Phosphoinositide 3′-Kinase γ Facilitates Polyomavirus Infection

 and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Cells and Viruses
2.2. Immunoblotting
2.3. Transient shRNA Knockdown and JCPsV Infectivity Assay
2.4. Stable shRNA Knockdown and JCPsV Infectivity Assay
2.5. Luciferase Activity in SVGA Cells Transfected with Gluc Plasmid
2.6. siRNA Knockdown and JCPsV Infectivity Assay
2.7. Construction of PI3Kγ and PIK3R5 Knockout Cells
2.8. Infection of CRISPR Knockout Cells with JCPyV
2.9. Rescue of JCPyV Infection in PI3Kγ Knockout Cells
2.10. Effect of PI3Kγ Pathway on Infection by Other Polyomaviruses
2.11. Virus Binding and Internalization Studies
3. Results and Discussion
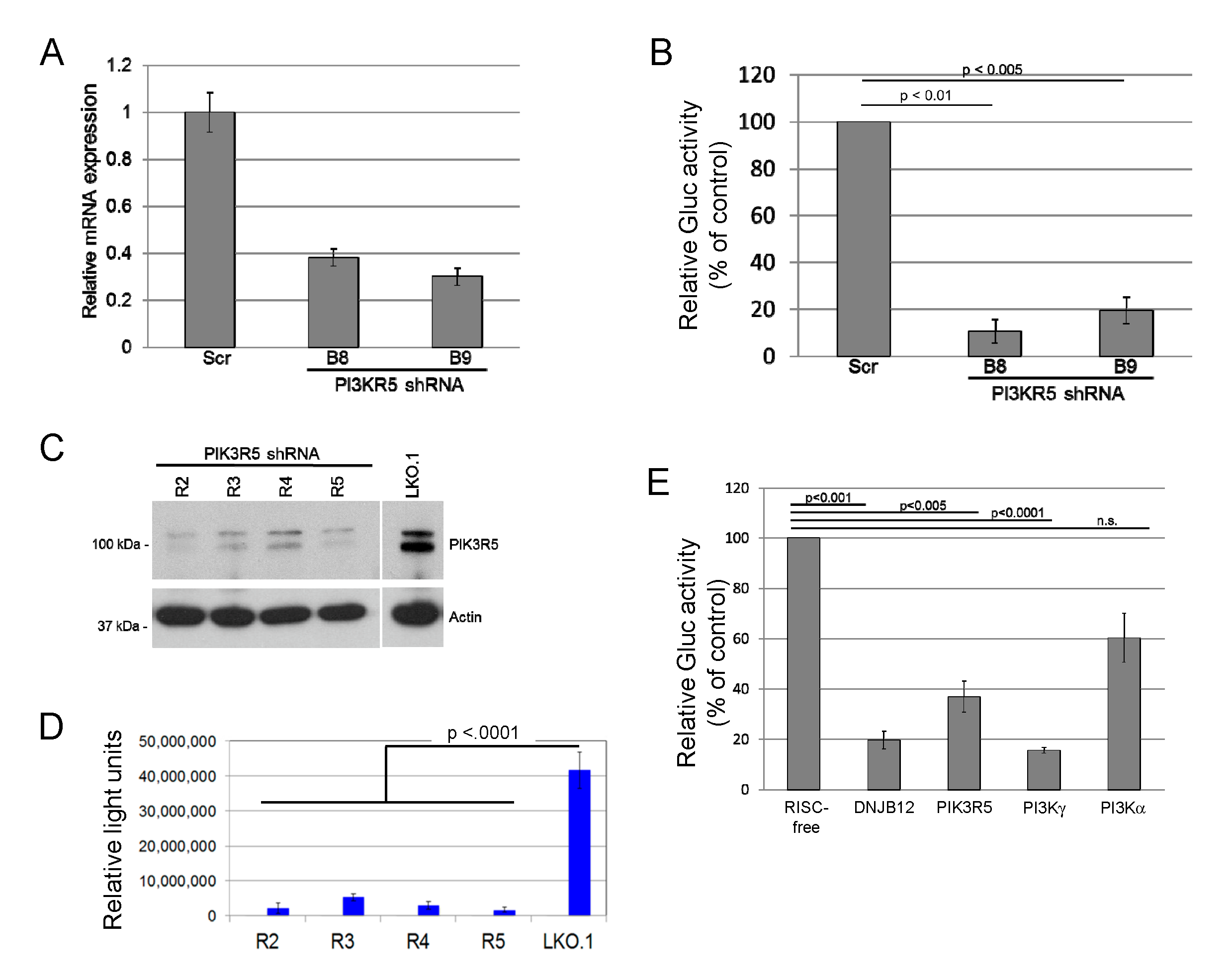
3.1. PIK3R5 and PI3Kγ Knockdown Inhibits JCV Infection
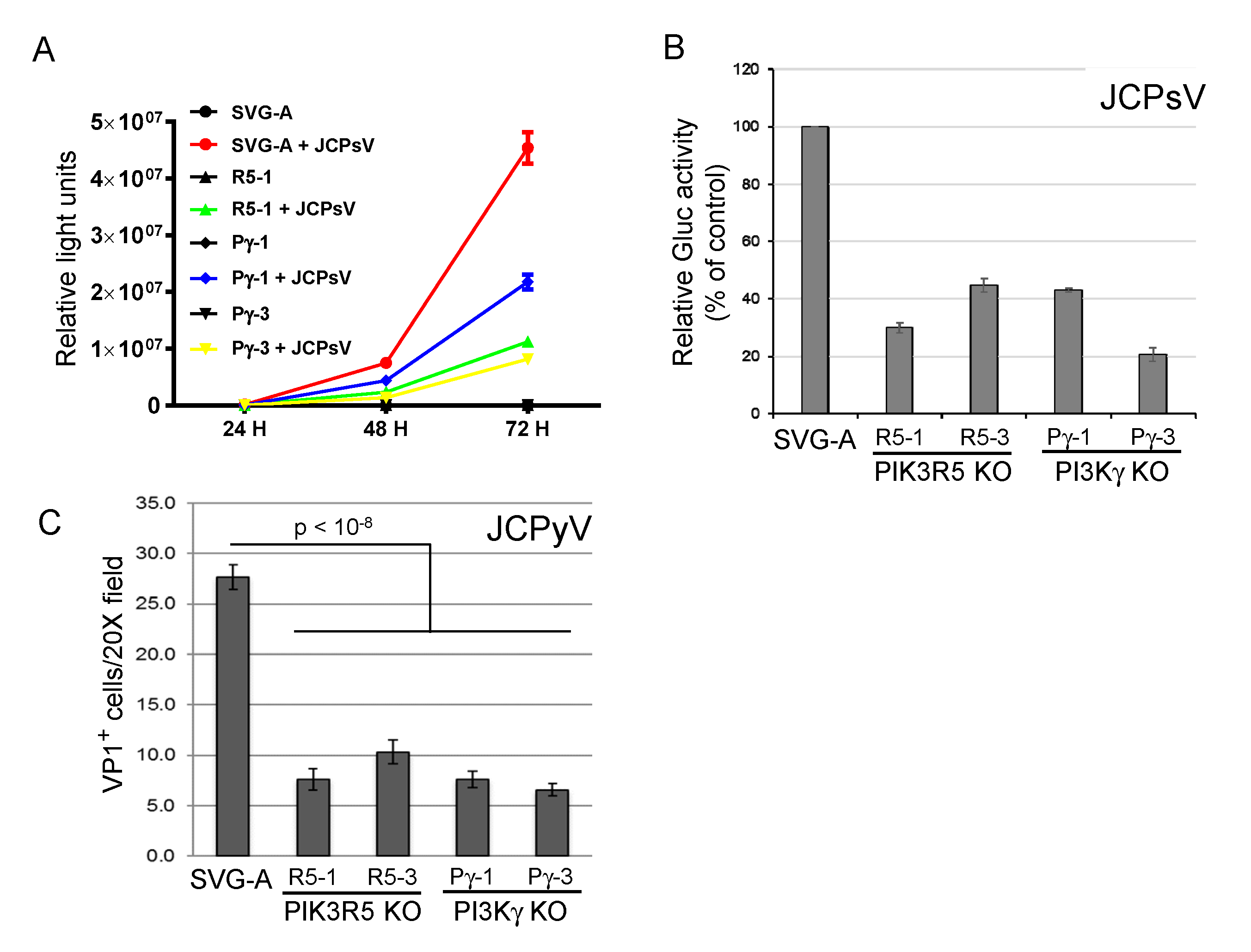
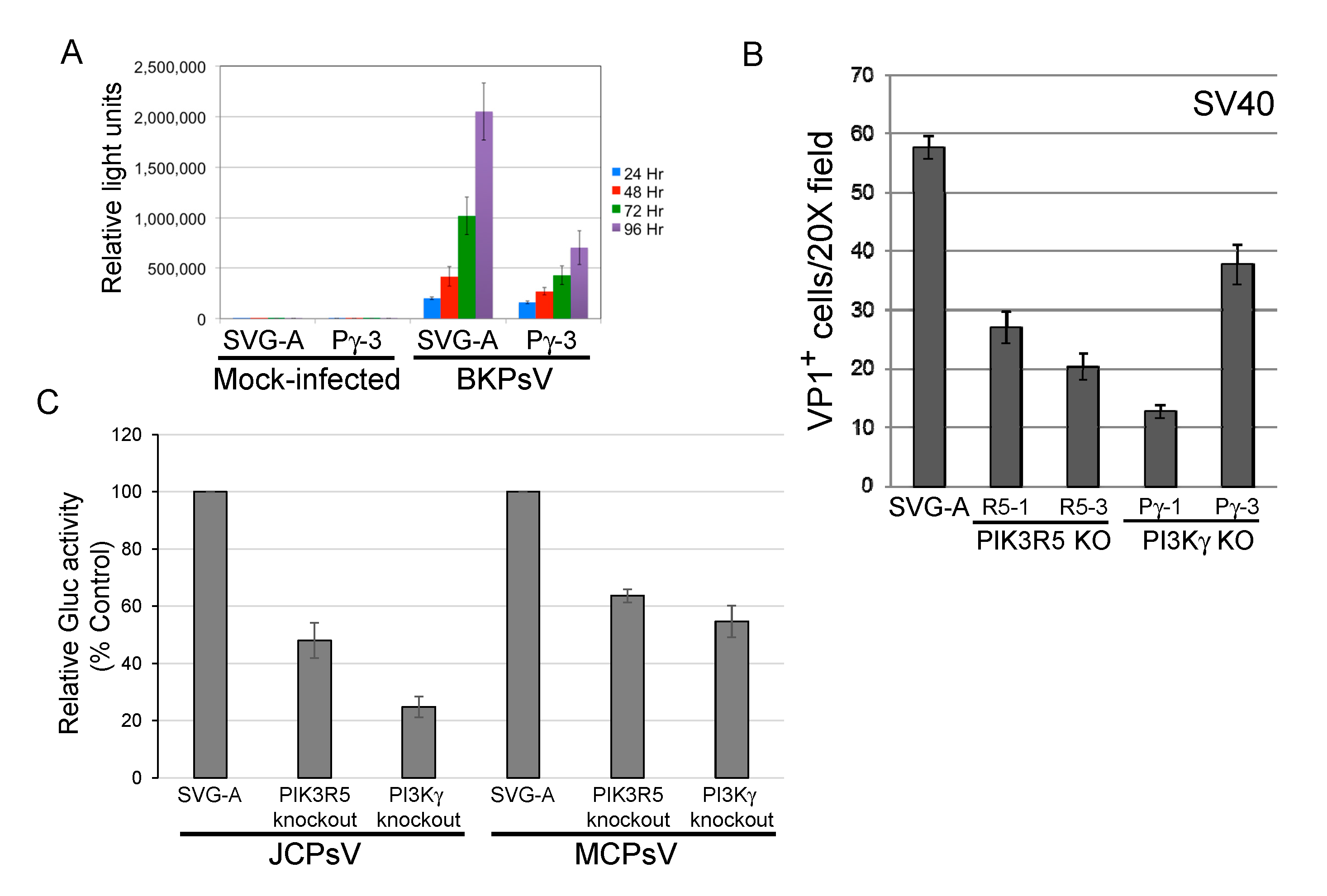
3.2. PIK3R5 and PI3Kγ Knockout Inhibits Infection by JCPsV
3.3. PI3Kγ Expression Rescues JCPyV Infection in Knock-Out Cells
3.4. PI3Kγ Facilitates Infection by other Polyomaviruses
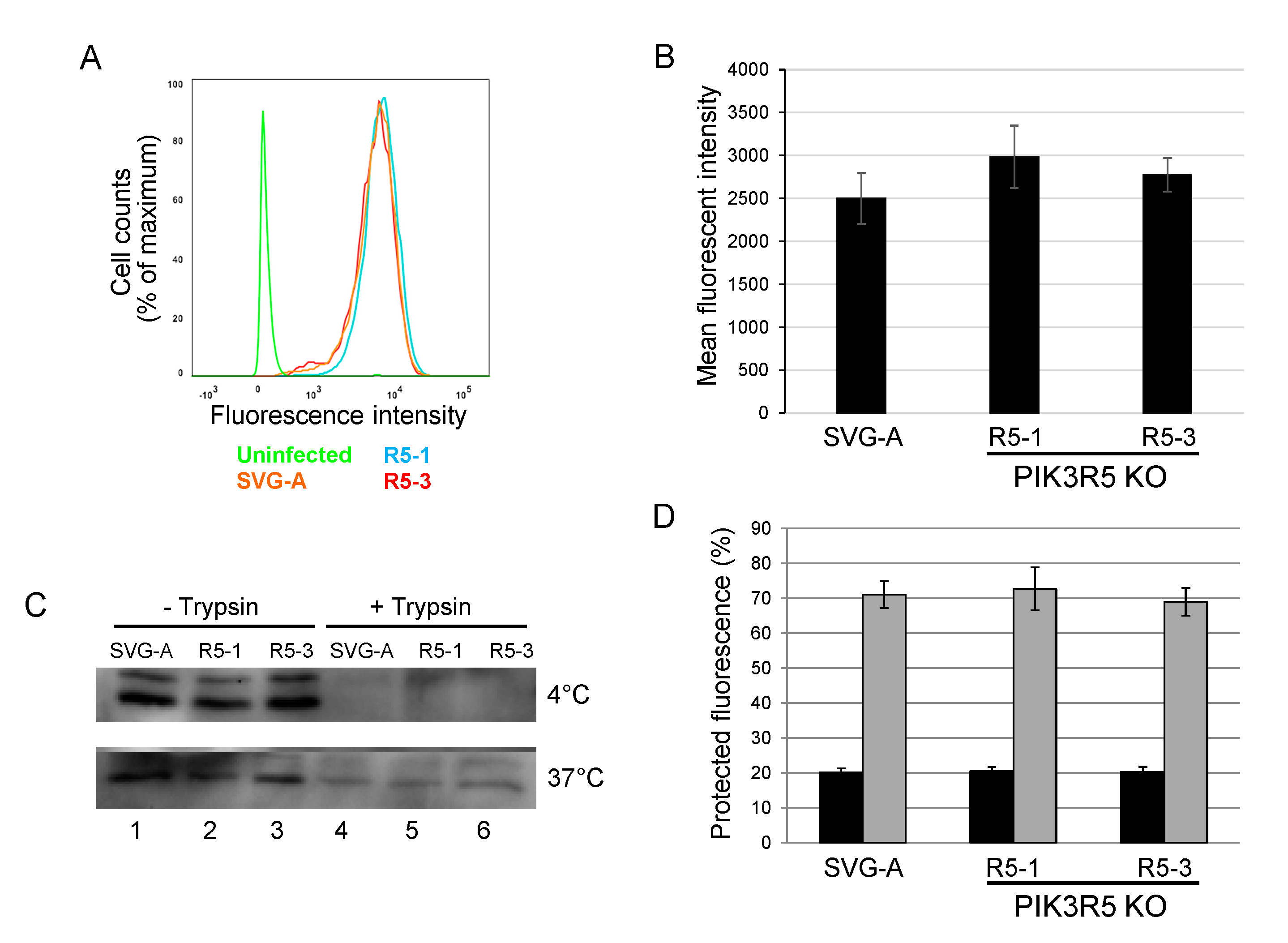
3.5. JCPyV Binding and Internalization Is not Inhibited in PIK3R5 Knockout SVG-A Cells
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Assetta, B.; Atwood, W.J. The biology of JC polyomavirus. Biol. Chem. 2017, 398, 839–855. [Google Scholar] [CrossRef]
- Haley, S.A.; Atwood, W.J. Progressive multifocal Leukoencephalopathy: Endemic viruses and lethal brain disease. Annu. Rev. Virol. 2017, 4, 349–367. [Google Scholar] [CrossRef] [PubMed]
- Cinque, P.; Koralnik, I.J.; Gerevini, S.; Miro, J.M.; Price, R.W. Progressive multifocal leukoencephalopathy in HIV-1 infection. Lancet Infect. Dis. 2009, 9, 625–636. [Google Scholar] [CrossRef]
