virus replication. RNA 2009, 15, 1971–1979. [Google Scholar] [CrossRef] [PubMed]
- Luo, G.X.; Chao, M.; Hsieh, S.Y.; Sureau, C.; Nishikura, K.; Taylor, J. A specific base transition occurs on replicating hepatitis delta virus RNA. J. Virol. 1990, 64, 1021–1027. [Google Scholar] [CrossRef] [PubMed]
- Glenn, J.S.; White, J.M. trans-dominant inhibition of human hepatitis delta virus genome replication. J. Virol. 1991, 65, 2357–2361. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.P.; Lai, M.M. Oligomerization of hepatitis delta antigen is required for both the trans-activating and trans-dominant inhibitory activities of the delta antigen. J. Virol. 1992, 66, 6641–6648. [Google Scholar] [CrossRef] [PubMed]
- Chang, F.L.; Chen, P.J.; Tu, S.J.; Wang, C.J.; Chen, D.S. The large form of hepatitis delta antigen is crucial for assembly of hepatitis delta virus. Proc. Natl. Acad. Sci. USA 1991, 88, 8490–8494. [Google Scholar] [CrossRef]
- Lee, C.H.; Chang, S.C.; Wu, C.H.; Chang, M.F. A novel chromosome region maintenance 1-independent nuclear export signal of the large form of hepatitis delta antigen that is required for the viral assembly. J. Biol. Chem. 2001, 276, 8142–8148. [Google Scholar] [CrossRef]
- Sureau, C.; Negro, F. The hepatitis delta virus: Replication and pathogenesis. J. Hepatol. 2016, 64, S102–S116. [Google Scholar] [CrossRef]
- O’Malley, B.; Lazinski, D.W. Roles of carboxyl-terminal and farnesylated residues in the functions of the large hepatitis delta antigen. J. Virol. 2005, 79, 1142–1153. [Google Scholar] [CrossRef]
- Hwang, S.B.; Lai, M.M. Isoprenylation mediates direct protein-protein interactions between hepatitis large delta antigen and hepatitis B virus surface antigen. J. Virol. 1993, 67, 7659–7662. [Google Scholar] [CrossRef]
- Lucifora, J.; Delphin, M. Current knowledge on Hepatitis Delta Virus replication. Antivir. Res. 2020, 179, 104812. [Google Scholar] [CrossRef]
- Lempp, F.A.; Ni, Y.; Urban, S. Hepatitis delta virus: Insights into a peculiar pathogen and novel treatment options. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 580–589. [Google Scholar] [CrossRef]
- Watashi, K.; Wakita, T. Hepatitis B Virus and Hepatitis D Virus Entry, Species Specificity, and Tissue Tropism. Cold Spring Harb. Perspect. Med. 2015, 5, a021378. [Google Scholar] [CrossRef] [PubMed]
- Herrscher, C.; Roingeard, P.; Blanchard, E. Hepatitis B Virus Entry into Cells. Cells 2020, 9, 1486. [Google Scholar] [CrossRef]
- Wei, L.; Ploss, A. Core components of DNA lagging strand synthesis machinery are essential for hepatitis B virus cccDNA formation. Nat. Microbiol. 2020, 5, 715–726. [Google Scholar] [CrossRef]
- Wooddell, C.I.; Yuen, M.F.; Chan, H.L.; Gish, R.G.; Locarnini, S.A.; Chavez, D.; Ferrari, C.; Given, B.D.; Hamilton, J.; Kanner, S.B.; et al. RNAi-based treatment of chronically infected patients and chimpanzees reveals that integrated hepatitis B virus DNA is a source of HBsAg. Sci. Transl. Med. 2017, 9, eaan0241. [Google Scholar] [CrossRef]
- Gripon, P.; Cannie, I.; Urban, S. Efficient inhibition of hepatitis B virus infection by acylated peptides derived from the large viral surface protein. J. Virol. 2005, 79, 1613–1622. [Google Scholar] [CrossRef] [PubMed]
- Glebe, D.; Urban, S.; Knoop, E.V.; Cag, N.; Krass, P.; Grun, S.; Bulavaite, A.; Sasnauskas, K.; Gerlich, W.H. Mapping of the hepatitis B virus attachment site by use of infection-inhibiting preS1 lipopeptides and tupaia hepatocytes. Gastroenterology 2005, 129, 234–245. [Google Scholar] [CrossRef] [PubMed]
- Volz, T.; Allweiss, L.; Ben, M.M.; Warlich, M.; Lohse, A.W.; Pollok, J.M.; Alexandrov, A.; Urban, S.; Petersen, J.; Lutgehetmann, M.; et al. The entry inhibitor Myrcludex-B efficiently blocks intrahepatic virus spreading in humanized mice previously infected with hepatitis B virus. J. Hepatol. 2013, 58, 861–867. [Google Scholar] [CrossRef]
