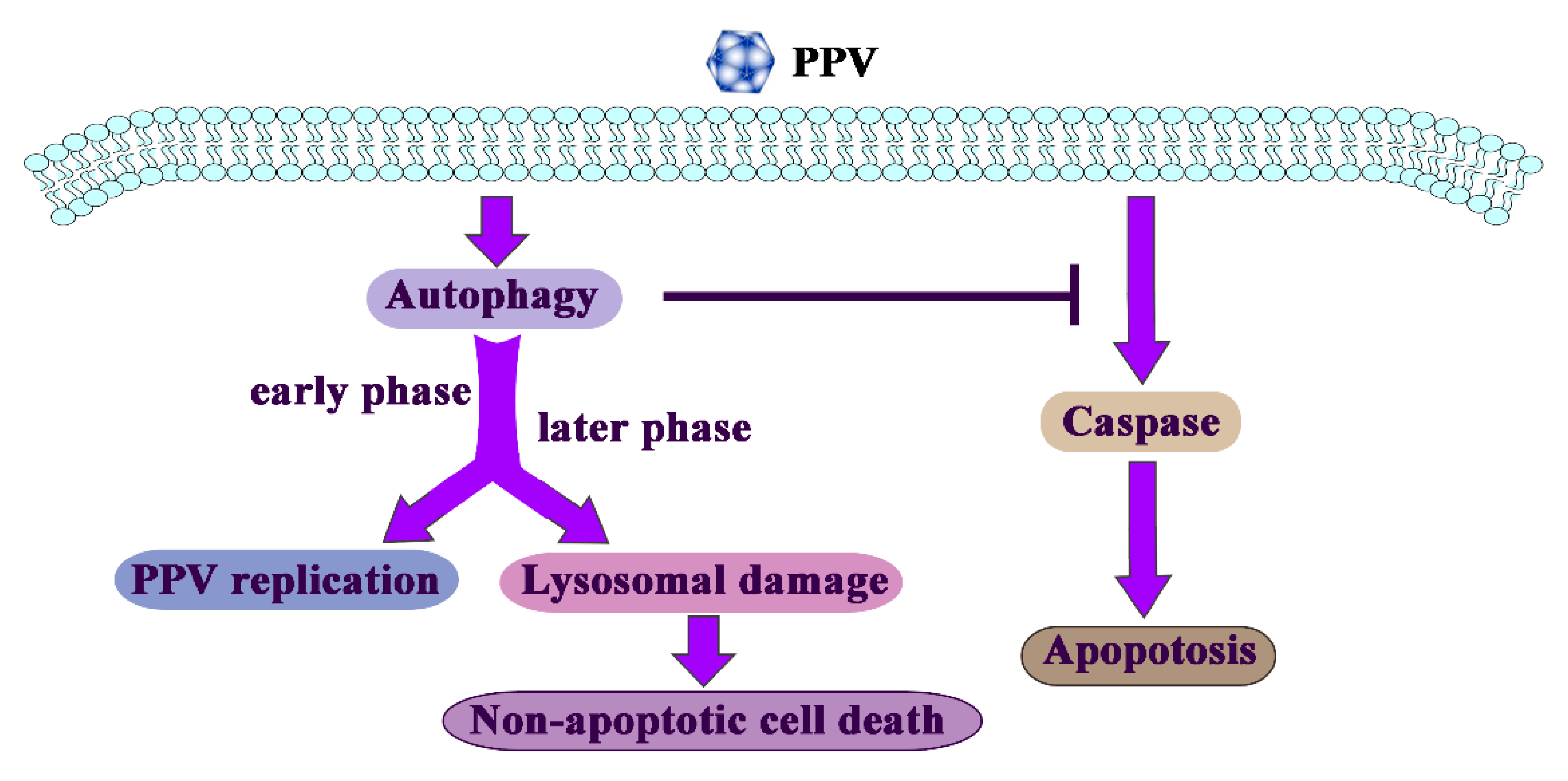
Autophagy Promotes Porcine Parvovirus Replication and Induces Non-Apoptotic Cell Death in Porcine Placental Trophoblasts

 and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Virus Preparation
2.2. Antibodies and Inhibitors
2.3. Transmission Electron Microscopy
2.4. Confocal Microscopy
2.5. siRNA Transfection
2.6. Quantitative PCR
2.7. Flow Cytometry Analysis
2.8. Caspase Activity Assay
2.9. Extraction of Cytosol
2.10. Western Blotting
2.11. Statistical Analysis
3. Results
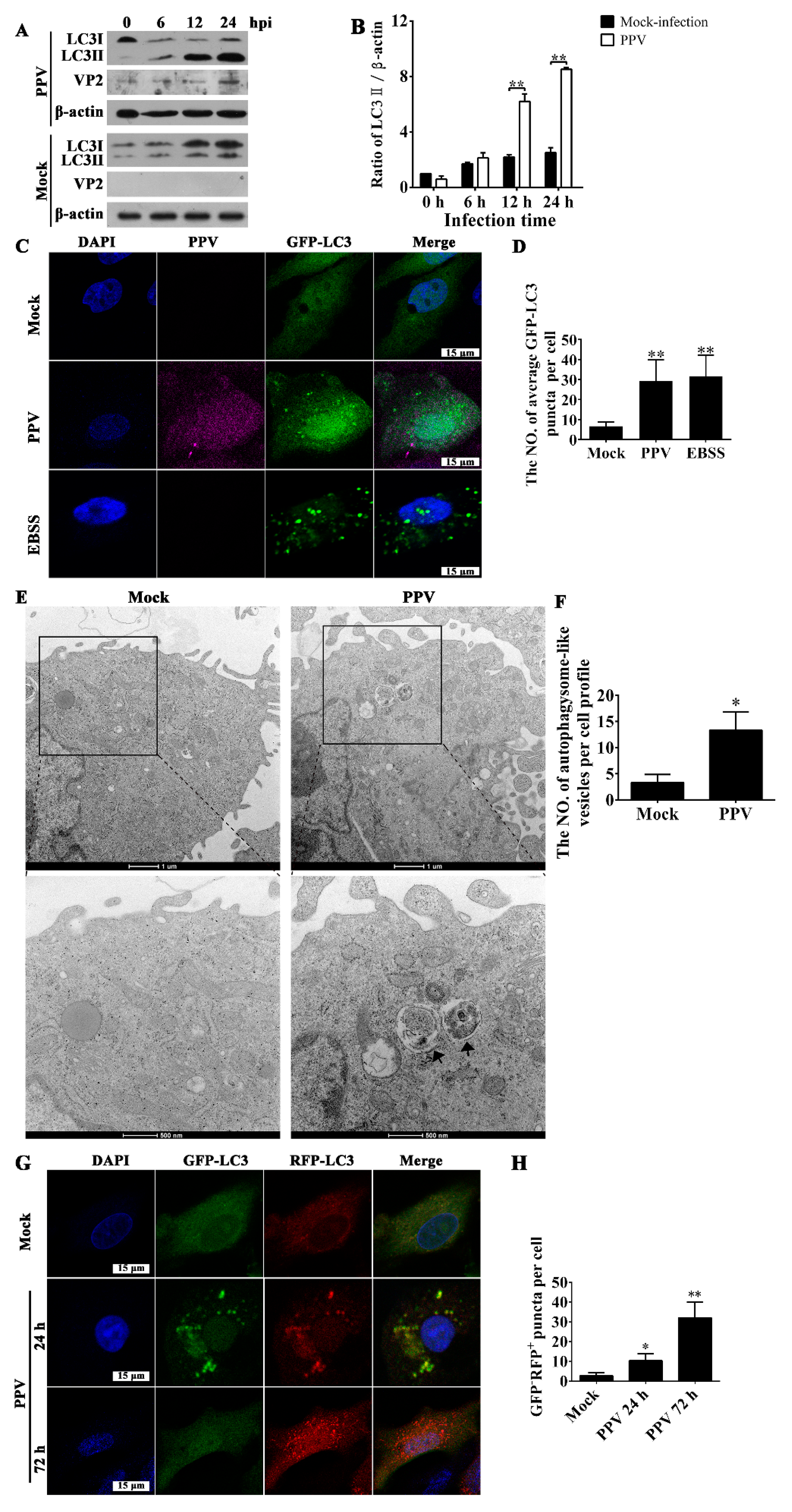
3.1. PPV Infection Triggers the Accumulation of Autophagosomes
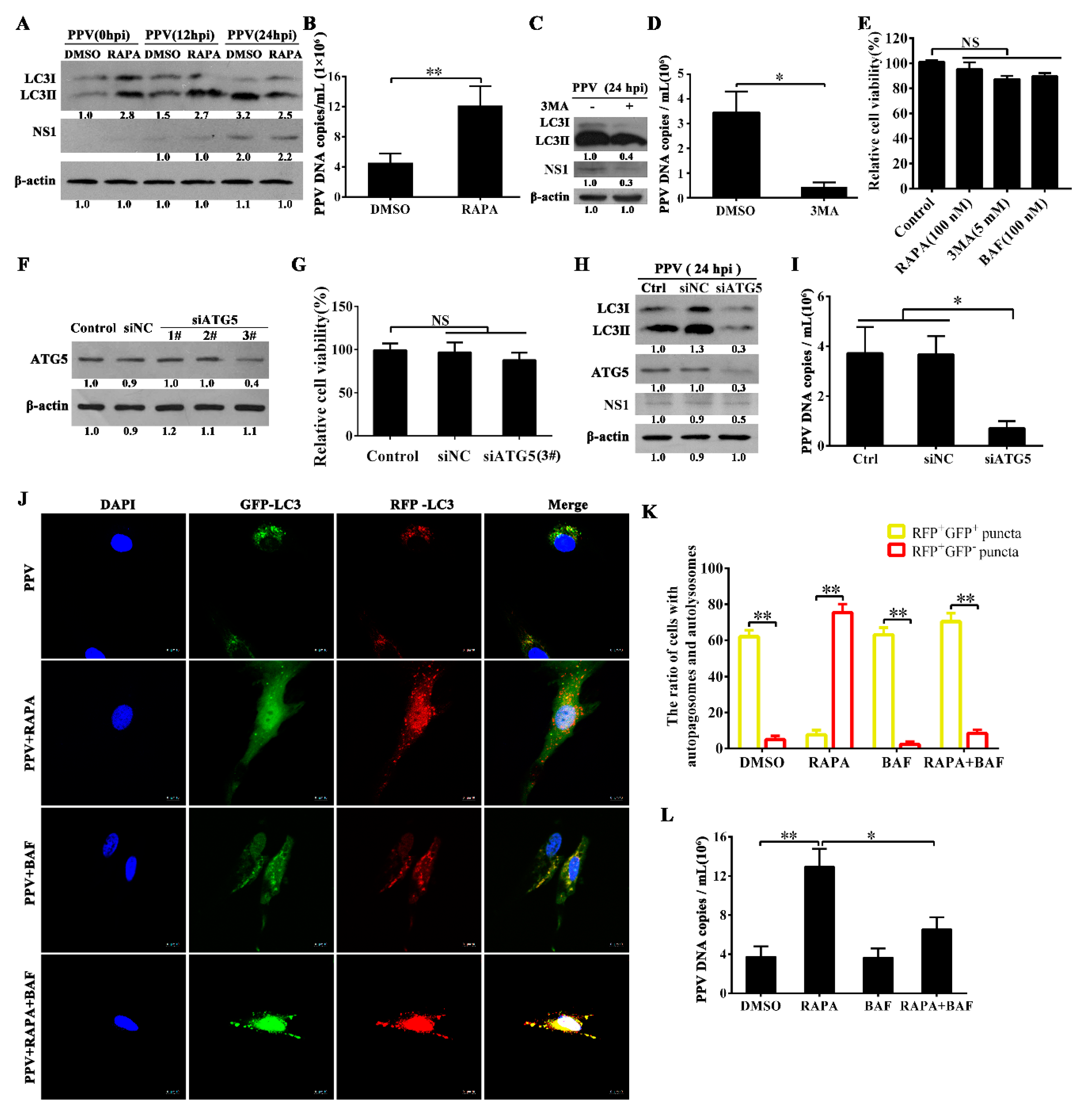
3.2. Autophagy Promotes PPV Replication
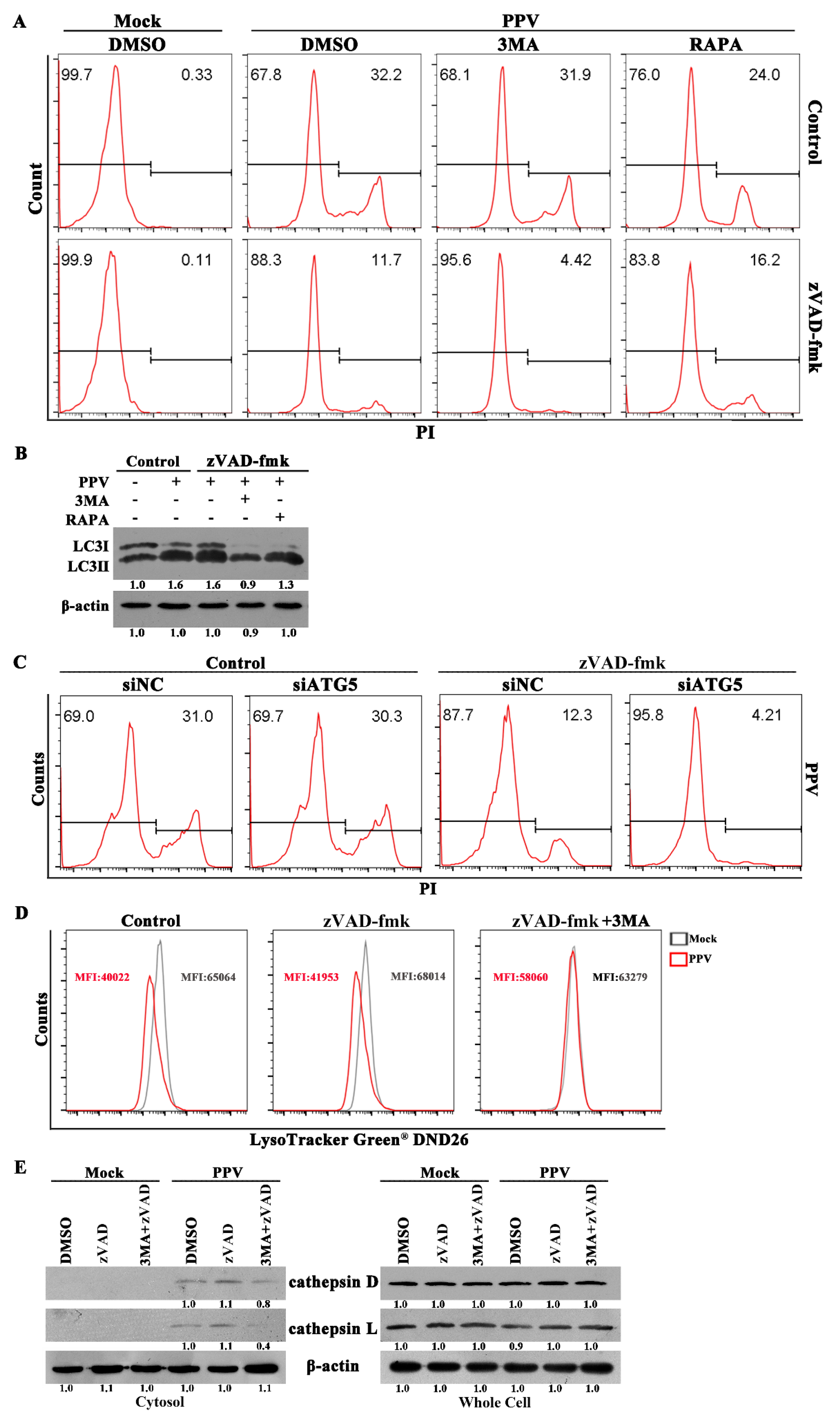
3.3. Autophagy Blocks the Occurrence of Cell Apoptosis in PPV-Infected Cells
3.4. PPV Can Induce Non-Apoptotic Death in PTCs Characteristic of Lysosome Damage
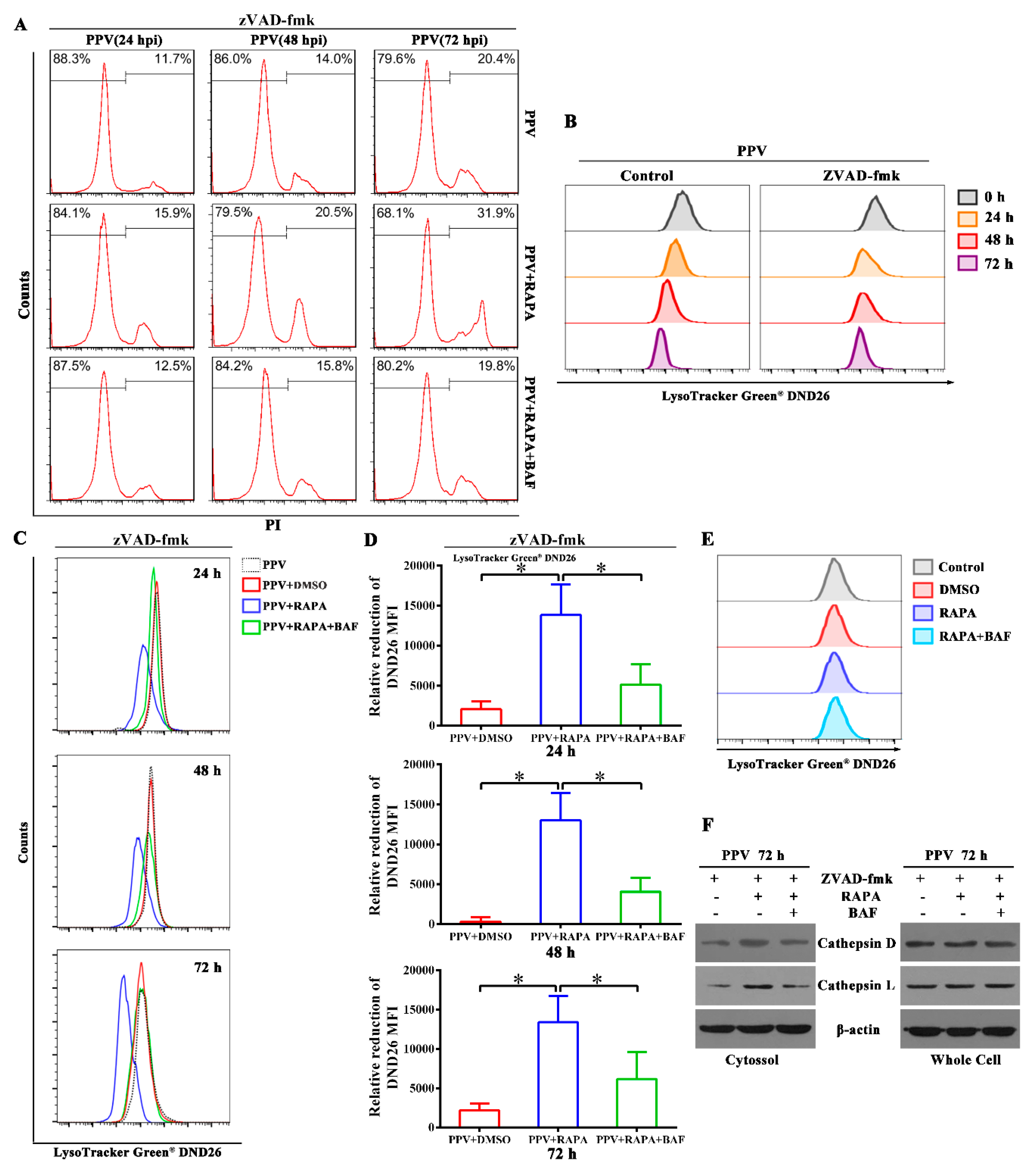
3.5. Autophagy Flux Increases the Ratio of Non-Apoptotic Cell Death in the Later Phase of PPV Infection
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cotmore, S.F.; Agbandje-McKenna, M.; Chiorini, J.A.; Mukha, D.V.; Pintel, D.J.; Qiu, J.; Soderlund-Venermo, M.; Tattersall, P.; Tijssen, P.; Gatherer, D.; et al. The family Parvoviridae. Arch. Virol. 2014, 159, 1239–1247. [Google Scholar] [CrossRef]
- Mengeling, W.L.; Cutlip, R.C. Reproductive disease experimentally induced by exposing pregnant gilts to porcine parvovirus. Am. J. Vet. Res. 1976, 37, 1393–1400. [Google Scholar] [PubMed]