- Ferenczy, M.W.; Marshall, L.J.; Nelson, C.D.S.; Atwood, W.J.; Nath, A.; Khalili, K.; Major, E.O. Molecular biology, epidemiology, and pathogenesis of progressive multifocal Leukoencephalopathy, the JC virus-induced demyelinating disease of the human brain. Clin. Microbiol. Rev. 2012, 25, 471–506. [Google Scholar] [CrossRef] [PubMed]
- Frisque, R.J.; Bream, G.L.; Cannella, M.T. Human polyomavirus JC virus genome. J. Virol. 1984, 51, 458–469. [Google Scholar] [CrossRef] [PubMed]
- Major, E.O. Reemergence of PML in natalizumab-treated patients--new cases, same concerns. N. Engl. J. Med. 2009, 361, 1041–1043. [Google Scholar] [CrossRef]
- Adelman, B.; Sandrock, A.; Panzara, M.A. Natalizumab and progressive multifocal Leukoencephalopathy. N. Engl. J. Med. 2005, 353, 432–433. [Google Scholar] [CrossRef]
- Berger, J.R.; Fox, R.J. Reassessing the risk of natalizumab-associated PML. J. NeuroVirology 2016, 22, 533–535. [Google Scholar] [CrossRef]
- Major, E.O.; Nath, A. A link between long-term natalizumab dosing in MS and PML: Putting the puzzle together. Neurol. Neuroimmunol. Neuroinflammation 2016, 3, e235. [Google Scholar] [CrossRef]
- Patera, A.C.; on behalf of the PML Consortium; Butler, S.L.; Cinque, P.; Clifford, D.B.; Elston, R.; Garcea, R.L.; Major, E.O.; Pavlovic, D.; Peterson, I.S.; et al. 2nd International Conference on Progressive Multifocal Leukoencephalopathy (PML) 2015: JCV virology, progressive multifocal leukoencephalopathy pathogenesis, diagnosis and risk stratification, and new approaches to prevention and treatment. J. NeuroVirology 2015, 21, 702–705. [Google Scholar] [CrossRef]
- Williamson, E.M.L.; Berger, J.R. Central nervous system infections with Immunomodulatory therapies. Contin. Lifelong Learn. Neurol. 2015, 21, 1577–1598. [Google Scholar] [CrossRef]
- Williamson, E.M.; Berger, J.R. Infection risk in patients on multiple sclerosis therapeutics. CNS Drugs 2015, 29, 229–244. [Google Scholar] [CrossRef]