- Lutgehetmann, M.; Mancke, L.V.; Volz, T.; Helbig, M.; Allweiss, L.; Bornscheuer, T.; Pollok, J.M.; Lohse, A.W.; Petersen, J.; Urban, S.; et al. Humanized chimeric uPA mouse model for the study of hepatitis B and D virus interactions and preclinical drug evaluation. Hepatology 2012, 55, 685–694. [Google Scholar] [CrossRef]
- Koh, C.; Canini, L.; Dahari, H.; Zhao, X.; Uprichard, S.L.; Haynes-Williams, V.; Winters, M.A.; Subramanya, G.; Cooper, S.L.; Pinto, P.; et al. Oral prenylation inhibition with lonafarnib in chronic hepatitis D infection: A proof-of-concept randomised, double-blind, placebo-controlled phase 2A trial. Lancet Infect. Dis. 2015, 15, 1167–1174. [Google Scholar] [CrossRef]
- Yurdaydin, C.; Keskin, O.; Kalkan, C.; Karakaya, F.; Caliskan, A.; Karatayli, E.; Karatayli, S.; Bozdayi, A.M.; Koh, C.; Heller, T.; et al. Optimizing lonafarnib treatment for the management of chronic delta hepatitis: The LOWR HDV-1 study. Hepatology 2018, 67, 1224–1236. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Walther, T.; Lempp, F.A.; Ni, Y.; Urban, S. Synergistic suppression of HDV persistence in vitro by co-treatment with investigational drugs targeting both extracellular and cell division mediated spreading pathways. Hepatology 2019, 70, 176A–177A. [Google Scholar]
- Paraskevopoulou, S.; Pirzer, F.; Goldmann, N.; Schmid, J.; Corman, V.M.; Gottula, L.T.; Schroeder, S.; Rasche, A.; Muth, D.; Drexler, J.F.; et al. Mammalian deltavirus without hepadnavirus coinfection in the neotropical rodent Proechimys semispinosus. Proc. Natl. Acad. Sci. USA 2020, 117, 17977–17983. [Google Scholar] [CrossRef] [PubMed]
- Hetzel, U.; Szirovicza, L.; Smura, T.; Prahauser, B.; Vapalahti, O.; Kipar, A.; Hepojoki, J. Identification of a Novel Deltavirus in Boa Constrictors. mBio 2019, 10, e00014-19. [Google Scholar] [CrossRef]
- Wille, M.; Netter, H.J.; Littlejohn, M.; Yuen, L.; Shi, M.; Eden, J.S.; Klaassen, M.; Holmes, E.C.; Hurt, A.C. A Divergent Hepatitis D-Like Agent in Birds. Viruses 2018, 10, 720. [Google Scholar] [CrossRef]
- Chang, W.S.; Pettersson, J.H.; Le Lay, C.; Shi, M.; Lo, N.; Wille, M.; Eden, J.S.; Holmes, E.C. Novel hepatitis D-like agents in vertebrates and invertebrates. Virus Evol. 2019, 5, vez021. [Google Scholar]
- Perez-Vargas, J.; Amirache, F.; Boson, B.; Mialon, C.; Freitas, N.; Sureau, C.; Fusil, F.; Cosset, F.L. Enveloped viruses distinct from HBV induce dissemination of hepatitis D virus in vivo. Nat. Commun. 2019, 10, 2098. [Google Scholar] [CrossRef]
- Chemin, I.; Pujol, F.H.; Scholtes, C.; Loureiro, C.L.; Amirache, F.; Levrero, M.; Zoulim, F.; Perez-Vargas, J.; Cosset, F.L. Preliminary evidence for hdv exposure in apparently non-HBV-infected patients. Hepatology 2020. [Google Scholar] [CrossRef]
- Pfluger, L.S.; Schulze Zur Wiesch, J.; Polywka, S.; Lutgehetmann, M. Hepatitis delta virus propagation enabled by hepatitis C virus—Scientifically intriguing; but is it relevant to clinical practice? J. Viral Hepat. 2020. [Google Scholar] [CrossRef]
- Cappy, P.; Lucas, Q.; Kankarafou, N.; Sureau, C.; Laperche, S. No Evidence of HCV-Assisted HDV Propagation in a Large Cohort of Hepatitis C Positive Blood Donors. J. Infect. Dis. 2020. [Google Scholar] [CrossRef]
- Bichko, V.V.; Taylor, J.M. Redistribution of the delta antigens in cells replicating the genome of hepatitis delta virus. J. Virol. 1996, 70, 8064–8070. [Google Scholar] [CrossRef] [PubMed]
- Medzhitov, R.; Janeway, C., Jr. Innate immunity. N. Engl. J. Med. 2000, 343, 338–344. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.; Pasare, C. Innate Control of Adaptive Immunity: Beyond the Three-Signal Paradigm. J. Immunol. 2017, 198, 3791–3800. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, K.; Zou, Z.Q. Crosstalk between innate and adaptive immunity in hepatitis B virus infection. World J. Hepatol. 2015, 7, 2980–2991. [Google Scholar] [CrossRef]