- Bergeron, J.; Hebert, B.; Tijssen, P. Genome organization of the Kresse strain of porcine parvovirus: Identification of the allotropic determinant and comparison with those of NADL-2 and field isolates. J. Virol. 1996, 70, 2508–2515. [Google Scholar] [PubMed]
- Simpson, A.A.; Hebert, B.; Sullivan, G.M.; Parrish, C.R.; Zadori, Z.; Tijssen, P.; Rossmann, M.G. The structure of porcine parvovirus: Comparison with related viruses. J. Mol. Biol. 2002, 315, 1189–1198. [Google Scholar] [CrossRef]
- Zadori, Z.; Szelei, J.; Tijssen, P. SAT: A late NS protein of porcine parvovirus. J. Virol. 2005, 79, 13129–13138. [Google Scholar] [CrossRef]
- Molitor, T.W.; Joo, H.S.; Collett, M.S. Porcine parvovirus: Virus purification and structural and antigenic properties of virion polypeptides. J. Virol. 1983, 45, 842–854. [Google Scholar] [PubMed]
- Ros, C.; Gerber, M.; Kempf, C. Conformational changes in the VP1-unique region of native human parvovirus B19 lead to exposure of internal sequences that play a role in virus neutralization and infectivity. J. Virol. 2006, 80, 12017–12024. [Google Scholar] [CrossRef] [PubMed]
- Boisvert, M.; Fernandes, S.; Tijssen, P. Multiple pathways involved in porcine parvovirus cellular entry and trafficking toward the nucleus. J. Virol. 2010, 84, 7782–7792. [Google Scholar] [CrossRef]
