Taking the Scenic Route: Polyomaviruses Utilize Multiple Pathways to Reach the Same Destination

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
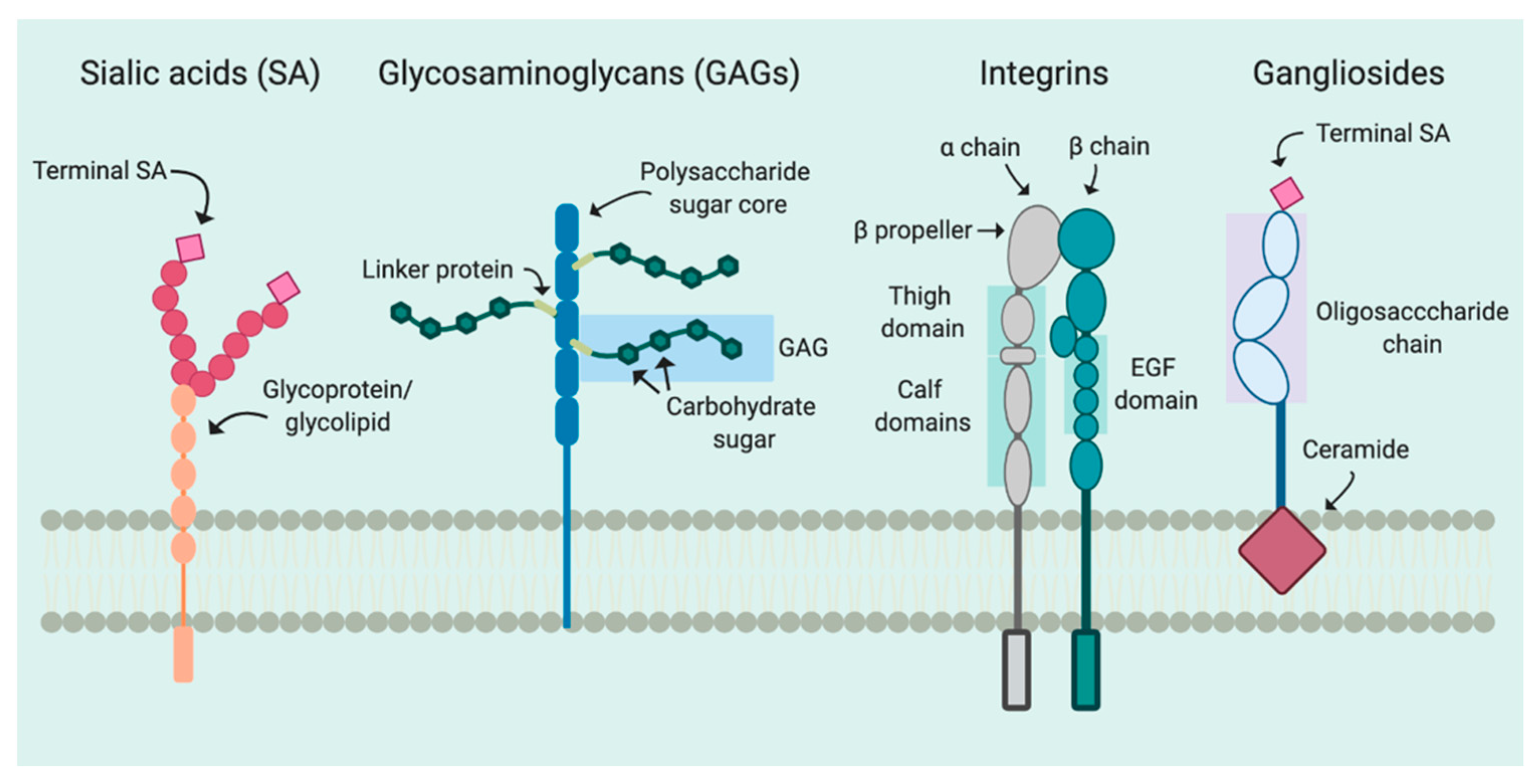
2. Attachment
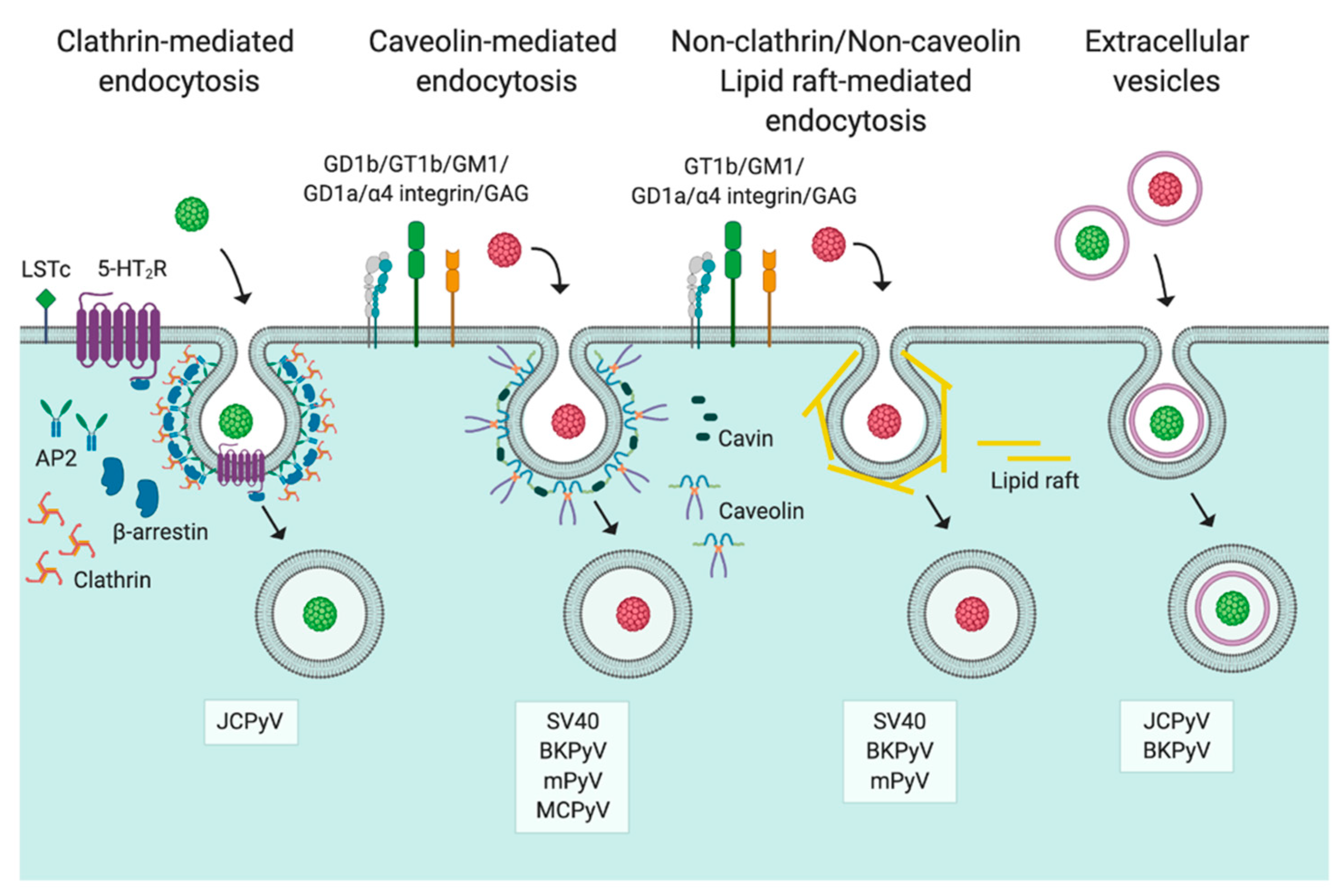
3. Entry
3.1. Caveolin-/Lipid Raft-Mediated Endocytosis
3.2. Clathrin-Mediated Endocytosis
3.3. Non-Clathrin/Non-Caveolin Endocytosis
3.4. Extracellular Vesicles
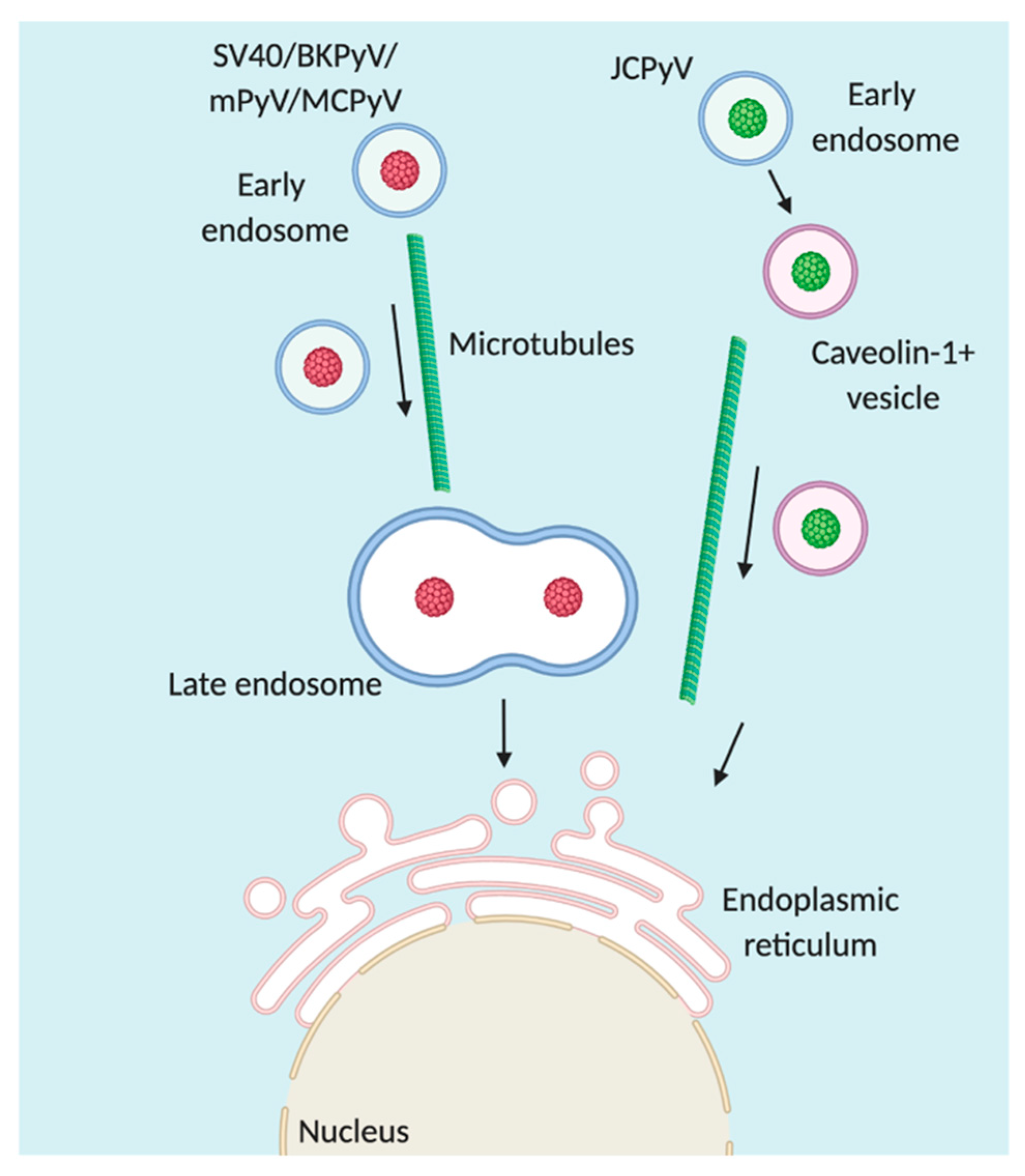
4. Trafficking
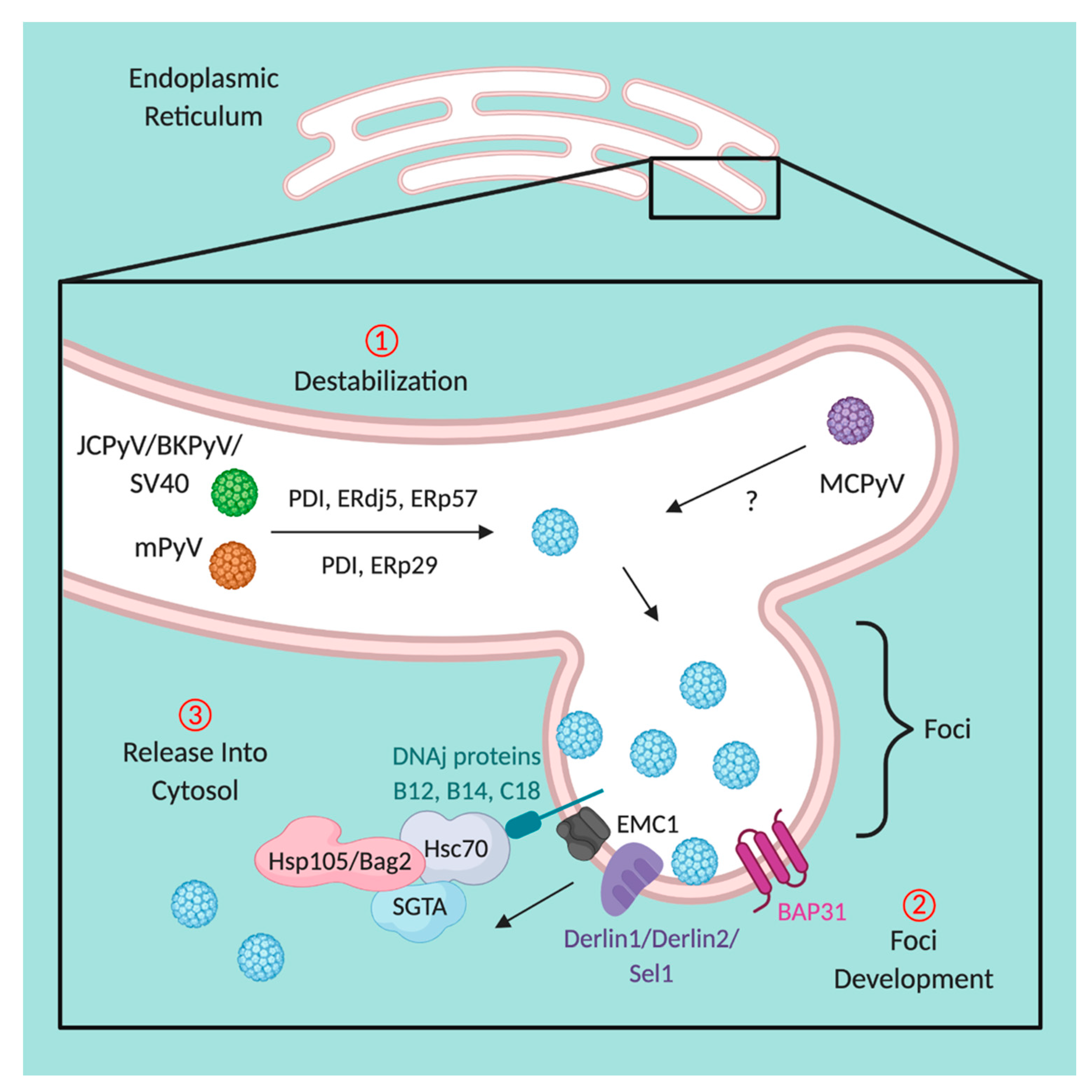
5. Disassembly within the Endoplasmic Reticulum
6. Nuclear Transport of Polyomaviruses
7. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Marsh, M.; Helenius, A. Virus Entry: Open Sesame. Cell 2006, 124, 729–740. [Google Scholar] [CrossRef]
- Mercer, J.; Schelhaas, M.; Helenius, A. Virus Entry by Endocytosis. Annu. Rev. Biochem. 2010, 79, 803–833. [Google Scholar] [CrossRef]
- Moens, U.; Calvignac-Spencer, S.; Lauber, C.; Ramqvist, T.; Feltkamp, M.C.W.; Daugherty, M.D.; Verschoor, E.J.; Ehlers, B. ICTV Report Consortium ICTV Virus Taxonomy Profile: Polyomaviridae. J. Gen. Virol. 2017, 98, 1159–1160. [Google Scholar] [CrossRef] [PubMed]
- Liddington, R.C.; Yan, Y.; Moulai, J.; Sahli, R.; Benjamin, T.L.; Harrison, S.C. Structure of simian virus 40 at 3.8-Å resolution. Nat. Cell Biol. 1991, 354, 278–284. [Google Scholar] [CrossRef] [PubMed]
- Maginnis, M.S. Virus–Receptor Interactions: The Key to Cellular Invasion. J. Mol. Biol. 2018, 430, 2590–2611. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, H.H.; Kardas, P.; Kranz, D.; Leboeuf, C. The human JC polyomavirus (JCPyV): Virological background and clinical implications. APMIS 2013, 121, 685–727. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.S.; Stehle, T.; Harrison, S.C. Interaction of polyomavirus internal protein VP2 with the major capsid protein VP1 and implications for participation of VP2 in viral entry. EMBO J. 1998, 17, 3233–3240. [Google Scholar] [CrossRef] [PubMed]
- Tsai, B.; Qian, M. Cellular Entry of Polyomaviruses. Curr. Top. Microbiol. Immunol. 2010, 343, 177–194. [Google Scholar] [CrossRef] [PubMed]
- Van Der Meijden, E.; Janssens, R.W.; Lauber, C.; Bavinck, J.N.B.; Gorbalenya, A.E.; Feltkamp, M.C. Discovery of a New Human Polyomavirus Associated with Trichodysplasia Spinulosa in an Immunocompromized Patient. PLoS Pathog. 2010, 6, e1001024. [Google Scholar] [CrossRef]
- Schrama, D.; Buck, C.B.; Houben, R.; Becker, J.C. No Evidence for Association of HPyV6 or HPyV7 with Different Skin Cancers. J. Investig. Dermatol. 2012, 132, 239–241. [Google Scholar] [CrossRef]
- Bergallo, M.; Daprà, V.; Fava, P.; Ponti, R.; Calvi, C.; Montanari, P.; Novelli, M.; Quaglino, P.; Galliano, I.; Fierro, M.T. DNA from Human Polyomaviruses, MWPyV, HPyV6, HPyV7, HPyV9 and HPyV12 in Cutaneous T-cell Lymphomas. Anticancer Res. 2018, 38, 4111–4114. [Google Scholar] [CrossRef] [PubMed]
- Mishra, N.; Pereira, M.; Rhodes, R.H.; An, P.; Pipas, J.M.; Jain, K.; Kapoor, A.; Briese, T.; Faust, P.L.; Lipkin, W.I. Identification of a Novel Polyomavirus in a Pancreatic Transplant Recipient With Retinal Blindness and Vasculitic Myopathy. J. Infect. Dis. 2014, 210, 1595–1599. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, H.H.; Randhawa, P. The AST Infectious Diseases Community of Practice BK Polyomavirus in Solid Organ Transplantation. Arab. Archaeol. Epigr. 2013, 13, 179–188. [Google Scholar] [CrossRef]
- Feng, H.; Shuda, M.; Chang, Y.; Moore, P.S. Clonal Integration of a Polyomavirus in Human Merkel Cell Carcinoma. Science 2008, 319, 1096–1100. [Google Scholar] [CrossRef]
- Kartenbeck, J.; Stukenbrok, H.; Helenius, A. Endocytosis of simian virus 40 into the endoplasmic reticulum. J. Cell Biol. 1989, 109 Pt 1, 2721–2729. [Google Scholar] [CrossRef]
- Pelkmans, L.; Kartenbeck, J.; Helenius, A. Caveolar endocytosis of simian virus 40 reveals a new two-step vesicular-transport pathway to the ER. Nat. Cell Biol. 2001, 3, 473. [Google Scholar] [CrossRef]
- Richards, A.A.; Stang, E.; Pepperkok, R.; Parton, R.G. Inhibitors of COP-mediated Transport and Cholera Toxin Action Inhibit Simian Virus 40 Infection. Mol. Biol. Cell 2002, 13, 1750–1764. [Google Scholar] [CrossRef]
- Bennett, S.M.; Jiang, M.; Imperiale, M.J. Role of Cell-Type-Specific Endoplasmic Reticulum-Associated Degradation in Polyomavirus Trafficking. J. Virol. 2013, 87, 8843–8852. [Google Scholar] [CrossRef]
- Inoue, T.; Tsai, B. A Large and Intact Viral Particle Penetrates the Endoplasmic Reticulum Membrane to Reach the Cytosol. PLoS Pathog. 2011, 7, e1002037. [Google Scholar] [CrossRef]
- Nelson, C.D.; Derdowski, A.; Maginnis, M.S.; O’Hara, B.A.; Atwood, W.J. The VP1 subunit of JC polyomavirus recapitulates early events in viral trafficking and is a novel tool to study polyomavirus entry. Virology 2012, 428, 30–40. [Google Scholar] [CrossRef]
- Becker, M.; Dominguez, M.; Greune, L.; Soria-Martinez, L.; Pfleiderer, M.M.; Schowalter, R.; Buck, C.B.; Blaum, B.S.; Schmidt, M.A.; Schelhaas, M. Infectious Entry of Merkel Cell Polyomavirus. J. Virol. 2019, 93. [Google Scholar] [CrossRef]
- Sobhy, H. A comparative review of viral entry and attachment during large and giant dsDNA virus infections. Arch. Virol. 2017, 162, 3567–3585. [Google Scholar] [CrossRef]
- Haywood, A.M. Virus receptors: Binding, adhesion strengthening, and changes in viral structure. J. Virol. 1994, 68, 1–5. [Google Scholar] [CrossRef]
