H-1 Parvovirus as a Cancer-Killing Agent: Past, Present, and Future

Abstract
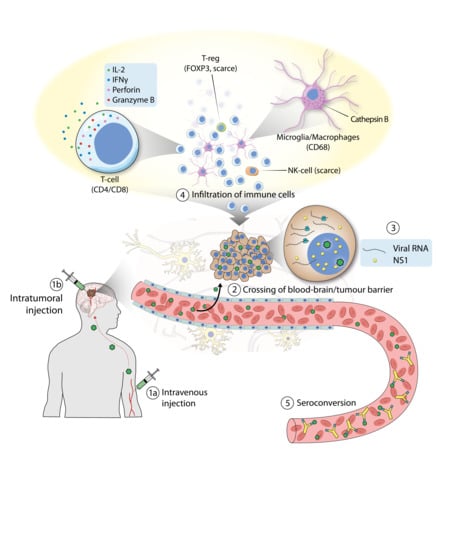
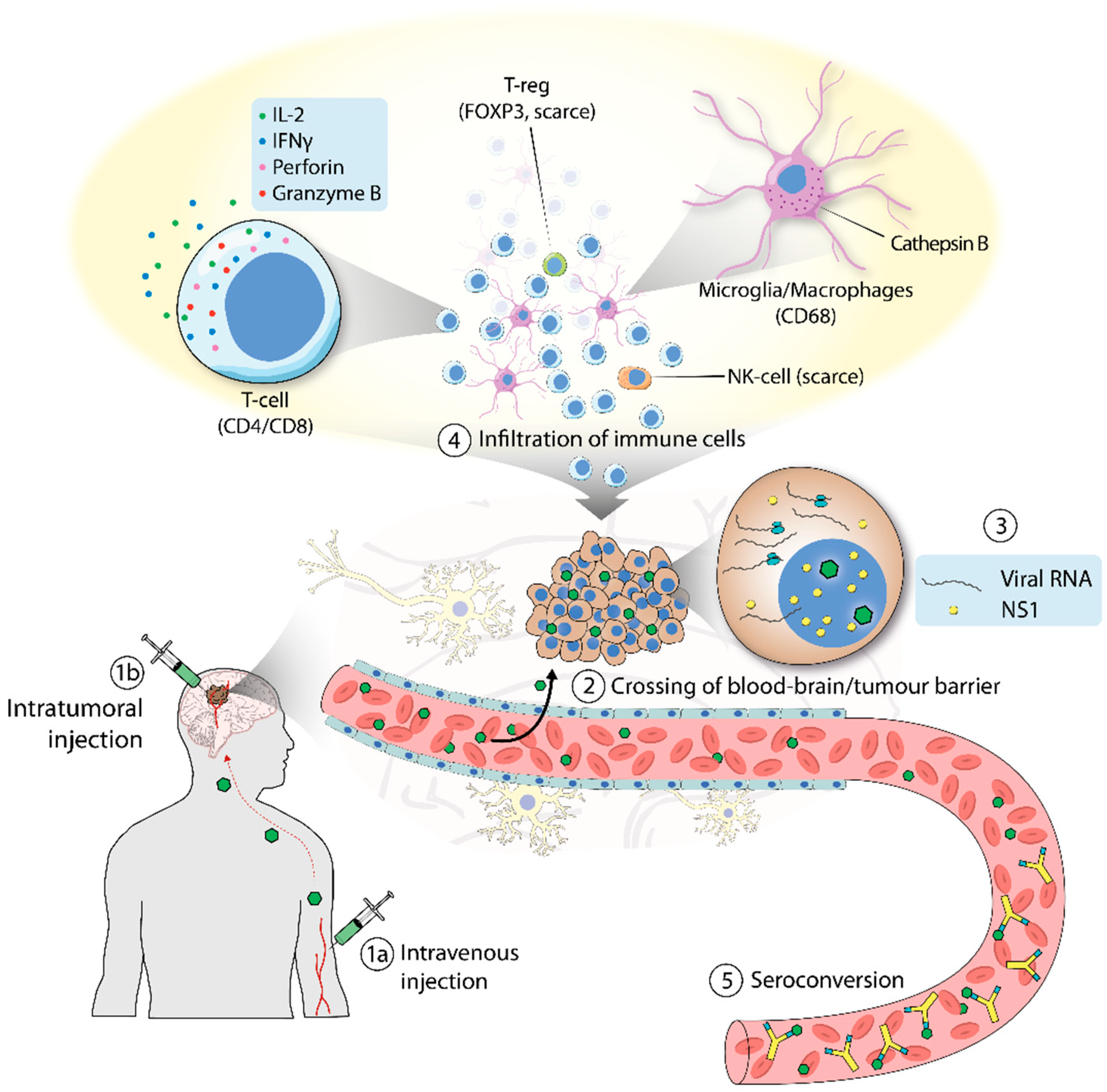
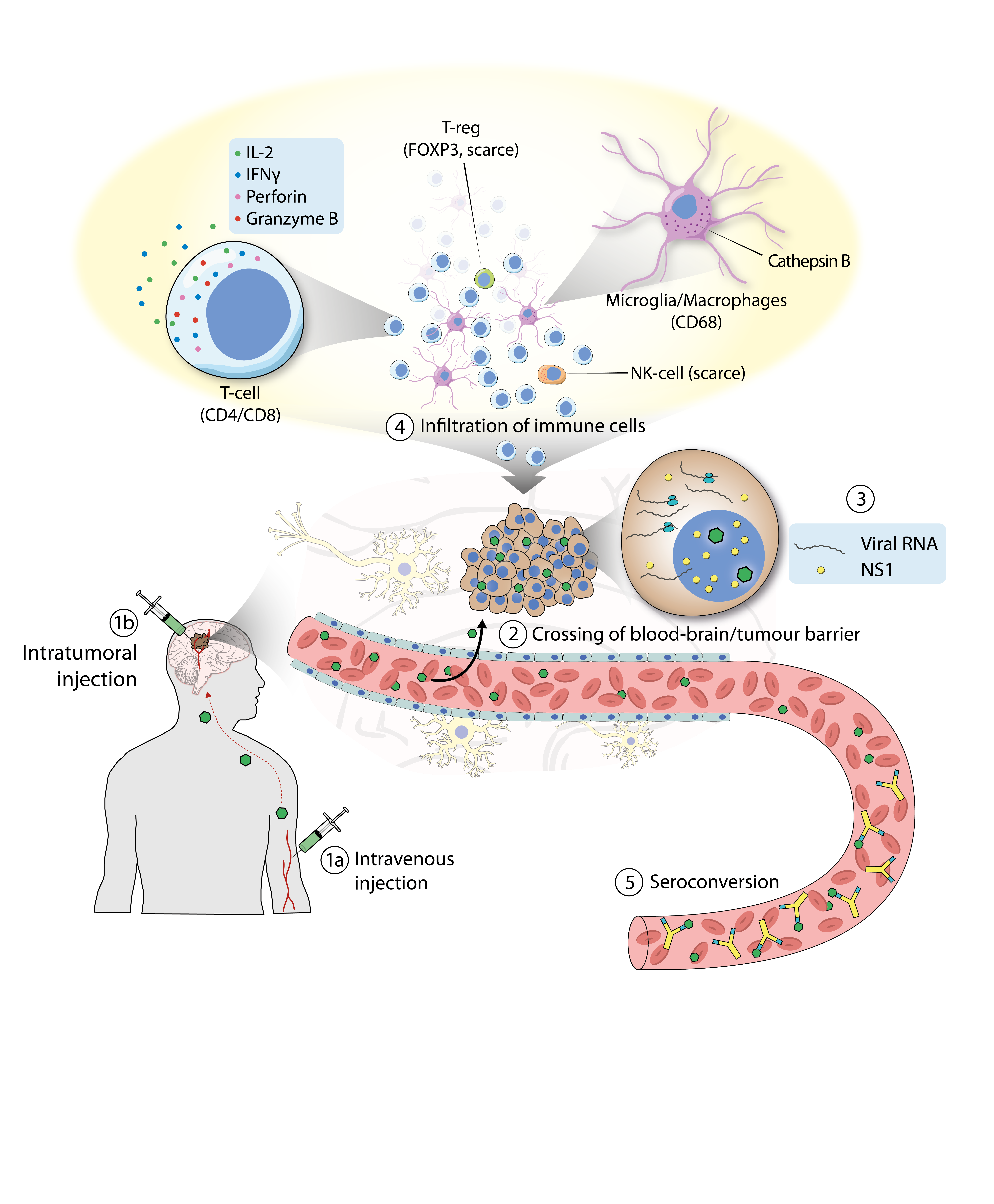{kind=link}