- Christensen, J.; Tattersall, P. Parvovirus Initiator Protein NS1 and RPA Coordinate Replication Fork Progression in a Reconstituted DNA Replication System. J. Virol. 2002, 76, 6518–6531. [Google Scholar] [CrossRef] [PubMed]
- Bashir, T.; Horlein, R.; Rommelaere, J.; Willwand, K. Cyclin A activates the DNA polymerase delta -dependent elongation machinery in vitro: A parvovirus DNA replication model. Proc. Natl. Acad. Sci. USA 2000, 97, 5522–5527. [Google Scholar] [CrossRef] [PubMed]
- Rautou, P.E.; Cazals-Hatem, D.; Feldmann, G.; Mansouri, A.; Grodet, A.; Barge, S.; Martinot-Peignoux, M.; Duces, A.; Bieche, I.; Lebrec, D.; et al. Changes in autophagic response in patients with chronic hepatitis C virus infection. Am. J. Pathol. 2011, 178, 2708–2715. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.S.; Li, J.; Liu, Y.; Wang, F.P.; Wang, S.Q.; She, W.M.; Wu, S.D.; Qi, X.L.; Zhou, Y.P.; Jiang, W. HMGB1-induced autophagy: A new pathway to maintain Treg function during chronic hepatitis B virus infection. Clin. Sci. 2017, 131, 381–394. [Google Scholar] [CrossRef] [PubMed]
- White, E.; Mehnert, J.M.; Chan, C.S. Autophagy, Metabolism, and Cancer. Clin. Cancer Res. 2015, 21, 5037–5046. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Marino, G.; Levine, B. Autophagy and the integrated stress response. Mol. Cell 2010, 40, 280–293. [Google Scholar] [CrossRef] [PubMed]
- Deretic, V. Autophagy in leukocytes and other cells: Mechanisms, subsystem organization, selectivity, and links to innate immunity. J. Leukoc. Biol. 2016, 100, 969–978. [Google Scholar] [CrossRef]