- Arthur, R.R.; Shah, K.V.; Baust, S.J.; Santos, G.W.; Saral, R. Association of BK Viruria with hemorrhagic cystitis in recipients of bone marrow transplants. N. Engl. J. Med. 1986, 315, 230–234. [Google Scholar] [CrossRef]
- Gardner, S.; Field, A.; Coleman, D.; Hulme, B. New human Papovavirus (B.K.) isolated from urine after renal transplantation. Lancet 1971, 297, 1253–1257. [Google Scholar] [CrossRef]
- Drachenberg, C.B.; Papadimitriou, J.C.; Ramos, E. Histologic versus molecular diagnosis of BK polyomavirus-associated nephropathy: A shifting paradigm? Clin. J. Am. Soc. Nephrol. 2006, 1, 374–379. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, H.H. Polyomavirus BK nephropathy: A (Re-)emerging complication in renal transplantation. Arab. Archaeol. Epigr. 2002, 2, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Nickeleit, V.; Hirsch, H.H.; Zeiler, M.; Gudat, F.; Prince, O.; Thiel, G.; Mihatsch, M.J. BK-virus nephropathy in renal transplants—Tubular necrosis, MHC-class II expression and rejection in a puzzling game. Nephrol. Dial. Transplant. 2000, 15, 324–332. [Google Scholar] [CrossRef]
- Kantarci, G.; Eren, Z.; Demirag, A.; Dogan, I.; Çakalaǧaoǧlu, F.; Yilmaz, G.; Çakalaǧaoǧlu, F. JC virus-associated nephropathy in a renal transplant recipient and comparative analysis of previous cases. Transpl. Infect. Dis. 2010, 13, 89–92. [Google Scholar] [CrossRef]
- Kazory, A.; Ducloux, D.; Chalopin, J.-M.; Angonin, R.; Fontanière, B.; Moret, H. The first case of JC virus allograft nephropathy. Transplantation 2003, 76, 1653–1655. [Google Scholar] [CrossRef]
- Lautenschlager, I.; Jahnukainen, T.; Kardas, P.; Lohi, J.; Auvinen, E.; Mannonen, L.; Dumoulin, A.; Hirsch, H.H.; Jalanko, H. A case of primary JC Polyomavirus infection-associated nephropathy. Arab. Archaeol. Epigr. 2014, 14, 2887–2892. [Google Scholar] [CrossRef]