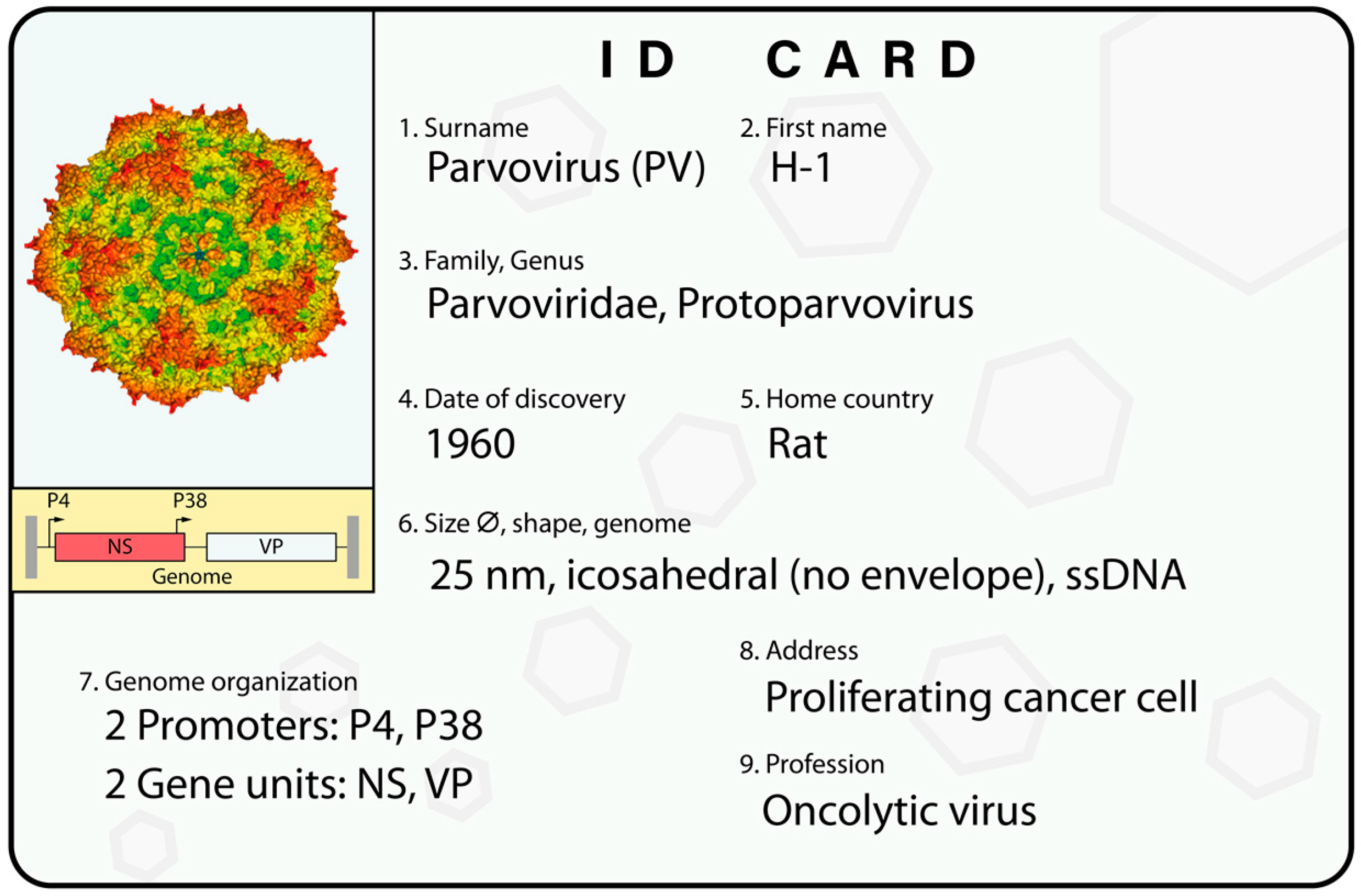{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Oncolytic Viruses: A General Introduction