- Ma, Y.; Galluzzi, L.; Zitvogel, L.; Kroemer, G. Autophagy and cellular immune responses. Immunity 2013, 39, 211–227. [Google Scholar] [CrossRef]
- Puleston, D.J.; Simon, A.K. Autophagy in the immune system. Immunology 2014, 141, 1–8. [Google Scholar] [CrossRef]
- Deretic, V.; Saitoh, T.; Akira, S. Autophagy in infection, inflammation and immunity. Nat. Rev. Immunol. 2013, 13, 722–737. [Google Scholar] [CrossRef]
- Pei, J.; Zhao, M.; Ye, Z.; Gou, H.; Wang, J.; Yi, L.; Dong, X.; Liu, W.; Luo, Y.; Liao, M.; et al. Autophagy enhances the replication of classical swine fever virus in vitro. Autophagy 2014, 10, 93–110. [Google Scholar] [CrossRef]
- Huang, W.; Zhao, F.; Huang, Y.; Li, X.; Zhu, S.; Hu, Q.; Chen, W. Rapamycin enhances HBV production by inducing cellular autophagy. Hepat. Mon. 2014, 14, e20719. [Google Scholar] [CrossRef]
- Fitzwalter, B.E.; Thorburn, A. Recent insights into cell death and autophagy. FEBS J. 2015, 282, 4279–4288. [Google Scholar] [CrossRef]
- Levine, B.; Yuan, J. Autophagy in cell death: An innocent convict? J. Clin. Investig. 2005, 115, 2679–2688. [Google Scholar] [CrossRef]
- Booth, L.A.; Tavallai, S.; Hamed, H.A.; Cruickshanks, N.; Dent, P. The role of cell signalling in the crosstalk between autophagy and apoptosis. Cell. Signal. 2014, 26, 549–555. [Google Scholar] [CrossRef] [PubMed]