- Wen, M.-C.; Wang, C.-L.; Wang, M.; Cheng, C.-H.; Wu, M.-J.; Chen, C.-H.; Shu, K.-H.; Chang, D. Association of JC virus with tubulointerstitial nephritis in a renal allograft recipient. J. Med. Virol. 2004, 72, 675–678. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.S.; Bohl, D.L.; Storch, G.A.; Ryschkewitsch, C.; Gaudreault-Keener, M.; Major, E.O.; Randhawa, P.; Hardinger, K.L.; Brennan, D.C. Inhibitory interactions between BK and JC virus among kidney transplant recipients. J. Am. Soc. Nephrol. 2011, 22, 825–831. [Google Scholar] [CrossRef]
- Drachenberg, C.B.; Hirsch, H.H.; Papadimitriou, J.C.; Gosert, R.; Wali, R.K.; Munivenkatappa, R.; Nogueira, J.; Cangro, C.B.; Haririan, A.; Mendley, S.; et al. Polyomavirus BK versus JC replication and nephropathy in renal transplant recipients: A prospective evaluation. Transplantation 2007, 84, 323–330. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.; Shuda, M.; Chang, Y.; Moore, P.S. Clonal Integration of a Polyomavirus in human merkel cell carcinoma. Science 2008, 319, 1096–1100. [Google Scholar] [CrossRef] [PubMed]
- Schowalter, R.M.; Pastrana, D.V.; Pumphrey, K.A.; Moyer, A.L.; Buck, C.B. Merkel cell Polyomavirus and two previously unknown Polyomaviruses are chronically shed from human skin. Cell Host Microbe 2010, 7, 509–515. [Google Scholar] [CrossRef] [PubMed]
- Shuda, M.; Feng, H.; Kwun, H.J.; Rosen, S.T.; Gjoerup, O.; Moore, P.S.; Chang, Y. T antigen mutations are a human tumor-specific signature for Merkel cell polyomavirus. Proc. Natl. Acad. Sci. USA 2008, 105, 16272–16277. [Google Scholar] [CrossRef] [PubMed]