- Brito-Arias, M. Glycoconjugates. In Synthesis and Characterization of Glycosides; Springer: Boston, MA, USA, 2007. [Google Scholar]
- Varki, A. Multiple changes in sialic acid biology during human evolution. Glycoconj. J. 2008, 26, 231–245. [Google Scholar] [CrossRef]
- Bush, C.A.; Martín-Pastor, M.; Imberty, A. Structure and Conformation of Complex Carbohydrates of Glycoproteins, Glycolipids, and Bacterial Polysaccharides. Annu. Rev. Biophys. Biomol. Struct. 1999, 28, 269–293. [Google Scholar] [CrossRef]
- Yu, R.K.; Tsai, Y.-T.; Ariga, T.; Yanagisawa, M. Structures, Biosynthesis, and Functions of Gangliosides—An Overview. J. Oleo Sci. 2011, 60, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Maccioni, H.J.F.; Quiroga, R.; Ferrari, M.L. Cellular and molecular biology of glycosphingolipid glycosylation. J. Neurochem. 2011, 117. [Google Scholar] [CrossRef] [PubMed]
- Varki, A.; Cummings, R.D.; Esko, J.D.; Freeze, H.H.; Stanley, P.; Hart, G.W.; Aebi, M.; Darvill, A.G.; Kinoshita, T.; Packer, N.H. Sialic Acids. In Essentials of Glycobiology; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2009. [Google Scholar]
- Altheide, T.K.; Hayakawa, T.; Mikkelsen, T.S.; Diaz, S.; Varki, N.; Varki, A. System-wide Genomic and Biochemical Comparisons of Sialic Acid Biology Among Primates and Rodents: Evidence for two modes of rapid evolution. J. Biol. Chem. 2006, 281, 25689–25702. [Google Scholar] [CrossRef] [PubMed]
- Schauer, R. Sialic acids as regulators of molecular and cellular interactions. Curr. Opin. Struct. Biol. 2009, 19, 507–514. [Google Scholar] [CrossRef]
- Höök, M.; Kjellén, L.; Johansson, S.; Robinson, J. Cell-Surface Glycosaminoglycans. Annu. Rev. Biochem. 1984, 53, 847–869. [Google Scholar] [CrossRef] [PubMed]
- Triantafilou, K.; Triantafilou, M. Mechanisms of Integrin-Mediated Virus Attachment and Internalization Process. Crit. Rev. Immunol. 2001, 21, 12. [Google Scholar] [CrossRef]
- Schneider-Schaulies, J. Cellular receptors for viruses: Links to tropism and pathogenesis. J. Gen. Virol. 2000, 81 Pt 6, 1413–1429. [Google Scholar] [CrossRef]
- Ströh, L.J.; Stehle, T. Glycan Engagement by Viruses: Receptor Switches and Specificity. Annu. Rev. Virol. 2014, 1, 285–306. [Google Scholar] [CrossRef]
- O’Hara, S.D.; Stehle, T.; Garcea, R.L. Glycan receptors of the Polyomaviridae: Structure, function, and pathogenesis. Curr. Opin. Virol. 2014, 7, 73–78. [Google Scholar] [CrossRef]
- Fried, H.; Cahan, L.D.; Paulson, J.C. Polyoma virus recognizes specific sialyloligosaccharide receptors on host cells. Virology 1981, 109, 188–192. [Google Scholar] [CrossRef]
- Cahan, L.D.; Singh, R.; Paulson, J.C. Sialyloligosaccharide receptors of binding variants of polyoma virus. Virology 1983, 130, 281–289. [Google Scholar] [CrossRef]
- Stehle, T.; Harrison, S.C. High-resolution structure of a polyomavirus VP1-oligosaccharide complex: Implications for assembly and receptor binding. EMBO J. 1997, 16, 5139–5148. [Google Scholar] [CrossRef] [PubMed]
- Neu, U.; Hengel, H.; Blaum, B.S.; Schowalter, R.M.; Macejak, D.; Gilbert, M.; Wakarchuk, W.W.; Imamura, A.; Ando, H.; Kiso, M.; et al. Structures of Merkel Cell Polyomavirus VP1 Complexes Define a Sialic Acid Binding Site Required for Infection. PLoS Pathog. 2012, 8, e1002738. [Google Scholar] [CrossRef]
- Liu, C.K.; Hope, A.P.; Atwood, W.J. The human polyomavirus, JCV, does not share receptor specificity with SV40 on human glial cells. J. Neurovirol. 1998, 4, 49–58. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Liu, C.K.; Wei, G.; Atwood, W.J. Infection of Glial Cells by the Human Polyomavirus JC Is Mediated by an N-Linked Glycoprotein Containing Terminal α(2-6)-Linked Sialic Acids. J. Virol. 1998, 72, 4643–4649. [Google Scholar] [CrossRef]
- Komagome, R.; Sawa, H.; Suzuki, T.; Suzuki, Y.; Tanaka, S.; Atwood, W.J.; Nagashima, K. Oligosaccharides as Receptors for JC Virus. J. Virol. 2002, 76, 12992–13000. [Google Scholar] [CrossRef]
- Dugan, A.S.; Gasparovic, M.L.; Atwood, W.J. Direct Correlation between Sialic Acid Binding and Infection of Cells by Two Human Polyomaviruses (JC Virus and BK Virus). J. Virol. 2007, 82, 2560–2564. [Google Scholar] [CrossRef] [PubMed]
- Neu, U.; Maginnis, M.S.; Palma, A.S.; Ströh, L.J.; Nelson, C.D.; Feizi, T.; Atwood, W.J.; Stehle, T. Structure-Function Analysis of the Human JC Polyomavirus Establishes the LSTc Pentasaccharide as a Functional Receptor Motif. Cell Host Microbe 2010, 8, 309–319. [Google Scholar] [CrossRef] [PubMed]
- Gorelik, L.; Reid, C.; Testa, M.; Brickelmaier, M.; Bossolasco, S.; Pazzi, A.; Bestetti, A.; Carmillo, P.; Wilson, E.; McAuliffe, M.; et al. Progressive Multifocal Leukoencephalopathy (PML) Development Is Associated With Mutations in JC Virus Capsid Protein VP1 That Change Its Receptor Specificity. J. Infect. Dis. 2011, 204, 103–114. [Google Scholar] [CrossRef]
- Low, J.; Humes, H.D.; Szczypka, M.; Imperiale, M.J. BKV and SV40 infection of human kidney tubular epithelial cells in vitro. Virology 2004, 323, 182–188. [Google Scholar] [CrossRef] [PubMed]
- Low, J.A.; Magnuson, B.; Tsai, B.; Imperiale, M.J. Identification of Gangliosides GD1b and GT1b as Receptors for BK Virus. J. Virol. 2006, 80, 1361–1366. [Google Scholar] [CrossRef]
- Buch, M.H.C.; Liaci, A.M.; O’Hara, S.D.; Garcea, R.L.; Neu, U.; Stehle, T. Structural and Functional Analysis of Murine Polyomavirus Capsid Proteins Establish the Determinants of Ligand Recognition and Pathogenicity. PLoS Pathog. 2015, 11, e1005104. [Google Scholar] [CrossRef]