- Tsujimoto, Y.; Shimizu, S. Another way to die: Autophagic programmed cell death. Cell Death Differ. 2005, 12 (Suppl. 2), 1528–1534. [Google Scholar] [CrossRef] [PubMed]
- Gozuacik, D.; Kimchi, A. Autophagy as a cell death and tumor suppressor mechanism. Oncogene 2004, 23, 2891–2906. [Google Scholar] [CrossRef] [PubMed]
- Denton, D.; Xu, T.; Kumar, S. Autophagy as a pro-death pathway. Immunol. Cell Biol. 2015, 93, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Puleston, D.J.; Simon, A.K. Autophagy and Immune Senescence. Trends Mol. Med. 2016, 22, 671–686. [Google Scholar] [CrossRef]
- Zhuang, H.; Tian, W.; Li, W.; Zhang, X.; Wang, J.; Yang, Y.; Liu, X.; Xia, Z.; Feng, D.; Zhang, L. Autophagic Cell Death and Apoptosis Jointly Mediate Cisatracurium Besylate-Induced Cell Injury. Int. J. Mol. Sci. 2016, 17, 515. [Google Scholar] [CrossRef]
- Di Piazza, M.; Mader, C.; Geletneky, K.; Herrero, Y.C.M.; Weber, E.; Schlehofer, J.; Deleu, L.; Rommelaere, J. Cytosolic activation of cathepsins mediates parvovirus H-1-induced killing of cisplatin and TRAIL-resistant glioma cells. J. Virol. 2007, 81, 4186–4198. [Google Scholar] [CrossRef]
- Angelova, A.L.; Grekova, S.P.; Heller, A.; Kuhlmann, O.; Soyka, E.; Giese, T.; Aprahamian, M.; Bour, G.; Ruffer, S.; Cziepluch, C.; et al. Complementary induction of immunogenic cell death by oncolytic parvovirus H-1PV and gemcitabine in pancreatic cancer. J. Virol. 2014, 88, 5263–5276. [Google Scholar] [CrossRef]