- Berrios, C.; Padi, M.; Keibler, M.A.; Park, D.E.; Molla, V.; Cheng, J.; Lee, S.M.; Stephanopoulos, G.; Quackenbush, J.; DeCaprio, J.A. Merkel cell Polyomavirus small T antigen promotes pro-glycolytic metabolic perturbations required for transformation. PLoS Pathog. 2016, 12, e1006020. [Google Scholar] [CrossRef]
- Cheng, J.; Rozenblatt-Rosen, O.; Paulson, K.G.; Nghiem, P.; DeCaprio, J.A. Merkel cell Polyomavirus large T antigen has growth-promoting and inhibitory activities. J. Virol. 2013, 87, 6118–6126. [Google Scholar] [CrossRef]
- Park, D.E.; Cheng, J.; Berrios, C.; Montero, J.; Cortés-Cros, M.; Ferretti, S.; Arora, R.; Tillgren, M.L.; Gokhale, P.C.; DeCaprio, J.A. Dual inhibition of MDM2 and MDM4 in virus-positive Merkel cell carcinoma enhances the p53 response. Proc. Natl. Acad. Sci. USA 2018, 116, 1027–1032. [Google Scholar] [CrossRef]
- An, P.; Robles, M.T.S.; Duray, A.M.; Cantalupo, P.G.; Pipas, J.M. Human polyomavirus BKV infection of endothelial cells results in interferon pathway induction and persistence. PLoS Pathog. 2019, 15, e1007505. [Google Scholar] [CrossRef]
- Assetta, B.; De Cecco, M.; O’Hara, B.; Atwood, W.J. JC Polyomavirus infection of primary human renal epithelial cells is controlled by a type I IFN-induced response. mBio 2016, 7, e00903-16. [Google Scholar] [CrossRef]
- Stehle, T.; Gamblin, S.J.; Yan, Y.; Harrison, S.C. The structure of simian virus 40 refined at 3.1 å resolution. Structure 1996, 4, 165–182. [Google Scholar] [CrossRef]
- Schelhaas, M.; Malmström, J.; Pelkmans, L.; Haugstetter, J.; Ellgaard, L.; Grünewald, K.; Helenius, A. Simian virus 40 depends on ER protein folding and quality control factors for entry into host cells. Cell 2007, 131, 516–529. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-J.; Liu, X.; Tsai, B. SV40 hijacks cellular transport, membrane penetration, and disassembly machineries to promote infection. Viruses 2019, 11, 917. [Google Scholar] [CrossRef] [PubMed]