- Frisque, R.J.; Bream, G.L.; Cannella, M.T. Human polyomavirus JC virus genome. J. Virol. 1984, 51, 458–469. [Google Scholar] [CrossRef]
- Yan, Y.; Stehle, T.; Liddington, R.C.; Zhao, H.; Harrison, S.C. Structure determination of simian virus 40 and murine polyomavirus by a combination of 30-fold and 5-fold electron-density averaging. Structure 1996, 4, 157–164. [Google Scholar] [CrossRef]
- Caruso, M.; Cavaldesi, M.; Gentile, M.; Sthandier, O.; Amati, P.; Garcia, M.-I. Role of sialic acid-containing molecules and the α4β1 integrin receptor in the early steps of polyomavirus infection. J. Gen. Virol. 2003, 84 Pt 11, 2927–2936. [Google Scholar] [CrossRef]
- Bauer, P.H.; Cui, C.; Stehle, T.; Harrison, S.C.; DeCaprio, J.A.; Benjamin, T.L. Discrimination between Sialic Acid-Containing Receptors and Pseudoreceptors Regulates Polyomavirus Spread in the Mouse. J. Virol. 1999, 73, 5826–5832. [Google Scholar] [CrossRef] [PubMed]
- Vogt, D.; Ott, M. Membrane Flotation Assay. Bio-Protocol 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.E.; Lilie, H.; Helenius, A. Ganglioside-dependent cell attachment and endocytosis of murine polyomavirus-like particles. FEBS Lett. 2003, 555, 199–203. [Google Scholar] [CrossRef]
- Tsai, B.; Gilbert, J.M.; Stehle, T.; Lencer, W.; Benjamin, T.L.; Rapoport, T.A. Gangliosides are receptors for murine polyoma virus and SV40. EMBO J. 2003, 22, 4346–4355. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, J.; Dahl, J.; Riney, C.; You, J.; Cui, C.; Holmes, R.; Lencer, W.I.; Benjamin, T.L. Ganglioside GD1a Restores Infectibility to Mouse Cells Lacking Functional Receptors for Polyomavirus. J. Virol. 2005, 79, 615–618. [Google Scholar] [CrossRef]
- Breau, W.C.; Atwood, W.J.; Norkin, L.C. Class I major histocompatibility proteins are an essential component of the simian virus 40 receptor. J. Virol. 1992, 66, 2037–2045. [Google Scholar] [CrossRef]
- Stang, E.; Kartenbeck, J.; Parton, R.G. Major histocompatibility complex class I molecules mediate association of SV40 with caveolae. Mol. Biol. Cell 1997, 8, 47–57. [Google Scholar] [CrossRef]
- Anderson, H.A.; Norkin, L.C.; Chen, Y. MHC class I molecules are enriched in caveolae but do not enter with simian virus. J. Gen. Virol. 1998, 79 Pt 6, 1469–1477. [Google Scholar] [CrossRef][Green Version]
- Norkin, L.C. Simian virus 40 infection via MHC class I molecules and caveolae. Immunol. Rev. 1999, 168, 13–22. [Google Scholar] [CrossRef]
- Basak, S.; Turner, H.; Compans, R.W. Expression of SV40 receptors on apical surfaces of polarized epithelial cells. Virology 1992, 190, 393–402. [Google Scholar] [CrossRef]
- Neu, U.; Woellner, K.; Gauglitz, G.; Stehle, T. Structural basis of GM1 ganglioside recognition by simian virus. Proc. Natl. Acad. Sci. USA 2008, 105, 5219–5224. [Google Scholar] [CrossRef]
- Ewers, H.; Roemer, W.; Smith, A.E.; Bacia, K.; Dmitrieff, S.; Chai, W.; Mancini, R.; Kartenbeck, J.; Chambon, V.; Berland, L.; et al. GM1 structure determines SV40-induced membrane invagination and infection. Nat. Cell Biol. 2009, 12, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Caruso, M.; Belloni, L.; Sthandier, O.; Amati, P.; Garcia, M.-I. α4β1 Integrin Acts as a Cell Receptor for Murine Polyomavirus at the Postattachment Level. J. Virol. 2003, 77, 3913–3921. [Google Scholar] [CrossRef] [PubMed]
- Caruso, M.; Busanello, A.; Sthandier, O.; Cavaldesi, M.; Gentile, M.; Garcia, M.I.; Amati, P. Mutation in the VP1-LDV Motif of the Murine Polyomavirus Affects Viral Infectivity and Conditions Virus Tissue Tropism in vivo. J. Mol. Biol. 2007, 367, 54–64. [Google Scholar] [CrossRef] [PubMed]
- O’Hara, S.D.; Garcea, R.L. Murine Polyomavirus Cell Surface Receptors Activate Distinct Signaling Pathways Required for Infection. MBio 2016, 7, e01836-16. [Google Scholar] [CrossRef]
- Dugan, A.S.; Eash, S.; Atwood, W.J. An N-Linked Glycoprotein with α(2,3)-Linked Sialic Acid Is a Receptor for BK Virus. J. Virol. 2005, 79, 14442–14445. [Google Scholar] [CrossRef]
- Neu, U.; Ramdial, S.-A.A.A.; Blaum, B.S.; Liu, Y.; Frank, M.; Palma, A.S.; Ströh, L.J.; Feizi, T.; Peters, T.; Atwood, W.J.; et al. A Structure-Guided Mutation in the Major Capsid Protein Retargets BK Polyomavirus. PLoS Pathog. 2013, 9, e1003688. [Google Scholar] [CrossRef]
- Pastrana, D.V.; Ray, U.; Magaldi, T.G.; Schowalter, R.M.; Çuburu, N.; Buck, C.B. BK Polyomavirus Genotypes Represent Distinct Serotypes with Distinct Entry Tropism. J. Virol. 2013, 87, 10105–10113. [Google Scholar] [CrossRef]
- Erickson, K.D.; Garcea, R.L.; Tsai, B. Ganglioside GT1b Is a Putative Host Cell Receptor for the Merkel Cell Polyomavirus. J. Virol. 2009, 83, 10275–10279. [Google Scholar] [CrossRef]
- Schowalter, R.M.; Buck, C.B. The Merkel Cell Polyomavirus Minor Capsid Protein. PLoS Pathog. 2013, 9, e1003558. [Google Scholar] [CrossRef]
- Geoghegan, E.M.; Pastrana, D.V.; Schowalter, R.M.; Ray, U.; Gao, W.; Ho, M.; Pauly, G.T.; Sigano, D.M.; Kaynor, C.; Cahir-McFarland, E.; et al. Infectious Entry and Neutralization of Pathogenic JC Polyomaviruses. Cell Rep. 2017, 21, 1169–1179. [Google Scholar] [CrossRef]
- Ströh, L.J.; Maginnis, M.S.; Blaum, B.S.; Nelson, C.D.S.; Neu, U.; Gee, G.V.; O’Hara, B.A.; Motamedi, N.; DiMaio, D.; Atwood, W.J.; et al. The Greater Affinity of JC Polyomavirus Capsid for α2,6-Linked Lactoseries Tetrasaccharide c than for Other Sialylated Glycans Is a Major Determinant of Infectivity. J. Virol. 2015, 89, 6364–6375. [Google Scholar] [CrossRef]