2. The Rat Protoparvovirus (PV) H-1PV: A Biosketch
3. H-1PV at the Preclinical Level: Acquiring License to Kill Cancer Cells
3.1. Non-Pathogenicity in Humans
3.2. Natural Oncotropism
3.2.1. Uncontrolled Proliferation
3.2.2. Dysregulated Signaling Pathways
3.2.3. Impairments of Innate Antiviral Immunity
3.3. Oncolytic Activities
3.4. Oncosuppressive Activities
3.4.1. Glioma Models
3.4.2. Pancreatic Ductal Adenocarcinoma (PDAC) Models
4. H-1PV Goes to Patients: Meeting the First Endpoints
5. H-1PV Back to the Bench: Further Improving Its Anticancer Profile
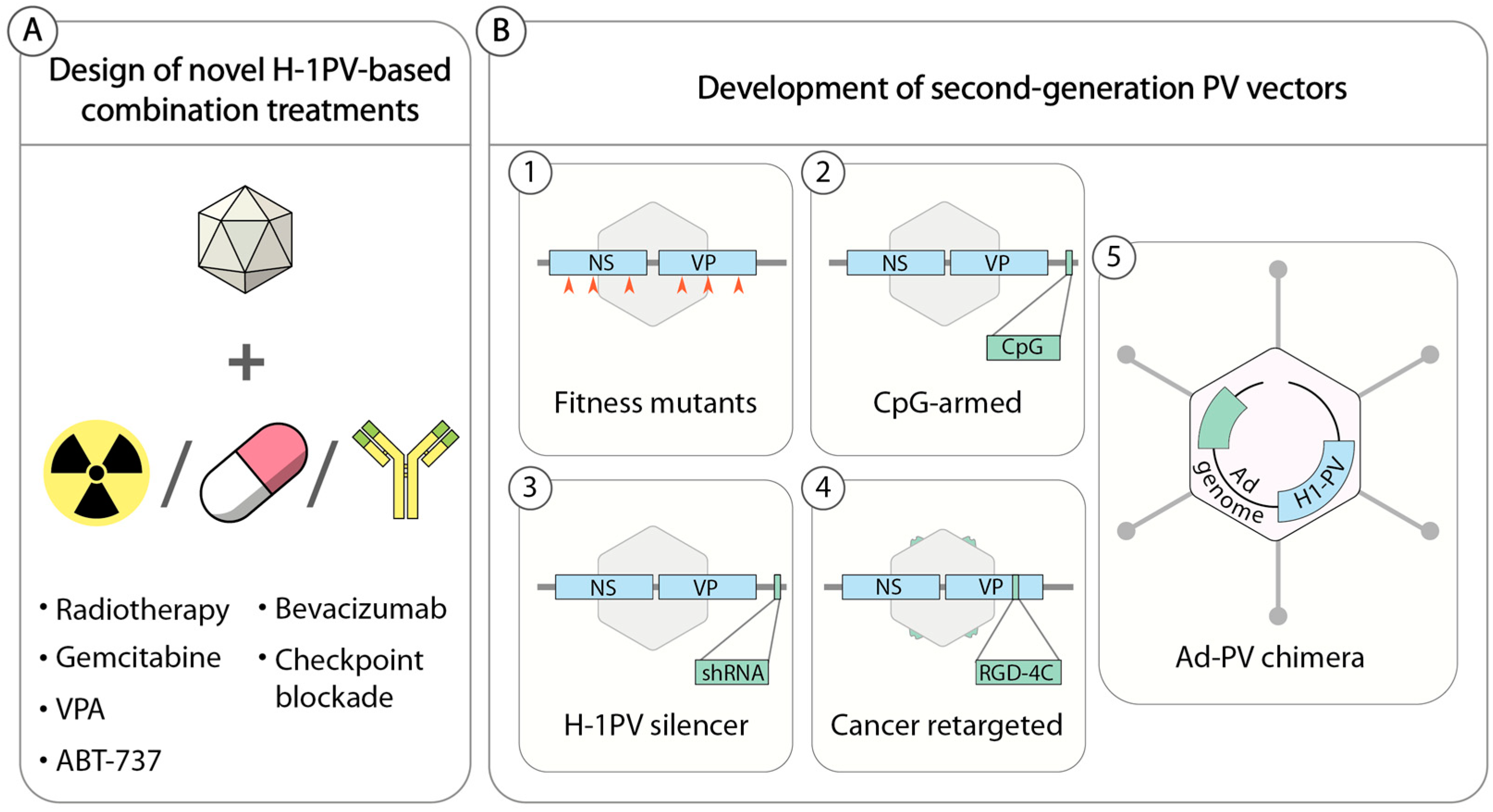
5.1. H-1PV-Based Combination Therapies
5.1.1. H-1PV in Combination with Conventional Treatments
5.1.2. H-1PV in Combination with Epigenetic Modulators
5.1.3. H-1PV in Combination with Apoptosis Inducers
5.1.4. H-1PV in Combination with Antiangiogenic and Immune-Modulating Drugs
5.2. Second-Generation Propagation-Competent H-1PV-Based Vectors
5.2.1. H-1PV Fitness Mutants
5.2.2. H-1PVs Armed with Immune Stimulators
5.2.3. H-1PVs Armed with RNA Interference Triggers
5.2.4. Cancer Retargeted H-1PVs
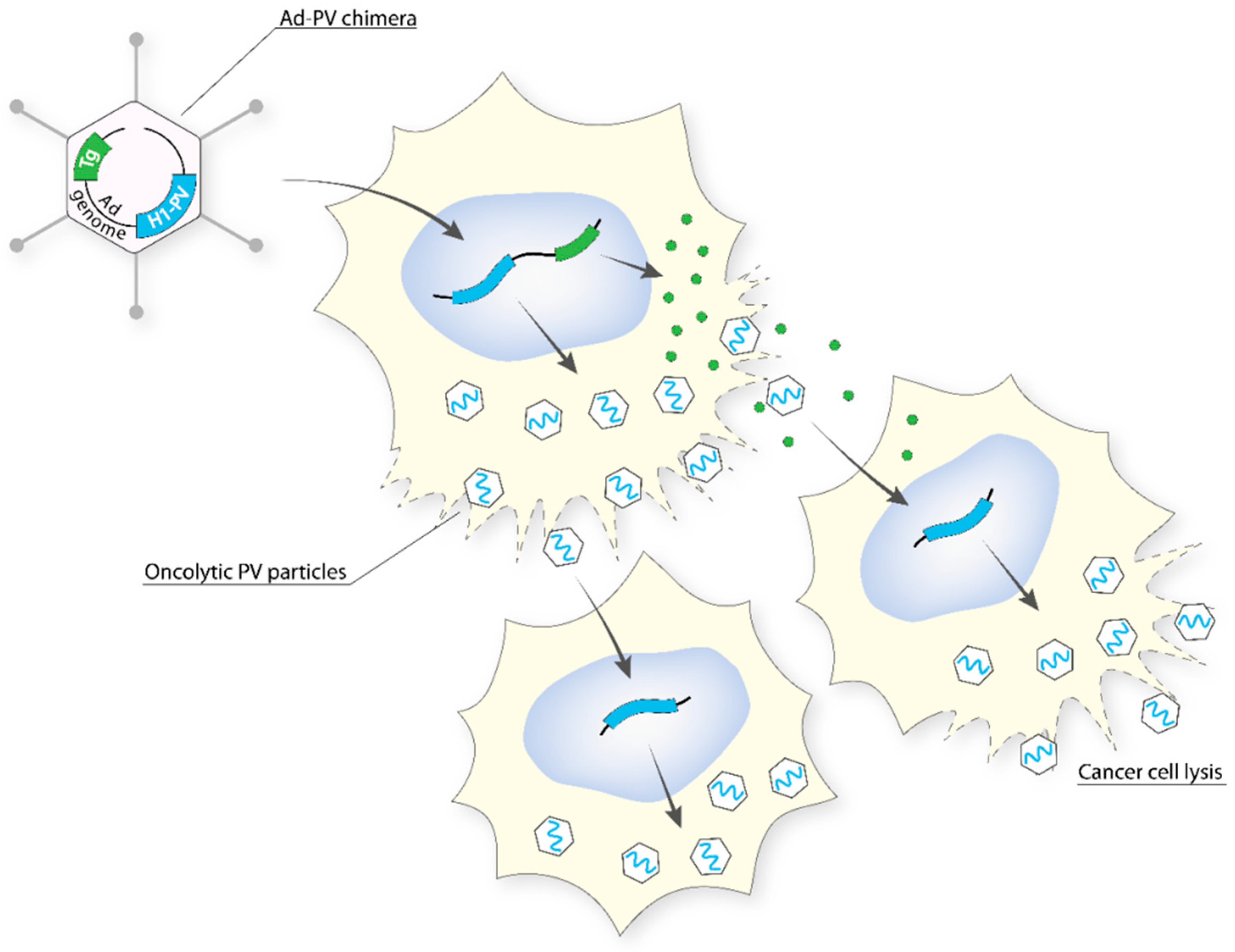
5.2.5. Adenovirus (Ad)–PV Chimera
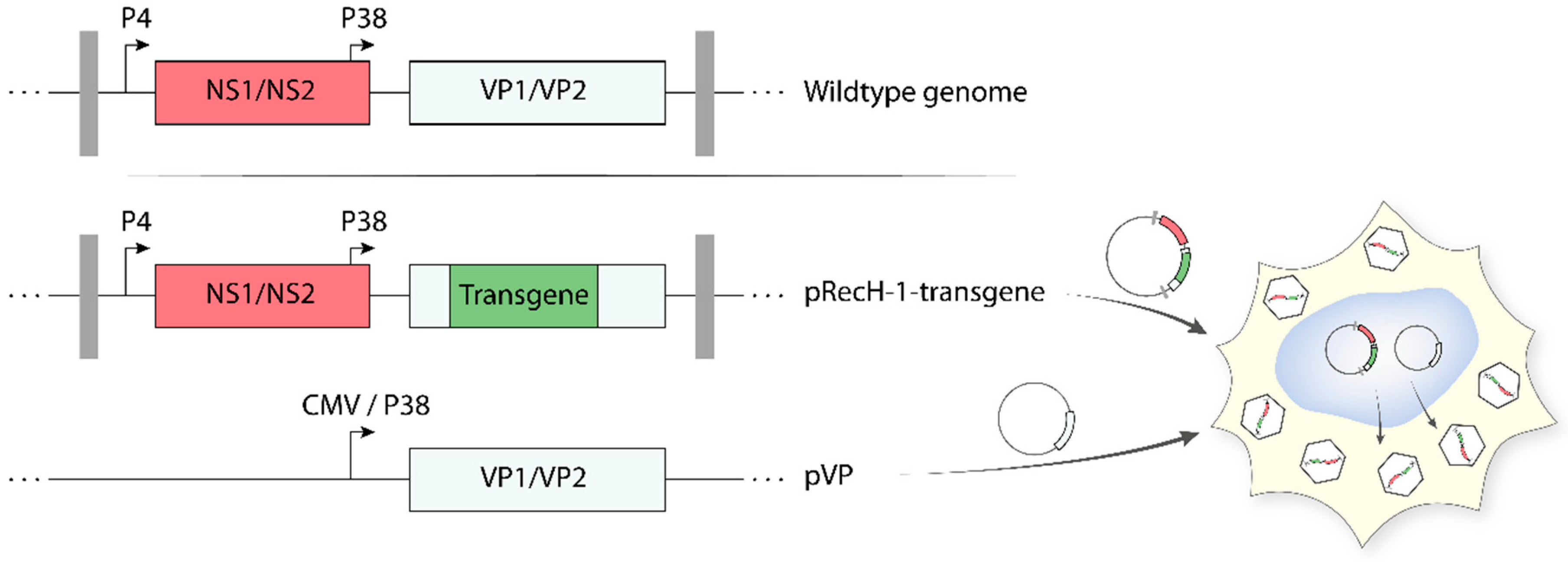
5.3. Recombinant Propagation-Deficient H-1PV-Based Vectors
6. Where Next for H-1PV?