- Geiger, R.; Andritschke, D.; Friebe, S.; Herzog, F.; Luisoni, S.; Heger, T.; Helenius, A. BAP31 and BiP are essential for dislocation of SV40 from the endoplasmic reticulum to the cytosol. Nat. Cell Biol. 2011, 13, 1305–1314. [Google Scholar] [CrossRef]
- Brucher, P.; Piéchaud, T.; Brunier, G.; Ferrière, J.M.; Deminière, C.; Grenier, N.; Le Guillou, M. Oncocytic tumors of the kidney. 11 cases (1978–1987). Ann. d’Urologie 1989, 23, 459–462. [Google Scholar]
- Goodwin, E.C.; Lipovsky, A.; Inoue, T.; Magaldi, T.G.; Edwards, A.P.B.; Van Goor, K.E.Y.; Paton, A.W.; Paton, J.C.; Atwood, W.J.; Tsai, B.; et al. BiP and multiple DNAJ molecular chaperones in the endoplasmic reticulum are required for efficient simian virus 40 infection. mBio 2011, 2, e00101-11. [Google Scholar] [CrossRef]
- Chen, K.; Healy, M.D.; Collins, B.M. Towards a molecular understanding of endosomal trafficking by Retromer and Retriever. Traffic 2019, 20, 465–478. [Google Scholar] [CrossRef]
- Nelson, C.D.S.; Carney, D.W.; Derdowski, A.; Lipovsky, A.; Gee, G.V.; O’Hara, B.; Williard, P.; DiMaio, D.; Sello, J.K.; Atwood, W.J. A retrograde trafficking inhibitor of ricin and shiga-like toxins inhibits infection of cells by human and monkey Polyomaviruses. mBio 2013, 4, e00729-13. [Google Scholar] [CrossRef]
- Dushane, J.K.; Maginnis, M.S. Human DNA virus exploitation of the MAPK-ERK cascade. Int. J. Mol. Sci. 2019, 20, 3427. [Google Scholar] [CrossRef]
- Dushane, J.K.; Mayberry, C.L.; Wilczek, M.P.; Nichols, S.L.; Maginnis, M.S. JCPyV-Induced MAPK signaling activates transcription factors during infection. Int. J. Mol. Sci. 2019, 20, 4779. [Google Scholar] [CrossRef]
- Dushane, J.K.; Wilczek, M.P.; Mayberry, C.L.; Maginnis, M.S. ERK is a critical regulator of JC Polyomavirus infection. J. Virol. 2018, 92, 01529-17. [Google Scholar] [CrossRef] [PubMed]