- Neu, U.; Khan, Z.M.; Schuch, B.; Palma, A.S.; Liu, Y.; Pawlita, M.; Feizi, T.; Stehle, T. Structures of B-Lymphotropic Polyomavirus VP1 in Complex with Oligosaccharide Ligands. PLoS Pathog. 2013, 9, e1003714. [Google Scholar] [CrossRef] [PubMed]
- Ströh, L.J.; Neu, U.; Blaum, B.S.; Buch, M.H.C.; Garcea, R.L.; Stehle, T. Structure Analysis of the Major Capsid Proteins of Human Polyomaviruses 6 and 7 Reveals an Obstructed Sialic Acid Binding Site. J. Virol. 2014, 88, 10831–10839. [Google Scholar] [CrossRef] [PubMed]
- Ströh, L.J.; Rustmeier, N.H.; Blaum, B.S.; Botsch, J.; Rößler, P.; Wedekink, F.; Lipkin, W.I.; Mishra, N.; Stehle, T. Structural Basis and Evolution of Glycan Receptor Specificities within the Polyomavirus Family. MBio 2020, 11. [Google Scholar] [CrossRef]
- Hausen, H.Z.; Gissmann, L. Lymphotropic papovaviruses isolated from African green monkey and human cells. Med. Microbiol. Immunol. 1979, 167, 137–153. [Google Scholar] [CrossRef]
- Khan, Z.M.; Liu, Y.; Neu, U.; Gilbert, M.; Ehlers, B.; Feizi, T.; Stehle, T. Crystallographic and Glycan Microarray Analysis of Human Polyomavirus 9 VP1 Identifies N-Glycolyl Neuraminic Acid as a Receptor Candidate. J. Virol. 2014, 88, 6100–6111. [Google Scholar] [CrossRef]
- Anderson, H.A.; Chen, Y.; Norkin, L.C. Bound simian virus 40 translocates to caveolin-enriched membrane domains, and its entry is inhibited by drugs that selectively disrupt caveolae. Mol. Biol. Cell 1996, 7, 1825–1834. [Google Scholar] [CrossRef]
- Richterová, Z.; Liebl, D.; Horák, M.; Palková, Z.; Štokrová, J.; Hozák, P.; Korb, J.; Forstova, J. Caveolae Are Involved in the Trafficking of Mouse Polyomavirus Virions and Artificial VP1 Pseudocapsids toward Cell Nuclei. J. Virol. 2001, 75, 10880–10891. [Google Scholar] [CrossRef]
- Pelkmans, L.; Helenius, A. Insider information: What viruses tell us about endocytosis. Curr. Opin. Cell Biol. 2003, 15, 414–422. [Google Scholar] [CrossRef]
- Damm, E.-M.; Pelkmans, L.; Kartenbeck, J.; Mezzacasa, A.; Kurzchalia, T.; Helenius, A. Clathrin- and caveolin-1–independent endocytosis: Entry of simian virus 40 into cells devoid of caveolae. J. Cell Biol. 2005, 168, 477–488. [Google Scholar] [CrossRef] [PubMed]
- Dugan, A.; Eash, S.; Atwood, W.J. Update on BK virus entry and intracellular trafficking. Transpl. Infect. Dis. 2006, 8, 62–67. [Google Scholar] [CrossRef] [PubMed]
- Parton, R.G.; Simons, K. The multiple faces of caveolae. Nat. Rev. Mol. Cell Biol. 2007, 8, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Rothberg, K.G.; Heuser, J.E.; Donzell, W.C.; Ying, Y.-S.; Glenney, J.R.; Anderson, R.G. Caveolin, a protein component of caveolae membrane coats. Cell 1992, 68, 673–682. [Google Scholar] [CrossRef]
- Thomsen, P.; Roepstorff, K.; Stahlhut, M.; Van Deurs, B. Caveolae Are Highly Immobile Plasma Membrane Microdomains, Which Are not Involved in Constitutive Endocytic Trafficking. Mol. Biol. Cell 2002, 13, 238–250. [Google Scholar] [CrossRef]
- Kurzchalia, T.V.; DuPree, P.; Parton, R.G.; Kellner, R.; Virta, H.; Lehnert, M.; Simons, K. VIP21, a 21-kD membrane protein is an integral component of trans-Golgi-network-derived transport vesicles. J. Cell Biol. 1992, 118, 1003–1014. [Google Scholar] [CrossRef]
- DuPree, P.; Parton, R.G.; Raposo, G.; Kurzchalia, T.; Simons, K. Caveolae and sorting in the trans-Golgi network of epithelial cells. EMBO J. 1993, 12, 1597–1605. [Google Scholar] [CrossRef]
- Smart, E.J.; Ying, Y.S.; Conrad, P.A.; Anderson, R.G. Caveolin moves from caveolae to the Golgi apparatus in response to cholesterol oxidation. J. Cell Biol. 1994, 127, 1185–1197. [Google Scholar] [CrossRef]
- Carozzi, A.J.; Ikonen, E.; Lindsay, M.R.; Parton, R.G. Role of cholesterol in developing T-tubules: Analogous mechanisms for T-tubule and caveolae biogenesis. Traffic 2000, 1, 326–341. [Google Scholar] [CrossRef]
- Parton, R.G.; Richards, A.A. Lipid Rafts and Caveolae as Portals for Endocytosis: New Insights and Common Mechanisms. Traffic 2003, 4, 724–738. [Google Scholar] [CrossRef]
- Lilley, B.N.; Gilbert, J.M.; Ploegh, H.L.; Benjamin, T.L. Murine Polyomavirus Requires the Endoplasmic Reticulum Protein Derlin-2 To Initiate Infection. J. Virol. 2006, 80, 8739–8744. [Google Scholar] [CrossRef] [PubMed]
- Moriyama, T.; Marquez, J.P.; Wakatsuki, T.; Sorokin, A. Caveolar Endocytosis Is Critical for BK Virus Infection of Human Renal Proximal Tubular Epithelial Cells. J. Virol. 2007, 81, 8552–8562. [Google Scholar] [CrossRef]
- Nelson, C.D.; Carney, D.W.; Derdowski, A.; Lipovsky, A.; Gee, G.V.; O’Hara, B.; Williard, P.; DiMaio, D.; Sello, J.K.; Atwood, W.J. A Retrograde Trafficking Inhibitor of Ricin and Shiga-Like Toxins Inhibits Infection of Cells by Human and Monkey Polyomaviruses. MBio 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- Hummeler, K.; Tomassini, N.; Sokol, F. Morphological Aspects of the Uptake of Simian Virus 40 by Permissive Cells. J. Virol. 1970, 6, 87–93. [Google Scholar] [CrossRef]
- Eash, S.; Querbes, W.; Atwood, W.J. Infection of Vero Cells by BK Virus Is Dependent on Caveolae. J. Virol. 2004, 78, 11583–11590. [Google Scholar] [CrossRef]
- Eash, S.; Atwood, W.J. Involvement of Cytoskeletal Components in BK Virus Infectious Entry. J. Virol. 2005, 79, 11734–11741. [Google Scholar] [CrossRef]
- Zhao, L.; Marciano, A.T.; Rivet, C.R.; Imperiale, M.J. Caveolin- and clathrin-independent entry of BKPyV into primary human proximal tubule epithelial cells. Virology 2016, 492, 66–72. [Google Scholar] [CrossRef]