- Nakashima, A.; Tanaka, N.; Tamai, K.; Kyuuma, M.; Ishikawa, Y.; Sato, H.; Yoshimori, T.; Saito, S.; Sugamura, K. Survival of parvovirus B19-infected cells by cellular autophagy. Virology 2006, 349, 254–263. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Huang, Y.; Wang, L.; Yu, T.; Wang, Z.; Chang, L.; Zhao, X.; Luo, X.; Zhang, L.; Tong, D. Immortalization of porcine placental trophoblast cells through reconstitution of telomerase activity. Theriogenology 2016, 85, 1446–1456. [Google Scholar] [PubMed]
- Zhang, H.; Huang, Y.; Du, Q.; Luo, X.; Zhang, L.; Zhao, X.; Tong, D. Porcine parvovirus infection induces apoptosis in PK-15 cells through activation of p53 and mitochondria-mediated pathway. Biochem. Biophys. Res. Commun. 2015, 456, 649–655. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Acevedo Arozena, A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2016, 12, 1–222. [Google Scholar] [PubMed]
- Cui, B.; Liu, W.; Wang, X.; Chen, Y.; Du, Q.; Zhao, X.; Zhang, H.; Liu, S.L.; Tong, D.; Huang, Y. Brucella Omp25 Upregulates miR-155, miR-21-5p, and miR-23b to Inhibit Interleukin-12 Production via Modulation of Programmed Death-1 Signaling in Human Monocyte/Macrophages. Front. Immunol. 2017, 8, 708. [Google Scholar] [CrossRef] [PubMed]
- Johansson, A.C.; Steen, H.; Ollinger, K.; Roberg, K. Cathepsin D mediates cytochrome c release and caspase activation in human fibroblast apoptosis induced by staurosporine. Cell Death Differ. 2003, 10, 1253–1259. [Google Scholar] [CrossRef]