- Goubau, D.; Deddouche, S.; Reis e Sousa, C. Cytosolic sensing of viruses. Immunity 2013, 38, 855–869. [Google Scholar] [CrossRef]
- Hartmann, G. Nucleic Acid Immunity. Adv. Immunol. 2017, 133, 121–169. [Google Scholar]
- Gitlin, L.; Barchet, W.; Gilfillan, S.; Cella, M.; Beutler, B.; Flavell, R.A.; Diamond, M.S.; Colonna, M. Essential role of mda-5 in type I IFN responses to polyriboinosinic:polyribocytidylic acid and encephalomyocarditis picornavirus. Proc. Natl. Acad. Sci. USA 2006, 103, 8459–8464. [Google Scholar] [CrossRef]
- Kato, H.; Takeuchi, O.; Sato, S.; Yoneyama, M.; Yamamoto, M.; Matsui, K.; Uematsu, S.; Jung, A.; Kawai, T.; Ishii, K.J.; et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 2006, 441, 101–105. [Google Scholar] [CrossRef]
- Kotenko, S.V.; Gallagher, G.; Baurin, V.V.; Lewis-Antes, A.; Shen, M.; Shah, N.K.; Langer, J.A.; Sheikh, F.; Dickensheets, H.; Donnelly, R.P. IFN-lambdas mediate antiviral protection through a distinct class II cytokine receptor complex. Nat. Immunol. 2003, 4, 69–77. [Google Scholar] [CrossRef]
- Sheppard, P.; Kindsvogel, W.; Xu, W.; Henderson, K.; Schlutsmeyer, S.; Whitmore, T.E.; Kuestner, R.; Garrigues, U.; Birks, C.; Roraback, J.; et al. IL-28, IL-29 and their class II cytokine receptor IL-28R. Nat. Immunol. 2003, 4, 63–68. [Google Scholar] [CrossRef]
- De Weerd, N.A.; Nguyen, T. The interferons and their receptors--distribution and regulation. Immunol. Cell Biol. 2012, 90, 483–491. [Google Scholar] [CrossRef] [PubMed]
- Mesev, E.V.; LeDesma, R.A.; Ploss, A. Decoding type I and III interferon signalling during viral infection. Nat. Microbiol. 2019, 4, 914–924. [Google Scholar] [CrossRef] [PubMed]
- Stark, G.R.; Darnell, J.E., Jr. The JAK-STAT pathway at twenty. Immunity 2012, 36, 503–514. [Google Scholar] [CrossRef]
- Schneider, W.M.; Chevillotte, M.D.; Rice, C.M. Interferon-stimulated genes: A complex web of host defenses. Annu. Rev. Immunol. 2014, 32, 513–545. [Google Scholar] [CrossRef] [PubMed]
- Rehwinkel, J.; Gack, M.U. RIG-I-like receptors: Their regulation and roles in RNA sensing. Nat. Rev. Immunol. 2020, 20, 537–551. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Filzmayer, C.; Ni, Y.; Sultmann, H.; Mutz, P.; Hiet, M.S.; Vondran, F.W.R.; Bartenschlager, R.; Urban, S. Hepatitis D virus replication is sensed by MDA5 and induces IFN-beta/lambda responses in hepatocytes. J. Hepatol. 2018, 69, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Brisse, M.; Ly, H. Comparative Structure and Function Analysis of the RIG-I-Like Receptors: RIG-I and MDA5. Front. Immunol. 2019, 10, 1586. [Google Scholar] [CrossRef]
- Yoneyama, M.; Kikuchi, M.; Matsumoto, K.; Imaizumi, T.; Miyagishi, M.; Taira, K.; Foy, E.; Loo, Y.M.; Gale, M., Jr.; Akira, S.; et al. Shared and unique functions of the DExD/H-box helicases RIG-I, MDA5, and LGP2 in antiviral innate immunity. J. Immunol. 2005, 175, 2851–2858. [Google Scholar] [CrossRef]
- Schlee, M.; Roth, A.; Hornung, V.; Hagmann, C.A.; Wimmenauer, V.; Barchet, W.; Coch, C.; Janke, M.; Mihailovic, A.; Wardle, G.; et al. Recognition of 5′ triphosphate by RIG-I helicase requires short blunt double-stranded RNA as contained in panhandle of negative-strand virus. Immunity 2009, 31, 25–34. [Google Scholar] [CrossRef]
- Schmidt, A.; Schwerd, T.; Hamm, W.; Hellmuth, J.C.; Cui, S.; Wenzel, M.; Hoffmann, F.S.; Michallet, M.C.; Besch, R.; Hopfner, K.P.; et al. 5′-triphosphate RNA requires base-paired structures to activate antiviral signaling via RIG-I. Proc. Natl. Acad. Sci. USA 2009, 106, 12067–12072. [Google Scholar] [CrossRef]
- Goubau, D.; Schlee, M.; Deddouche, S.; Pruijssers, A.J.; Zillinger, T.; Goldeck, M.; Schuberth, C.; Van der Veen, A.G.; Fujimura, T.; Rehwinkel, J.; et al. Antiviral immunity via RIG-I-mediated recognition of RNA bearing 5’-diphosphates. Nature 2014, 514, 372–375. [Google Scholar] [CrossRef] [PubMed]