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kaufman, H.L.; Kohlhapp, F.J.; Zloza, A. Oncolytic viruses: A new class of immunotherapy drugs. Nat. Rev. Drug Discov. 2015, 14, 642–662. [Google Scholar] [CrossRef] [PubMed]
- Achard, C.; Surendran, A.; Wedge, M.E.; Ungerechts, G.; Bell, J.; Ilkow, C.S. Lighting a Fire in the Tumor Microenvironment Using Oncolytic Immunotherapy. EBioMedicine 2018, 31, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Najafi, M.; Goradel, N.H.; Farhood, B.; Salehi, E.; Solhjoo, S.; Toolee, H.; Kharazinejad, E.; Mortezaee, K. Tumor microenvironment: Interactions and therapy. J. Cell. Physiol. 2019, 234, 5700–5721. [Google Scholar] [CrossRef] [PubMed]
- Balkwill, F.R.; Capasso, M.; Hagemann, T. The tumor microenvironment at a glance. J. Cell Sci. 2012, 125 Pt 23, 5591–5596. [Google Scholar] [CrossRef]
- Breitbach, C.J.; Lichty, B.D.; Bell, J.C. Oncolytic Viruses: Therapeutics With an Identity Crisis. EBioMedicine 2016, 9, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Breitbach, C.J.; Arulanandam, R.; De Silva, N.; Thorne, S.H.; Patt, R.; Daneshmand, M.; Moon, A.; Ilkow, C.; Burke, J.; Hwang, T.H.; et al. Oncolytic vaccinia virus disrupts tumor-associated vasculature in humans. Cancer Res. 2013, 73, 1265–1275. [Google Scholar] [CrossRef]