- Repnik, U.; Cesen, M.H.; Turk, B. The Use of Lysosomotropic Dyes to Exclude Lysosomal Membrane Permeabilization. Cold Spring Harb. Protoc. 2016, 2016, pdb-prot087106. [Google Scholar] [CrossRef]
- Wang, L.; Tian, Y.; Ou, J.H. HCV induces the expression of Rubicon and UVRAG to temporally regulate the maturation of autophagosomes and viral replication. PLoS Pathog. 2015, 11, e1004764. [Google Scholar] [CrossRef]
- Ke, P.Y.; Chen, S.S. Autophagy in hepatitis C virus-host interactions: Potential roles and therapeutic targets for liver-associated diseases. World J. Gastroenterol. 2014, 20, 5773–5793. [Google Scholar] [CrossRef]
- Tabor-Godwin, J.M.; Tsueng, G.; Sayen, M.R.; Gottlieb, R.A.; Feuer, R. The role of autophagy during coxsackievirus infection of neural progenitor and stem cells. Autophagy 2012, 8, 938–953. [Google Scholar] [CrossRef]
- Walsh, C.M.; Edinger, A.L. The complex interplay between autophagy, apoptosis, and necrotic signals promotes T-cell homeostasis. Immunol. Rev. 2010, 236, 95–109. [Google Scholar] [CrossRef] [PubMed]
- Whelan, R.S.; Kaplinskiy, V.; Kitsis, R.N. Cell death in the pathogenesis of heart disease: Mechanisms and significance. Annu. Rev. Physiol. 2010, 72, 19–44. [Google Scholar] [CrossRef] [PubMed]
- Fulda, S.; Kogel, D. Cell death by autophagy: Emerging molecular mechanisms and implications for cancer therapy. Oncogene 2015, 34, 5105–5113. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| siRNA | Sense (5′-3′) | Antisense (5′-3′) |
|---|---|---|