- Baum, A.; Sachidanandam, R.; Garcia-Sastre, A. Preference of RIG-I for short viral RNA molecules in infected cells revealed by next-generation sequencing. Proc. Natl. Acad. Sci. USA 2010, 107, 16303–16308. [Google Scholar] [CrossRef] [PubMed]
- Sanchez David, R.Y.; Combredet, C.; Sismeiro, O.; Dillies, M.A.; Jagla, B.; Coppee, J.Y.; Mura, M.; Guerbois Galla, M.; Despres, P.; Tangy, F.; et al. Comparative analysis of viral RNA signatures on different RIG-I-like receptors. eLife 2016, 5, e11275. [Google Scholar] [CrossRef]
- Rehwinkel, J.; Tan, C.P.; Goubau, D.; Schulz, O.; Pichlmair, A.; Bier, K.; Robb, N.; Vreede, F.; Barclay, W.; Fodor, E.; et al. RIG-I detects viral genomic RNA during negative-strand RNA virus infection. Cell 2010, 140, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Park, H.S.; Pyo, H.M.; Liu, Q.; Zhou, Y. Influenza A Virus Panhandle Structure Is Directly Involved in RIG-I Activation and Interferon Induction. J. Virol. 2015, 89, 6067–6079. [Google Scholar] [CrossRef]
- Chazal, M.; Beauclair, G.; Gracias, S.; Najburg, V.; Simon-Loriere, E.; Tangy, F.; Komarova, A.V.; Jouvenet, N. RIG-I Recognizes the 5’ Region of Dengue and Zika Virus Genomes. Cell Rep. 2018, 24, 320–328. [Google Scholar] [CrossRef]
- Kato, H.; Takeuchi, O.; Mikamo-Satoh, E.; Hirai, R.; Kawai, T.; Matsushita, K.; Hiiragi, A.; Dermody, T.S.; Fujita, T.; Akira, S. Length-dependent recognition of double-stranded ribonucleic acids by retinoic acid-inducible gene-I and melanoma differentiation-associated gene 5. J. Exp. Med. 2008, 205, 1601–1610. [Google Scholar] [CrossRef]
- Pichlmair, A.; Schulz, O.; Tan, C.P.; Rehwinkel, J.; Kato, H.; Takeuchi, O.; Akira, S.; Way, M.; Schiavo, G.; Reis e Sousa, C. Activation of MDA5 requires higher-order RNA structures generated during virus infection. J. Virol. 2009, 83, 10761–10769. [Google Scholar] [CrossRef]
- Cao, X.; Ding, Q.; Lu, J.; Tao, W.; Huang, B.; Zhao, Y.; Niu, J.; Liu, Y.J.; Zhong, J. MDA5 plays a critical role in interferon response during hepatitis C virus infection. J. Hepatol. 2015, 62, 771–778. [Google Scholar] [CrossRef]
- Li, X.; Ranjith-Kumar, C.T.; Brooks, M.T.; Dharmaiah, S.; Herr, A.B.; Kao, C.; Li, P. The RIG-I-like receptor LGP2 recognizes the termini of double-stranded RNA. J. Biol. Chem. 2009, 284, 13881–13891. [Google Scholar] [CrossRef]
- Pippig, D.A.; Hellmuth, J.C.; Cui, S.; Kirchhofer, A.; Lammens, K.; Lammens, A.; Schmidt, A.; Rothenfusser, S.; Hopfner, K.P. The regulatory domain of the RIG-I family ATPase LGP2 senses double-stranded RNA. Nucleic Acids Res. 2009, 37, 2014–2025. [Google Scholar] [CrossRef] [PubMed]
- Uchikawa, E.; Lethier, M.; Malet, H.; Brunel, J.; Gerlier, D.; Cusack, S. Structural Analysis of dsRNA Binding to Anti-viral Pattern Recognition Receptors LGP2 and MDA5. Mol. Cell 2016, 62, 586–602. [Google Scholar] [CrossRef] [PubMed]
- Hei, L.; Zhong, J. Laboratory of genetics and physiology 2 (LGP2) plays an essential role in hepatitis C virus infection-induced interferon responses. Hepatology 2017, 65, 1478–1491. [Google Scholar] [CrossRef] [PubMed]
- Bruns, A.M.; Leser, G.P.; Lamb, R.A.; Horvath, C.M. The innate immune sensor LGP2 activates antiviral signaling by regulating MDA5-RNA interaction and filament assembly. Mol. Cell 2014, 55, 771–781. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Hirai, R.; Loo, Y.M.; Owen, D.; Johnson, C.L.; Sinha, S.C.; Akira, S.; Fujita, T.; Gale, M., Jr. Regulation of innate antiviral defenses through a shared repressor domain in RIG-I and LGP2. Proc. Natl. Acad. Sci. USA 2007, 104, 582–587. [Google Scholar] [CrossRef]
- Bamming, D.; Horvath, C.M. Regulation of signal transduction by enzymatically inactive antiviral RNA helicase proteins MDA5, RIG-I, and LGP2. J. Biol. Chem. 2009, 284, 9700–9712. [Google Scholar] [CrossRef]