- Nabi, I.R. Cavin fever: Regulating caveolae. Nat. Cell Biol. 2009, 11, 789–791. [Google Scholar] [CrossRef]
- Oh, P.; McIntosh, D.P.; Schnitzer, J.E. Dynamin at the Neck of Caveolae Mediates Their Budding to Form Transport Vesicles by GTP-driven Fission from the Plasma Membrane of Endothelium. J. Cell Biol. 1998, 141, 101–114. [Google Scholar] [CrossRef] [PubMed]
- Pelkmans, L.; Püntener, D.; Helenius, A. Local Actin Polymerization and Dynamin Recruitment in SV40-Induced Internalization of Caveolae. Science 2002, 296, 535–539. [Google Scholar] [CrossRef] [PubMed]
- Engel, S.; Heger, T.; Mancini, R.; Herzog, F.; Kartenbeck, J.; Hayer, A.; Helenius, A. Role of Endosomes in Simian Virus 40 Entry and Infection. J. Virol. 2011, 85, 4198–4211. [Google Scholar] [CrossRef] [PubMed]
- Hayer, A.; Stoeber, M.; Ritz, D.; Engel, S.; Meyer, H.H.; Helenius, A. Caveolin-1 is ubiquitinated and targeted to intralumenal vesicles in endolysosomes for degradation. J. Cell Biol. 2010, 191, 615–629. [Google Scholar] [CrossRef] [PubMed]
- Mercer, J.; Helenius, A. Virus entry by macropinocytosis. Nat. Cell Biol. 2009, 11, 510–520. [Google Scholar] [CrossRef] [PubMed]
- Querbes, W.; Benmerah, A.; Tosoni, D.; Di Fiore, P.P.; Atwood, W.J. A JC Virus-Induced Signal Is Required for Infection of Glial Cells by a Clathrin- and eps15-Dependent Pathway. J. Virol. 2004, 78, 250–256. [Google Scholar] [CrossRef]
- Gilbert, J.; Benjamin, T.L. Uptake Pathway of Polyomavirus via Ganglioside GD1a. J. Virol. 2004, 78, 12259–12267. [Google Scholar] [CrossRef]
- Liebl, D.; Difato, F.; Horníková, L.; Mannová, P.; Štokrová, J.; Forstova, J. Mouse Polyomavirus Enters Early Endosomes, Requires Their Acidic pH for Productive Infection, and Meets Transferrin Cargo in Rab11-Positive Endosomes. J. Virol. 2006, 80, 4610–4622. [Google Scholar] [CrossRef]
- Qian, M.; Cai, D.; Verhey, K.J.; Tsai, B. A Lipid Receptor Sorts Polyomavirus from the Endolysosome to the Endoplasmic Reticulum to Cause Infection. PLoS Pathog. 2009, 5, e1000465. [Google Scholar] [CrossRef]
- Pho, M.T.; Ashok, A.; Atwood, W.J. JC Virus Enters Human Glial Cells by Clathrin-Dependent Receptor-Mediated Endocytosis. J. Virol. 2000, 74, 2288–2292. [Google Scholar] [CrossRef]
- Mayberry, C.L.; Soucy, A.N.; Lajoie, C.R.; Dushane, J.K.; Maginnis, M.S. JC Polyomavirus Entry by Clathrin-Mediated Endocytosis Is Driven by β-Arrestin. J. Virol. 2019, 93. [Google Scholar] [CrossRef]
- Grove, J.; Marsh, M. The cell biology of receptor-mediated virus entry. J. Cell Biol. 2011, 195, 1071–1082. [Google Scholar] [CrossRef]
- Benmerah, A.; Lamaze, C.; Begue, B.; Schmid, S.L.; Dautry-Varsat, A.; Cerf-Bensussan, N. AP-2/Eps15 interaction is required for receptor-mediated endocytosis. J. Cell Biol. 1998, 140, 1055. [Google Scholar] [CrossRef] [PubMed]
- Marks, B.; Stowell, M.H.B.; Vallis, Y.; Mills, I.G.; Gibson, A.; Hopkins, C.R.; McMahon, H.T. GTPase activity of dynamin and resulting conformation change are essential for endocytosis. Nat. Cell Biol. 2001, 410, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Takei, K.; Haucke, V. Clathrin-mediated endocytosis: Membrane factors pull the trigger. Trends Cell Biol. 2001, 11, 385–391. [Google Scholar] [CrossRef]
- Kim, Y.-M.; Benovic, J.L. Differential Roles of Arrestin-2 Interaction with Clathrin and Adaptor Protein 2 in G Protein-coupled Receptor Trafficking. J. Biol. Chem. 2002, 277, 30760–30768. [Google Scholar] [CrossRef]
- Conner, S.D.; Schmid, S.L. Identification of an adaptor-associated kinase, AAK1, as a regulator of clathrin-mediated endocytosis. J. Cell Biol. 2002, 156, 921–929. [Google Scholar] [CrossRef]
- Motley, A.; Bright, N.A.; Seaman, M.N.; Robinson, M.S. Clathrin-mediated endocytosis in AP-2–depleted cells. J. Cell Biol. 2003, 162, 909–918. [Google Scholar] [CrossRef]
- Conner, S.D.; Schmid, S.L. Differential requirements for AP-2 in clathrin-mediated endocytosis. J. Cell Biol. 2003, 162, 773–780. [Google Scholar] [CrossRef]
- McMahon, H.T.; Boucrot, E. Molecular mechanism and physiological functions of clathrin-mediated endocytosis. Nat. Rev. Mol. Cell Biol. 2011, 12, 517–533. [Google Scholar] [CrossRef]
- Ashok, A.; Atwood, W.J. Contrasting Roles of Endosomal pH and the Cytoskeleton in Infection of Human Glial Cells by JC Virus and Simian Virus. J. Virol. 2003, 77, 1347–1356. [Google Scholar] [CrossRef]
- Querbes, W.; O’Hara, B.A.; Williams, G.; Atwood, W.J. Invasion of Host Cells by JC Virus Identifies a Novel Role for Caveolae in EndosomalSorting of Noncaveolar Ligands. J. Virol. 2006, 80, 9402–9413. [Google Scholar] [CrossRef][Green Version]
- Assetta, B.; Maginnis, M.S.; Ahufinger, I.G.; Haley, S.A.; Gee, G.V.; Nelson, C.D.S.; O’Hara, B.A.; Ramdial, S.-A.A.A.; Atwood, W.J. 5-HT2 Receptors Facilitate JC Polyomavirus Entry. J. Virol. 2013, 87, 13490–13498. [Google Scholar] [CrossRef]
- Suzuki, H.; Gen, K.; Inoue, Y. Comparison of the anti-dopamine D2 and anti-serotonin 5-HT2A activities of chlorpromazine, bromperidol, haloperidol and second-generation antipsychotics parent compounds and metabolites thereof. J. Psychopharmacol. 2013, 27, 396–400. [Google Scholar] [CrossRef]
- Elphick, G.F.; Querbes, W.; Jordan, J.A.; Gee, G.V.; Eash, S.; Manley, K.; Dugan, A.; Stanifer, M.; Bhatnagar, A.; Kroeze, W.K.; et al. The Human Polyomavirus, JCV, Uses Serotonin Receptors to Infect Cells. Science 2004, 306, 1380–1383. [Google Scholar] [CrossRef] [PubMed]