- Maginnis, M.S. Virus–receptor interactions: The key to cellular invasion. J. Mol. Biol. 2018, 430, 2590–2611. [Google Scholar] [CrossRef] [PubMed]
- Zullo, J.; Stiles, C.D.; Garcea, R.L. Regulation of c-myc and c-fos mRNA levels by polyomavirus: Distinct roles for the capsid protein VP1 and the viral early proteins. Proc. Natl. Acad. Sci. USA 1987, 84, 1210–1214. [Google Scholar] [CrossRef] [PubMed]
- Vacante, D.A.; Traub, R.; Major, E.O. Extension of JC virus host range to monkey cells by insertion of a simian virus 40 enhancer into the JC virus regulatory region. Virology 1989, 170, 353–361. [Google Scholar] [CrossRef]
- Liu, C.K.; Atwood, W.J. Propagation and assay of the JC virus. Methods Mol. Boil. (Clifton N.J.) 2001, 165, 9–17. [Google Scholar]
- Luo, Y.; Motamedi, N.; Magaldi, T.G.; Gee, G.V.; Atwood, W.J.; DiMaio, D. Interaction between Simian virus 40 major capsid protein VP1 and cell surface Ganglioside GM1 triggers vacuole formation. mBio 2016, 7. [Google Scholar] [CrossRef]
- Gee, G.V.; O’Hara, B.A.; Derdowski, A.; Atwood, W.J. Pseudovirus mimics cell entry and trafficking of the human polyomavirus JCPyV. Virus Res. 2013, 178, 281–286. [Google Scholar] [CrossRef]
- Pastrana, D.V.; Tolstov, Y.L.; Becker, J.C.; Moore, P.S.; Chang, Y.; Buck, C.B. Quantitation of human Seroresponsiveness to merkel cell Polyomavirus. PLoS Pathog. 2009, 5, e1000578. [Google Scholar] [CrossRef]
- Sanjana, N.E.; Shalem, O.; Zhang, F. Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 2014, 11, 783–784. [Google Scholar] [CrossRef]