- Kowalinski, E.; Lunardi, T.; McCarthy, A.A.; Louber, J.; Brunel, J.; Grigorov, B.; Gerlier, D.; Cusack, S. Structural basis for the activation of innate immune pattern-recognition receptor RIG-I by viral RNA. Cell 2011, 147, 423–435. [Google Scholar] [CrossRef]
- Potter, J.A.; Randall, R.E.; Taylor, G.L. Crystal structure of human IPS-1/MAVS/VISA/Cardif caspase activation recruitment domain. BMC Struct. Biol. 2008, 8, 11. [Google Scholar] [CrossRef]
- Lassig, C.; Hopfner, K.P. Discrimination of cytosolic self and non-self RNA by RIG-I-like receptors. J. Biol. Chem. 2017, 292, 9000–9009. [Google Scholar] [CrossRef]
- Alfaiate, D.; Lucifora, J.; Abeywickrama-Samarakoon, N.; Michelet, M.; Testoni, B.; Cortay, J.C.; Sureau, C.; Zoulim, F.; Deny, P.; Durantel, D. HDV RNA replication is associated with HBV repression and interferon-stimulated genes induction in super-infected hepatocytes. Antiviral Res. 2016, 136, 19–31. [Google Scholar] [CrossRef]
- He, W.; Ren, B.; Mao, F.; Jing, Z.; Li, Y.; Liu, Y.; Peng, B.; Yan, H.; Qi, Y.; Sun, Y.; et al. Hepatitis D Virus Infection of Mice Expressing Human Sodium Taurocholate Co-transporting Polypeptide. PLoS Pathog. 2015, 11, e1004840. [Google Scholar] [CrossRef] [PubMed]
- Giersch, K.; Allweiss, L.; Volz, T.; Helbig, M.; Bierwolf, J.; Lohse, A.W.; Pollok, J.M.; Petersen, J.; Dandri, M.; Lutgehetmann, M. Hepatitis Delta co-infection in humanized mice leads to pronounced induction of innate immune responses in comparison to HBV mono-infection. J. Hepatol. 2015, 63, 346–353. [Google Scholar] [CrossRef] [PubMed]
- Suarez-Amaran, L.; Usai, C.; Di Scala, M.; Godoy, C.; Ni, Y.; Hommel, M.; Palomo, L.; Segura, V.; Olague, C.; Vales, A.; et al. A new HDV mouse model identifies mitochondrial antiviral signaling protein (MAVS) as a key player in IFN-beta induction. J. Hepatol. 2017, 67, 669–679. [Google Scholar] [CrossRef] [PubMed]
- Winer, B.Y.; Shirvani-Dastgerdi, E.; Bram, Y.; Sellau, J.; Low, B.E.; Johnson, H.; Huang, T.; Hrebikova, G.; Heller, B.; Sharon, Y.; et al. Preclinical assessment of antiviral combination therapy in a genetically humanized mouse model for hepatitis delta virus infection. Sci. Transl. Med. 2018, 10, eaap9328. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Sigal, L.J.; Lerro, A.; Taylor, J. Replication of the human hepatitis delta virus genome Is initiated in mouse hepatocytes following intravenous injection of naked DNA or RNA sequences. J. Virol. 2001, 75, 3469–3473. [Google Scholar] [CrossRef]
- Winer, B.Y.; Gaska, J.M.; Lipkowitz, G.; Bram, Y.; Parekh, A.; Parsons, L.; Leach, R.; Jindal, R.; Cho, C.H.; Shrirao, A.; et al. Analysis of Host Responses to Hepatitis B and Delta Viral Infections in a Micro-scalable Hepatic Co-culture System. Hepatology 2020, 71, 14–30. [Google Scholar] [CrossRef]
- Weber, M.; Gawanbacht, A.; Habjan, M.; Rang, A.; Borner, C.; Schmidt, A.M.; Veitinger, S.; Jacob, R.; Devignot, S.; Kochs, G.; et al. Incoming RNA virus nucleocapsids containing a 5′-triphosphorylated genome activate RIG-I and antiviral signaling. Cell Host Microbe 2013, 13, 336–346. [Google Scholar] [CrossRef]
- Liu, G.; Lu, Y.; Thulasi Raman, S.N.; Xu, F.; Wu, Q.; Li, Z.; Brownlie, R.; Liu, Q.; Zhou, Y. Nuclear-resident RIG-I senses viral replication inducing antiviral immunity. Nat. Commun. 2018, 9, 3199. [Google Scholar] [CrossRef]
- Ali, S.; Mann-Nuttel, R.; Schulze, A.; Richter, L.; Alferink, J.; Scheu, S. Sources of Type I Interferons in Infectious Immunity: Plasmacytoid Dendritic Cells Not Always in the Driver’s Seat. Front. Immunol. 2019, 10, 778. [Google Scholar] [CrossRef]
- Ito, T.; Kanzler, H.; Duramad, O.; Cao, W.; Liu, Y.J. Specialization, kinetics, and repertoire of type 1 interferon responses by human plasmacytoid predendritic cells. Blood 2006, 107, 2423–2431. [Google Scholar] [CrossRef]