| ATG5-1# | CCCUCUAUCAGGAUGAGAUTT | AUCUCAUCCUGAUAGAGGGTT |
| ATG5-2# | GGAUGUAAUUGAAGCUCAUTT | AUGAGCUUCAAUUACAUCCTT |
| ATG5-3# | CCAUCAACCGGAAACUCAUTT | AUGAGUUUCCGGUUGAUGGTT |
| Non-targeting siNC | UUCUCCGAACGUGUCACGUTT | ACGUGACACGUUCGGAGAATT |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, X.; Xiong, Y.; Zhang, J.; Shao, T.; Chen, S.; Miao, B.; Wang, Z.; Du, Q.; Huang, Y.; Tong, D. Autophagy Promotes Porcine Parvovirus Replication and Induces Non-Apoptotic Cell Death in Porcine Placental Trophoblasts. Viruses 2020, 12, 15. https://doi.org/10.3390/v12010015
Zhang X, Xiong Y, Zhang J, Shao T, Chen S, Miao B, Wang Z, Du Q, Huang Y, Tong D. Autophagy Promotes Porcine Parvovirus Replication and Induces Non-Apoptotic Cell Death in Porcine Placental Trophoblasts. Viruses. 2020; 12(1):15. https://doi.org/10.3390/v12010015
Chicago/Turabian StyleZhang, Xiujuan, Yingli Xiong, Jie Zhang, Ting Shao, Songbiao Chen, Bichen Miao, Zhenyu Wang, Qian Du, Yong Huang, and Dewen Tong. 2020. "Autophagy Promotes Porcine Parvovirus Replication and Induces Non-Apoptotic Cell Death in Porcine Placental Trophoblasts" Viruses 12, no. 1: 15. https://doi.org/10.3390/v12010015
APA StyleZhang, X., Xiong, Y., Zhang, J., Shao, T., Chen, S., Miao, B., Wang, Z., Du, Q., Huang, Y., & Tong, D. (2020). Autophagy Promotes Porcine Parvovirus Replication and Induces Non-Apoptotic Cell Death in Porcine Placental Trophoblasts. Viruses, 12(1), 15. https://doi.org/10.3390/v12010015