- Bonhaus, D.W.; Bach, C.; DeSouza, A.; Salazar, F.R.; Matsuoka, B.D.; Zuppan, P.; Chan, H.W.; Eglen, R.M. The pharmacology and distribution of human 5-hydroxytryptamine2B (5-HT2b) receptor gene products: Comparison with 5-HT2a and 5-HT2c receptors. Br. J. Pharmacol. 1995, 115, 622–628. [Google Scholar] [CrossRef] [PubMed]
- Bockaert, J.; Claeysen, S.; Bécamel, C.; Dumuis, A.; Marin, P. Neuronal 5-HT metabotropic receptors: Fine-tuning of their structure, signaling, and roles in synaptic modulation. Cell Tissue Res. 2006, 326, 553–572. [Google Scholar] [CrossRef] [PubMed]
- Haley, S.A.; O’Hara, B.A.; Nelson, C.D.; Brittingham, F.L.; Henriksen, K.J.; Stopa, E.G.; Atwood, W.J. Human Polyomavirus Receptor Distribution in Brain Parenchyma Contrasts with Receptor Distribution in Kidney and Choroid Plexus. Am. J. Pathol. 2015, 185, 2246–2258. [Google Scholar] [CrossRef]
- Assetta, B.; Morris-Love, J.; Gee, G.V.; Atkinson, A.L.; O’Hara, B.A.; Maginnis, M.S.; Haley, S.A.; Atwood, W.J. Genetic and Functional Dissection of the Role of Individual 5-HT2 Receptors as Entry Receptors for JC Polyomavirus. Cell Rep. 2019, 27, 1960–1966. [Google Scholar] [CrossRef]
- Gelber, E.I.; Kroeze, W.K.; Roth, B.L.; Gray, J.A.; Sinar, C.A.; Hyde, E.G.; Gurevich, V.V.; Benovic, J. Structure and Function of the Third Intracellular Loop of the 5-Hydroxytryptamine2A Receptor: The Third Intracellular Loop Is α-Helical and Binds Purified Arrestins. J. Neurochem. 2008, 72, 2206–2214. [Google Scholar] [CrossRef]
- Marion, S.; Oakley, R.H.; Kim, K.-M.; Caron, M.G.; Barak, L.S. A β-Arrestin Binding Determinant Common to the Second Intracellular Loops of Rhodopsin Family G Protein-coupled Receptors. J. Biol. Chem. 2005, 281, 2932–2938. [Google Scholar] [CrossRef]
- McCorvy, J.D.; Roth, B.L. Structure and function of serotonin G protein-coupled receptors. Pharmacol. Ther. 2015, 150, 129–142. [Google Scholar] [CrossRef]
- Bhattacharya, A.; Sankar, S.; Panicker, M.M. Differences in the C-terminus contribute to variations in trafficking between rat and human 5-HT2Areceptor isoforms: Identification of a primate-specific tripeptide ASK motif that confers GRK-2 and β arrestin-2 interactions. J. Neurochem. 2010, 112, 723–732. [Google Scholar] [CrossRef]
- Aguet, F.; Antonescu, C.N.; Mettlen, M.; Schmid, S.L.; Danuser, G. Advances in Analysis of Low Signal-to-Noise Images Link Dynamin and AP2 to the Functions of an Endocytic Checkpoint. Dev. Cell 2013, 26, 279–291. [Google Scholar] [CrossRef] [PubMed]
- Hansen, C.G.; Nichols, B.J. Molecular mechanisms of clathrin-independent endocytosis. J. Cell Sci. 2009, 122 Pt 11, 1713–1721. [Google Scholar] [CrossRef]
- Tremblay, J.D.; Sachsenmeier, K.F.; Pipas, J.M. Propagation of Wild-Type and Mutant SV40; Humana Press: Totowa, NJ, USA, 2001. [Google Scholar]
- Ahuja, D.; Sáenz-Robles, M.T.; Pipas, J.M. SV40 large T antigen targets multiple cellular pathways to elicit cellular transformation. Oncogene 2005, 24, 7729–7745. [Google Scholar] [CrossRef]
- Frick, M.; Bright, N.A.; Riento, K.; Bray, A.; Merrified, C.; Nichols, B.J. Coassembly of Flotillins Induces Formation of Membrane Microdomains, Membrane Curvature, and Vesicle Budding. Curr. Biol. 2007, 17, 1151–1156. [Google Scholar] [CrossRef]
- Glebov, O.O.; Bright, N.A.; Nichols, B.J. Flotillin-1 defines a clathrin-independent endocytic pathway in mammalian cells. Nat. Cell Biol. 2005, 8, 46–54. [Google Scholar] [CrossRef]
- Stuermer, C.A.; Lang, D.M.; Kirsch, F.; Wiechers, M.; Deininger, S.-O.; Plattner, H. Glycosylphosphatidyl Inositol-anchored Proteins and fyn Kinase Assemble in Noncaveolar Plasma Membrane Microdomains Defined by Reggie-1 and -2. Mol. Biol. Cell 2001, 12, 3031–3045. [Google Scholar] [CrossRef]
- Neumann-Giesen, C.; Fernow, I.; Amaddii, M.; Tikkanen, R. Role of EGF-induced tyrosine phosphorylation of reggie-1/flotillin-2 in cell spreading and signaling to the actin cytoskeleton. J. Cell Sci. 2007, 120 Pt 3, 395–406. [Google Scholar] [CrossRef]
- Sverdlov, M.; Shajahan, A.N.; Minshall, R.D. Tyrosine phosphorylation-dependence of caveolae-mediated endocytosis. J. Cell Mol. Med. 2007, 11, 1239. [Google Scholar] [CrossRef]
- Altan-Bonnet, N.; Perales, C.; Domingo, E. Extracellular vesicles: Vehicles of en bloc viral transmission. Virus Res. 2019, 265, 143–149. [Google Scholar] [CrossRef]
- Raposo, G.; Stoorvogel, W. Extracellular vesicles: Exosomes, microvesicles, and friends. J. Cell Biol. 2013, 200, 373–383. [Google Scholar] [CrossRef]
- Chen, Y.-H.; Du, W.; Hagemeijer, M.C.; Takvorian, P.M.; Pau, C.; Cali, A.; Brantner, C.A.; Stempinski, E.S.; Connelly, P.S.; Ma, H.-C.; et al. Phosphatidylserine Vesicles Enable Efficient En Bloc Transmission of Enteroviruses. Cell 2015, 160, 619–630. [Google Scholar] [CrossRef] [PubMed]
- Santiana, M.; Ghosh, S.; Ho, B.A.; Rajasekaran, V.; Du, W.-L.; Mutsafi, Y.; De Jésus-Diaz, D.A.; Sosnovtsev, S.V.; Levenson, E.A.; Parra, G.I.; et al. Vesicle-Cloaked Virus Clusters Are Optimal Units for Inter-organismal Viral Transmission. Cell Host Microbe 2018, 24, 208–220. [Google Scholar] [CrossRef]
- Morris-Love, J.; Gee, G.V.; O’Hara, B.A.; Assetta, B.; Atkinson, A.L.; Dugan, A.S.; Haley, S.A.; Atwood, W.J. JC Polyomavirus Uses Extracellular Vesicles To Infect Target Cells. MBio 2019, 10. [Google Scholar] [CrossRef]
- Scribano, S.; Guerrini, M.; Arvia, R.; Guasti, D.; Nardini, P.; Romagnoli, P.; Giannecchini, S. Archetype JC polyomavirus DNA associated with extracellular vesicles circulates in human plasma samples. J. Clin. Virol. 2020, 128, 104435. [Google Scholar] [CrossRef]