- Benencia, F.; Courreges, M.C.; Conejo-Garcia, J.R.; Buckanovich, R.J.; Zhang, L.; Carroll, R.H.; Morgan, M.A.; Coukos, G. Oncolytic HSV exerts direct antiangiogenic activity in ovarian carcinoma. Hum. Gene Ther. 2005, 16, 765–778. [Google Scholar] [CrossRef]
- Cinatl, J., Jr.; Michaelis, M.; Driever, P.H.; Cinatl, J.; Hrabeta, J.; Suhan, T.; Doerr, H.W.; Vogel, J.U. Multimutated herpes simplex virus g207 is a potent inhibitor of angiogenesis. Neoplasia 2004, 6, 725–735. [Google Scholar] [CrossRef]
- Chiocca, E.A.; Rabkin, S.D. Oncolytic viruses and their application to cancer immunotherapy. Cancer Immunol. Res. 2014, 2, 295–300. [Google Scholar] [CrossRef]
- Russell, S.J.; Peng, K.W.; Bell, J.C. Oncolytic virotherapy. Nat. Biotechnol. 2012, 30, 658–670. [Google Scholar] [CrossRef]
- Ledford, H. Cancer-fighting viruses win approval. Nature 2015, 526, 622–623. [Google Scholar] [CrossRef] [PubMed]
- Conry, R.M.; Westbrook, B.; McKee, S.; Norwood, T.G. Talimogene laherparepvec: First in class oncolytic virotherapy. Hum. Vaccin Immunother. 2018, 14, 839–846. [Google Scholar] [CrossRef] [PubMed]
- Fountzilas, C.; Patel, S.; Mahalingam, D. Review: Oncolytic virotherapy, updates and future directions. Oncotarget 2017, 8, 102617–102639. [Google Scholar] [CrossRef] [PubMed]