- Kouwaki, T.; Okamoto, M.; Tsukamoto, H.; Fukushima, Y.; Oshiumi, H. Extracellular Vesicles Deliver Host and Virus RNA and Regulate Innate Immune Response. Int. J. Mol. Sci. 2017, 18, 666. [Google Scholar] [CrossRef] [PubMed]
- Kouwaki, T.; Fukushima, Y.; Daito, T.; Sanada, T.; Yamamoto, N.; Mifsud, E.J.; Leong, C.R.; Tsukiyama-Kohara, K.; Kohara, M.; Matsumoto, M.; et al. Extracellular Vesicles Including Exosomes Regulate Innate Immune Responses to Hepatitis B Virus Infection. Front. Immunol. 2016, 7, 335. [Google Scholar] [CrossRef] [PubMed]
- Dreux, M.; Garaigorta, U.; Boyd, B.; Decembre, E.; Chung, J.; Whitten-Bauer, C.; Wieland, S.; Chisari, F.V. Short-range exosomal transfer of viral RNA from infected cells to plasmacytoid dendritic cells triggers innate immunity. Cell Host Microbe 2012, 12, 558–570. [Google Scholar] [CrossRef] [PubMed]
- Assil, S.; Coleon, S.; Dong, C.; Decembre, E.; Sherry, L.; Allatif, O.; Webster, B.; Dreux, M. Plasmacytoid Dendritic Cells and Infected Cells Form an Interferogenic Synapse Required for Antiviral Responses. Cell Host Microbe 2019, 25, 730–745.e736. [Google Scholar] [CrossRef] [PubMed]
- Webster, B.; Werneke, S.W.; Zafirova, B.; This, S.; Coleon, S.; Decembre, E.; Paidassi, H.; Bouvier, I.; Joubert, P.E.; Duffy, D.; et al. Plasmacytoid dendritic cells control dengue and Chikungunya virus infections via IRF7-regulated interferon responses. eLife 2018, 7, e34273. [Google Scholar] [CrossRef] [PubMed]
- Webster, B.; Assil, S.; Dreux, M. Cell-Cell Sensing of Viral Infection by Plasmacytoid Dendritic Cells. J. Virol. 2016, 90, 10050–10053. [Google Scholar] [CrossRef]
- Jung, S.; Altstetter, M.S.; Wilsch, F.; Shein, M.; Schütz, A.K.; Protzer, U. Extracellular vesicles derived from Hepatitis-D Virus infected cells induce a proinflammatory cytokine response in human peripheral blood mononuclear cells and macrophages. Matters 2020, in press. [Google Scholar]
- Mentha, N.; Clement, S.; Negro, F.; Alfaiate, D. A review on hepatitis D: From virology to new therapies. J. Adv. Res. 2019, 17, 3–15. [Google Scholar] [CrossRef]
- Abbas, Z.; Khan, M.A.; Salih, M.; Jafri, W. Interferon alpha for chronic hepatitis D. Cochrane Database Syst Rev. 2011, 2011, CD006002. [Google Scholar] [CrossRef]
- Alavian, S.M.; Tabatabaei, S.V.; Behnava, B.; Rizzetto, M. Standard and pegylated interferon therapy of HDV infection: A systematic review and meta-analysis. J. Res. Med. Sci. 2012, 17, 967–974. [Google Scholar]
- Triantos, C.; Kalafateli, M.; Nikolopoulou, V.; Burroughs, A. Meta-analysis: Antiviral treatment for hepatitis D. Aliment. Pharmacol. Ther. 2012, 35, 663–673. [Google Scholar] [CrossRef] [PubMed]
- Giersch, K.; Homs, M.; Volz, T.; Helbig, M.; Allweiss, L.; Lohse, A.W.; Petersen, J.; Buti, M.; Pollicino, T.; Sureau, C.; et al. Both interferon alpha and lambda can reduce all intrahepatic HDV infection markers in HBV/HDV infected humanized mice. Sci. Rep. 2017, 7, 3757. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.; Nogusa, S.; Nicolas, E.; Balachandran, S.; Taylor, J. Interferon impedes an early step of hepatitis delta virus infection. PLoS ONE 2011, 6, e22415. [Google Scholar] [CrossRef] [PubMed]
- Bressy, C.; Droby, G.N.; Maldonado, B.D.; Steuerwald, N.; Grdzelishvili, V.Z. Cell Cycle Arrest in G2/M Phase Enhances Replication of Interferon-Sensitive Cytoplasmic RNA Viruses via Inhibition of Antiviral Gene Expression. J. Virol. 2019, 93, e01885-18. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Korutla, L.; Zhang, K. Cell cycle-dependent modulation of alpha-interferon-inducible gene expression and activation of signaling components in Daudi cells. J. Biol. Chem. 1994, 269, 25437–25441. [Google Scholar]
- Hsiung, C.C.; Bartman, C.R.; Huang, P.; Ginart, P.; Stonestrom, A.J.; Keller, C.A.; Face, C.; Jahn, K.S.; Evans, P.; Sankaranarayanan, L.; et al. A hyperactive transcriptional state marks genome reactivation at the mitosis-G1 transition. Genes Dev. 2016, 30, 1423–1439. [Google Scholar] [CrossRef]