- Handala, L.; Blanchard, E.; Raynal, P.-I.; Roingeard, P.; Morel, V.; Descamps, V.; Castelain, S.; Francois, C.; Duverlie, G.; Brochot, E.; et al. BK Polyomavirus Hijacks Extracellular Vesicles for En Bloc Transmission. J. Virol. 2020, 94. [Google Scholar] [CrossRef] [PubMed]
- O’Hara, B.A.; Morris-Love, J.; Gee, G.V.; Haley, S.A.; Atwood, W.J. JC Virus infected choroid plexus epithelial cells produce extracellular vesicles that infect glial cells independently of the virus attachment receptor. PLoS Pathog. 2020, 16, e1008371. [Google Scholar] [CrossRef]
- O’Hara, B.A.; Gee, G.V.; Atwood, W.J.; Haley, S.A. Susceptibility of Primary Human Choroid Plexus Epithelial Cells and Meningeal Cells to Infection by JC Virus. J. Virol. 2018, 92, e00105-18. [Google Scholar] [CrossRef]
- Maginnis, M.S.; Ströh, L.J.; Gee, G.V.; O’Hara, B.A.; Derdowski, A.; Stehle, T.; Atwood, W.J. Progressive Multifocal Leukoencephalopathy-Associated Mutations in the JC Polyomavirus Capsid Disrupt Lactoseries Tetrasaccharide c Binding. MBio 2013, 4, e00247-13. [Google Scholar] [CrossRef] [PubMed]
- Wandinger-Ness, A.; Zerial, M. Rab Proteins and the Compartmentalization of the Endosomal System. Cold Spring Harb. Perspect. Biol. 2014, 6, a022616. [Google Scholar] [CrossRef]
- Hu, Y.-B.; Dammer, E.B.; Ren, R.-J.; Wang, G. The endosomal-lysosomal system: From acidification and cargo sorting to neurodegeneration. Transl. Neurodegener. 2015, 4, 1–10. [Google Scholar] [CrossRef]
- Chaudhary, N.; Gomez, G.A.; Howes, M.T.; Lo, H.P.; McMahon, K.-A.; Rae, J.A.; Schieber, N.L.; Hill, M.M.; Gaus, K.; Yap, A.S.; et al. Endocytic Crosstalk: Cavins, Caveolins, and Caveolae Regulate Clathrin-Independent Endocytosis. PLoS Biol. 2014, 12, e1001832. [Google Scholar] [CrossRef]
- Le Roy, C.; Wrana, J.L. Clathrin- and non-clathrin-mediated endocytic regulation of cell signalling. Nat. Rev. Mol. Cell Biol. 2005, 6, 112–126. [Google Scholar] [CrossRef]
- Jiang, M.; Abend, J.R.; Tsai, B.; Imperiale, M.J. Early Events during BK Virus Entry and Disassembly. J. Virol. 2008, 83, 1350–1358. [Google Scholar] [CrossRef]
- Panou, M.-M.; Antoni, M.; Morgan, E.L.; Loundras, E.-A.; Wasson, C.W.; Welberry-Smith, M.; Mankouri, J.; Macdonald, A. Glibenclamide inhibits BK polyomavirus infection in kidney cells through CFTR blockade. Antivir. Res. 2020, 178. [Google Scholar] [CrossRef]
- Dobson, S.J.; Mankouri, J.; Whitehouse, A. Identification of potassium and calcium channel inhibitors as modulators of polyomavirus endosomal trafficking. Antivir. Res. 2020, 179, 104819. [Google Scholar] [CrossRef]
- Zhao, L.; Imperiale, M.J.; DiMaio, D.; Banks, L. Identification of Rab18 as an Essential Host Factor for BK Polyomavirus Infection Using a Whole-Genome RNA Interference Screen. Msphere 2017, 2, e00291-17. [Google Scholar] [CrossRef]
- Schelhaas, M.; Malmström, J.; Pelkmans, L.; Haugstetter, J.; Ellgaard, L.; Grünewald, K.; Helenius, A. Simian Virus 40 Depends on ER Protein Folding and Quality Control Factors for Entry into Host Cells. Cell 2007, 131, 516–529. [Google Scholar] [CrossRef] [PubMed]
- Magnuson, B.; Rainey, E.K.; Benjamin, T.; Baryshev, M.; Mkrtchian, S.; Tsai, B. ERp29 Triggers a Conformational Change in Polyomavirus to Stimulate Membrane Binding. Mol. Cell 2005, 20, 289–300. [Google Scholar] [CrossRef]
- Gilbert, J.; Ou, W.; Silver, J.; Benjamin, T.L. Downregulation of Protein Disulfide Isomerase Inhibits Infection by the Mouse Polyomavirus. J. Virol. 2006, 80, 10868–10870. [Google Scholar] [CrossRef]
- Walczak, C.P.; Tsai, B. A PDI Family Network Acts Distinctly and Coordinately with ERp29 To Facilitate Polyomavirus Infection. J. Virol. 2010, 85, 2386–2396. [Google Scholar] [CrossRef]
- Hebert, D.N.; Molinari, M. In and Out of the ER: Protein Folding, Quality Control, Degradation, and Related Human Diseases. Physiol. Rev. 2007, 87, 1377–1408. [Google Scholar] [CrossRef]
- Geiger, R.; Andritschke, D.; Friebe, S.; Herzog, F.; Luisoni, S.; Heger, T.; Helenius, A. BAP31 and BiP are essential for dislocation of SV40 from the endoplasmic reticulum to the cytosol. Nat. Cell Biol. 2011, 13, 1305–1314. [Google Scholar] [CrossRef]
- Walczak, C.P.; Ravindran, M.S.; Inoue, T.; Tsai, B. A Cytosolic Chaperone Complexes with Dynamic Membrane J-Proteins and Mobilizes a Nonenveloped Virus out of the Endoplasmic Reticulum. PLoS Pathog. 2014, 10, e1004007. [Google Scholar] [CrossRef]
- Ravindran, M.S.; Bagchi, P.; Inoue, T.; Tsai, B. A Non-enveloped Virus Hijacks Host Disaggregation Machinery to Translocate across the Endoplasmic Reticulum Membrane. PLoS Pathog. 2015, 11, e1005086. [Google Scholar] [CrossRef]
- Bagchi, P.; Walczak, C.P.; Tsai, B. The Endoplasmic Reticulum Membrane J Protein C18 Executes a Distinct Role in Promoting Simian Virus 40 Membrane Penetration. J. Virol. 2015, 89, 4058–4068. [Google Scholar] [CrossRef]
- Bagchi, P.; Inoue, T.; Tsai, B. EMC1-dependent stabilization drives membrane penetration of a partially destabilized non-enveloped virus. eLife 2016, 5, 4921. [Google Scholar] [CrossRef]
- Goodwin, E.C.; Lipovsky, A.; Inoue, T.; Magaldi, T.G.; Edwards, A.P.B.; Van Goor, K.E.Y.; Paton, A.W.; Paton, J.C.; Atwood, W.J.; Tsai, B.; et al. BiP and Multiple DNAJ Molecular Chaperones in the Endoplasmic Reticulum Are Required for Efficient Simian Virus 40 Infection. MBio 2011, 2, e00101-11. [Google Scholar] [CrossRef]