- Marchini, A.; Scott, E.M.; Rommelaere, J. Overcoming Barriers in Oncolytic Virotherapy with HDAC Inhibitors and Immune Checkpoint Blockade. Viruses 2016, 8, 9. [Google Scholar] [CrossRef] [PubMed]
- Bommareddy, P.K.; Shettigar, M.; Kaufman, H.L. Integrating oncolytic viruses in combination cancer immunotherapy. Nat. Rev. Immunol. 2018, 18, 498–513. [Google Scholar] [CrossRef] [PubMed]
- LaRocca, C.J.; Warner, S.G. Oncolytic viruses and checkpoint inhibitors: Combination therapy in clinical trials. Clin. Transl. Med. 2018, 7, 35. [Google Scholar] [CrossRef] [PubMed]
- Samson, A.; Scott, K.J.; Taggart, D.; West, E.J.; Wilson, E.; Nuovo, G.J.; Thomson, S.; Corns, R.; Mathew, R.K.; Fuller, M.J.; et al. Intravenous delivery of oncolytic reovirus to brain tumor patients immunologically primes for subsequent checkpoint blockade. Sci. Transl. Med. 2018, 10, eaam7577. [Google Scholar] [CrossRef]
- Marchini, A.; Bonifati, S.; Scott, E.M.; Angelova, A.L.; Rommelaere, J. Oncolytic parvoviruses: From basic virology to clinical applications. Virol. J. 2015, 12, 6. [Google Scholar] [CrossRef]
- Vollmers, E.M.; Tattersall, P. Distinct host cell fates for human malignant melanoma targeted by oncolytic rodent parvoviruses. Virology 2013, 446, 37–48. [Google Scholar] [CrossRef]