- Naumova, N.; Imakaev, M.; Fudenberg, G.; Zhan, Y.; Lajoie, B.R.; Mirny, L.A.; Dekker, J. Organization of the mitotic chromosome. Science 2013, 342, 948–953. [Google Scholar] [CrossRef]
- Bazinet, M.; Pantea, V.; Cebotarescu, V.; Cojuhari, L.; Jimbei, P.; Albrecht, J.; Schmid, P.; Le Gal, F.; Gordien, E.; Krawczyk, A.; et al. Safety and efficacy of REP 2139 and pegylated interferon alfa-2a for treatment-naive patients with chronic hepatitis B virus and hepatitis D virus co-infection (REP 301 and REP 301-LTF): A non-randomised, open-label, phase 2 trial. Lancet Gastroenterol. Hepatol. 2017, 2, 877–889. [Google Scholar] [CrossRef]
- Bazinet, M.; Pântea, V.; Cebotarescu, V.; Cojuhari, L.; Jimbei, P.; Krawczyk, A.; Dittmer, U.; Vaillant, A. Ongoing analysis of virologic control/functional cure of HBV and HDV infection following REP 2139-Ca and pegylated interferon alpha-2a therapy in patients with chronic HBV/HDV co-infection: 3.5-year follow-up results from the REP 301-LTF study. Hepatology 2019, 70, 440A. [Google Scholar]
- Koh, C.; Da, B.L.; Surana, P.; Huang, A.; Kapuria, D.; Rotman, Y.; Vittal, A.; Gilman, C.; Ben-Yakov, G.; Lai, C.; et al. A Phase 2 Study of Lonafarnib, Ritonavir and Peginterferon Lambda for 24 Weeks: Interim End-Of-Treatment Results from the LIFT HDV Study; WILEY: Hoboken, NJ, USA, 2019; Volume 70, p. 1483A. [Google Scholar]
- Wedemeyer, H.; Schöneweis, K.; Bogomolov, P.; Voronkova, N.; Chulanov, V.; Stepanova, T.; Bremer, B.; Allweiss, L.; Dandri, M.; Burhenne, J.; et al. Final results of a multicenter, open-label phase 2 clinical trial (MYR203) to assess safety and efficacy of myrcludex B in cwith PEG-interferon Alpha 2a in patients with chronic HBV/HDV co-infection. J. Hepatol. 2019, 70, e81. [Google Scholar] [CrossRef]
- Asselah, T.; Loureiro, D.; Tout, I.; Castelnau, C.; Boyer, N.; Marcellin, P.; Mansouri, A. Future treatments for hepatitis delta virus infection. Liver Int. 2020, 40, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Koh, C.; Heller, T.; Glenn, J.S. Pathogenesis of and New Therapies for Hepatitis D. Gastroenterology 2019, 156, 461–476.e461. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Taylor, J.M. Susceptibility of human hepatitis delta virus RNAs to small interfering RNA action. J. Virol. 2003, 77, 9728–9731. [Google Scholar] [CrossRef] [PubMed]
- Ryu, W.S.; Netter, H.J.; Bayer, M.; Taylor, J. Ribonucleoprotein complexes of hepatitis delta virus. J. Virol. 1993, 67, 3281–3287. [Google Scholar] [CrossRef]
- Griffin, B.L.; Chasovskikh, S.; Dritschilo, A.; Casey, J.L. Hepatitis delta antigen requires a flexible quasi-double-stranded RNA structure to bind and condense hepatitis delta virus RNA in a ribonucleoprotein complex. J. Virol. 2014, 88, 7402–7411. [Google Scholar] [CrossRef]
- Lazinski, D.W.; Taylor, J.M. Expression of hepatitis delta virus RNA deletions: Cis and trans requirements for self-cleavage, ligation, and RNA packaging. J. Virol. 1994, 68, 2879–2888. [Google Scholar] [CrossRef]
- Pugnale, P.; Pazienza, V.; Guilloux, K.; Negro, F. Hepatitis delta virus inhibits alpha interferon signaling. Hepatology 2009, 49, 398–406. [Google Scholar] [CrossRef]
- Mutz, P.; Metz, P.; Lempp, F.A.; Bender, S.; Qu, B.; Schoneweis, K.; Seitz, S.; Tu, T.; Restuccia, A.; Frankish, J.; et al. HBV Bypasses the Innate Immune Response and Does Not Protect HCV From Antiviral Activity of Interferon. Gastroenterology 2018, 154, 1791–1804.e1722. [Google Scholar] [CrossRef]
- Wieland, S.; Thimme, R.; Purcell, R.H.; Chisari, F.V. Genomic analysis of the host response to hepatitis B virus infection. Proc. Natl. Acad. Sci. USA 2004, 101, 6669–6674. [Google Scholar] [CrossRef]