- Dupzyk, A.; Williams, J.M.; Bagchi, P.; Inoue, T.; Tsai, B. SGTA-Dependent Regulation of Hsc70 Promotes Cytosol Entry of Simian Virus 40 from the Endoplasmic Reticulum. J. Virol. 2017, 91. [Google Scholar] [CrossRef]
- Dupzyk, A.; Tsai, B. Bag2 Is a Component of a Cytosolic Extraction Machinery That Promotes Membrane Penetration of a Nonenveloped Virus. J. Virol. 2018, 92, e00607-18. [Google Scholar] [CrossRef]
- Ravindran, M.S.; Engelke, M.F.; Verhey, K.J.; Tsai, B. Exploiting the kinesin-1 molecular motor to generate a virus membrane penetration site. Nat. Commun. 2017, 8, 15496. [Google Scholar] [CrossRef] [PubMed]
- Ravindran, M.S.; Spriggs, C.C.; Verhey, K.J.; Tsai, B. Dynein Engages and Disassembles Cytosol-Localized Simian Virus 40 To Promote Infection. J. Virol. 2018, 92, e00353-18. [Google Scholar] [CrossRef] [PubMed]
- Strunze, S.; Engelke, M.F.; Wang, I.-H.; Puntener, D.; Boucke, K.; Schleich, S.; Way, M.; Schoenenberger, P.; Burckhardt, C.J.; Greber, U.F. Kinesin-1-Mediated Capsid Disassembly and Disruption of the Nuclear Pore Complex Promote Virus Infection. Cell Host Microbe 2011, 10, 210–223. [Google Scholar] [CrossRef]
- Chromy, L.R.; Oltman, A.; Estes, P.A.; Garcea, R.L. Chaperone-Mediated In Vitro Disassembly of Polyoma- and Papillomaviruses. J. Virol. 2006, 80, 5086–5091. [Google Scholar] [CrossRef] [PubMed]
- Cripe, T.P.; Delos, S.E.; Estes, P.A.; Garcea, R.L. In vivo and in vitro association of hsc70 with polyomavirus capsid proteins. J. Virol. 1995, 69, 7807–7813. [Google Scholar] [CrossRef]
- Chen, Y.-J.; Williams, J.M.; Arvan, P.; Tsai, B. Reticulon protects the integrity of the ER membrane during ER escape of large macromolecular protein complexes. J. Cell Biol. 2020, 219. [Google Scholar] [CrossRef]
- Weis, K. Importins and exportins: How to get in and out of the nucleus. Trends Biochem. Sci. 1998, 23, 185–189. [Google Scholar] [CrossRef]
- Fulcher, A.J.; Jans, D.A. Regulation of nucleocytoplasmic trafficking of viral proteins: An integral role in pathogenesis? Biochim. Biophys. Acta 2011, 1813, 2176–2190. [Google Scholar] [CrossRef]
- Efay, N.; Panté, N. Nuclear entry of DNA viruses. Front. Microbiol. 2015, 6, 467. [Google Scholar] [CrossRef]
- Zhang, C.; Hutchins, J.R.; Mühlhäusser, P.; Kutay, U.; Clarke, P.R. Role of Importin-β in the Control of Nuclear Envelope Assembly by Ran. Curr. Biol. 2002, 12, 498–502. [Google Scholar] [CrossRef]
- Nakanishi, A.; Shum, D.; Morioka, H.; Otsuka, E.; Kasamatsu, H. Interaction of the Vp3 Nuclear Localization Signal with the Importin α2/β Heterodimer Directs Nuclear Entry of Infecting Simian Virus. J. Virol. 2002, 76, 9368–9377. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, A.; Li, P.P.; Qu, Q.; Jafri, Q.H.; Kasamatsu, H. Molecular dissection of nuclear entry-competent SV40 during infection. Virus Res. 2007, 124, 226–230. [Google Scholar] [CrossRef]
- Bennett, S.M.; Zhao, L.; Bosard, C.; Imperiale, M.J. Role of a nuclear localization signal on the minor capsid Proteins VP2 and VP3 in BKPyV nuclear entry. Virology 2015, 474, 110–116. [Google Scholar] [CrossRef] [PubMed]
- Qu, Q.; Sawa, H.; Suzuki, T.; Semba, S.; Henmi, C.; Okada, Y.; Tsuda, M.; Tanaka, K.; Atwood, W.J.; Nagashima, K. Nuclear Entry Mechanism of the Human Polyomavirus JC Virus-like Particle: Role of importins and the nuclear pore complex. J. Biol. Chem. 2004, 279, 27735–27742. [Google Scholar] [CrossRef]
- Soldatova, I.; Prilepskaja, T.; Abrahamyan, L.G.; Forstova, J.; Huérfano, S. Interaction of the Mouse Polyomavirus Capsid Proteins with Importins Is Required for Efficient Import of Viral DNA into the Cell Nucleus. Viruses 2018, 10, 165. [Google Scholar] [CrossRef]
- Butin-Israeli, V.; Ben-Nun-Shaul, O.; Kopatz, I.; Adam, S.A.; Shimi, T.; Goldman, R.D.; Oppenheim, A. Simian virus 40 induces lamin A/C fluctuations and nuclear envelope deformation during cell entry. Nucleus 2011, 2, 320–330. [Google Scholar] [CrossRef]
- Nakanishi, A.; Clever, J.; Yamada, M.; Li, P.P.; Kasamatsu, H. Association with capsid proteins promotes nuclear targeting of simian virus 40 DNA. Proc. Natl. Acad. Sci. USA 1996, 93, 96–100. [Google Scholar] [CrossRef]
- Chen, Y.-J.; Liu, X.; Tsai, B. SV40 Hijacks Cellular Transport, Membrane Penetration, and Disassembly Machineries to Promote Infection. Viruses 2019, 11, 917. [Google Scholar] [CrossRef]
- Giannecchini, S. Evidence of the Mechanism by Which Polyomaviruses Exploit the Extracellular Vesicle Delivery System during Infection. Viruses 2020, 12, 585. [Google Scholar] [CrossRef]
- Evans, G.L.; Caller, L.G.; Foster, V.; Crump, C.M. Anion homeostasis is important for non-lytic release of BK polyomavirus from infected cells. Open Biol. 2015, 5. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mayberry, C.L.; Maginnis, M.S. Taking the Scenic Route: Polyomaviruses Utilize Multiple Pathways to Reach the Same Destination. Viruses 2020, 12, 1168. https://doi.org/10.3390/v12101168
Mayberry CL, Maginnis MS. Taking the Scenic Route: Polyomaviruses Utilize Multiple Pathways to Reach the Same Destination. Viruses. 2020; 12(10):1168. https://doi.org/10.3390/v12101168
Chicago/Turabian StyleMayberry, Colleen L., and Melissa S. Maginnis. 2020. "Taking the Scenic Route: Polyomaviruses Utilize Multiple Pathways to Reach the Same Destination" Viruses 12, no. 10: 1168. https://doi.org/10.3390/v12101168
APA StyleMayberry, C. L., & Maginnis, M. S. (2020). Taking the Scenic Route: Polyomaviruses Utilize Multiple Pathways to Reach the Same Destination. Viruses, 12(10), 1168. https://doi.org/10.3390/v12101168