- Stephens, L.R.; Eguinoa, A.; Erdjument-Bromage, H.; Lui, M.; Cooke, F.; Coadwell, J.; Smrcka, A.S.; Thelen, M.; Cadwallader, K.; Tempst, P.; et al. The Gβγ sensitivity of a PI3K is dependent upon a tightly associated adaptor, p101. Cell 1997, 89, 105–114. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SVGA | SVGA + PI3K | PI3K KO | PI3K KO + PI3K | |||||
|---|---|---|---|---|---|---|---|---|
| High | Low | High/Low | High | Low | High/Low | |||
| Exp. 1a * | 4.3 | 10.9 | 2.4 | 4.54 | 1.9 | 2.3 | 1.2 | 1.92 |
| Exp. 1b | 4.4 | 11.9 | 2.4 | 4.96 | 1.9 | 2.3 | 1.1 | 2.09 |
| Exp. 2a | 2.3 | 10.8 | 2.5 | 4.32 | 1.2 | 2.2 | 0.9 | 2.44 |
| Exp. 2b | 2.0 | 11.3 | 2.4 | 4.71 | 1.1 | 1.4 | 0.9 | 1.56 |
| Exp. 3a | 7.9 | 15.9 | 6.6 | 2.41 | 2.1 | 4.1 | 2.1 | 1.95 |
| Exp. 3b | 8.2 | 15.4 | 7.1 | 2.17 | 2.2 | 3.6 | 2.2 | 1.64 |
| Exp. 4a | 3.9 | 12.2 | 3.1 | 3.94 | 2.3 | 3.9 | 1.4 | 2.79 |
| Exp. 4b | 4.3 | 13.0 | 3.1 | 4.12 | 1.9 | 3.0 | 1.6 | 1.88 |
| Average | 4.7 ± 2.3 | 3.9 ± 1.04 | 1.8 ± 0.44 | 2.03 ± 0.41 | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Clark, P.; Gee, G.V.; Albright, B.S.; Assetta, B.; Han, Y.; Atwood, W.J.; DiMaio, D. Phosphoinositide 3′-Kinase γ Facilitates Polyomavirus Infection. Viruses 2020, 12, 1190. https://doi.org/10.3390/v12101190
Clark P, Gee GV, Albright BS, Assetta B, Han Y, Atwood WJ, DiMaio D. Phosphoinositide 3′-Kinase γ Facilitates Polyomavirus Infection. Viruses. 2020; 12(10):1190. https://doi.org/10.3390/v12101190
Chicago/Turabian StyleClark, Paul, Gretchen V. Gee, Brandon S. Albright, Benedetta Assetta, Ying Han, Walter J. Atwood, and Daniel DiMaio. 2020. "Phosphoinositide 3′-Kinase γ Facilitates Polyomavirus Infection" Viruses 12, no. 10: 1190. https://doi.org/10.3390/v12101190
APA StyleClark, P., Gee, G. V., Albright, B. S., Assetta, B., Han, Y., Atwood, W. J., & DiMaio, D. (2020). Phosphoinositide 3′-Kinase γ Facilitates Polyomavirus Infection. Viruses, 12(10), 1190. https://doi.org/10.3390/v12101190