- Suslov, A.; Boldanova, T.; Wang, X.; Wieland, S.; Heim, M.H. Hepatitis B Virus Does Not Interfere With Innate Immune Responses in the Human Liver. Gastroenterology 2018, 154, 1778–1790. [Google Scholar] [CrossRef]
- Cheng, X.; Xia, Y.; Serti, E.; Block, P.D.; Chung, M.; Chayama, K.; Rehermann, B.; Liang, T.J. Hepatitis B virus evades innate immunity of hepatocytes but activates cytokine production by macrophages. Hepatology 2017, 66, 1779–1793. [Google Scholar] [CrossRef] [PubMed]
- Dunn, C.; Peppa, D.; Khanna, P.; Nebbia, G.; Jones, M.; Brendish, N.; Lascar, R.M.; Brown, D.; Gilson, R.J.; Tedder, R.J.; et al. Temporal analysis of early immune responses in patients with acute hepatitis B virus infection. Gastroenterology 2009, 137, 1289–1300. [Google Scholar] [CrossRef] [PubMed]
- Lauterbach-Riviere, L.; Bergez, M.; Monch, S.; Qu, B.; Riess, M.; Vondran, F.W.R.; Liese, J.; Hornung, V.; Urban, S.; Konig, R. Hepatitis B Virus DNA is a Substrate for the cGAS/STING Pathway but is not Sensed in Infected Hepatocytes. Viruses 2020, 12, 592. [Google Scholar] [CrossRef] [PubMed]
- Sato, S.; Li, K.; Kameyama, T.; Hayashi, T.; Ishida, Y.; Murakami, S.; Watanabe, T.; Iijima, S.; Sakurai, Y.; Watashi, K.; et al. The RNA sensor RIG-I dually functions as an innate sensor and direct antiviral factor for hepatitis B virus. Immunity 2015, 42, 123–132. [Google Scholar] [CrossRef]
- Luangsay, S.; Gruffaz, M.; Isorce, N.; Testoni, B.; Michelet, M.; Faure-Dupuy, S.; Maadadi, S.; Ait-Goughoulte, M.; Parent, R.; Rivoire, M.; et al. Early inhibition of hepatocyte innate responses by hepatitis B virus. J. Hepatol. 2015, 63, 1314–1322. [Google Scholar] [CrossRef]
- Shlomai, A.; Schwartz, R.E.; Ramanan, V.; Bhatta, A.; de Jong, Y.P.; Bhatia, S.N.; Rice, C.M. Modeling host interactions with hepatitis B virus using primary and induced pluripotent stem cell-derived hepatocellular systems. Proc. Natl. Acad. Sci. USA 2014, 111, 12193–12198. [Google Scholar] [CrossRef]
- Zhang, Z.; Trippler, M.; Real, C.I.; Werner, M.; Luo, X.; Schefczyk, S.; Kemper, T.; Anastasiou, O.E.; Ladiges, Y.; Treckmann, J.; et al. Hepatitis B Virus Particles Activate Toll-Like Receptor 2 Signaling Initially Upon Infection of Primary Human Hepatocytes. Hepatology 2020. [Google Scholar] [CrossRef]
- Lu, H.L.; Liao, F. Melanoma differentiation-associated gene 5 senses hepatitis B virus and activates innate immune signaling to suppress virus replication. J. Immunol. 2013, 191, 3264–3276. [Google Scholar] [CrossRef]
- Colombo, P.; Di Blasi, F.; Magrin, S.; Fabiano, C.; Di Marco, V.; D’Amelio, L.; Lojacono, F.; Spinelli, G.; Craxi, A. Smouldering hepatitis B virus replication in patients with chronic liver disease and hepatitis delta virus superinfection. J. Hepatol. 1991, 12, 64–69. [Google Scholar] [CrossRef]
- Sureau, C.; Taylor, J.; Chao, M.; Eichberg, J.W.; Lanford, R.E. Cloned hepatitis delta virus cDNA is infectious in the chimpanzee. J. Virol. 1989, 63, 4292–4297. [Google Scholar] [CrossRef]
- Cheng, X.; Uchida, T.; Xia, Y.; Umarova, R.; Liu, C.J.; Chen, P.J.; Gaggar, A.; Suri, V.; Mucke, M.M.; Vermehren, J.; et al. Diminished hepatic IFN response following HCV clearance triggers HBV reactivation in coinfection. J. Clin. Investig. 2020, 130, 3205–3220. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Z.; Urban, S. Interplay between Hepatitis D Virus and the Interferon Response. Viruses 2020, 12, 1334. https://doi.org/10.3390/v12111334
Zhang Z, Urban S. Interplay between Hepatitis D Virus and the Interferon Response. Viruses. 2020; 12(11):1334. https://doi.org/10.3390/v12111334
Chicago/Turabian StyleZhang, Zhenfeng, and Stephan Urban. 2020. "Interplay between Hepatitis D Virus and the Interferon Response" Viruses 12, no. 11: 1334. https://doi.org/10.3390/v12111334
APA StyleZhang, Z., & Urban, S. (2020). Interplay between Hepatitis D Virus and the Interferon Response. Viruses, 12(11), 1334. https://doi.org/10.3390/v12111334