- Paglino, J.C.; Andres, W.; van den Pol, A.N. Autonomous parvoviruses neither stimulate nor are inhibited by the type I interferon response in human normal or cancer cells. J. Virol. 2014, 88, 4932–4942. [Google Scholar] [CrossRef]
- Grekova, S.; Zawatzky, R.; Horlein, R.; Cziepluch, C.; Mincberg, M.; Davis, C.; Rommelaere, J.; Daeffler, L. Activation of an antiviral response in normal but not transformed mouse cells: A new determinant of minute virus of mice oncotropism. J. Virol. 2010, 84, 516–531. [Google Scholar] [CrossRef] [PubMed]
- Vollmers, E.M.; D’Abramo, A., Jr.; Cotmore, S.F.; Tattersall, P. Genome sequence of tumor virus x, a member of the genus protoparvovirus in the family parvoviridae. Genome Announc. 2014, 2, e00758-14. [Google Scholar] [CrossRef] [PubMed]
- Toolan, H.W.; Dalldore, G.; Barclay, M.; Chandra, S.; Moore, A.E. An Unidentified, Filtrable Agent Isolated from Transplanted Human Tumors. Proc. Natl. Acad. Sci. USA 1960, 46, 1256–1258. [Google Scholar] [CrossRef] [PubMed]
- Toolan, H.W. A virus associated with transplantable human tumors. Bull. N. Y. Acad. Med. 1961, 37, 305–310. [Google Scholar] [PubMed]
- Toolan, H.W. Lack of oncogenic effect of the H-viruses for hamsters. Nature 1967, 214, 1036. [Google Scholar] [CrossRef]
- Toolan, H.W.; Ledinko, N. Inhibition by H-1 virus of the incidence of tumors produced by adenovirus 12 in hamsters. Virology 1968, 35, 475–478. [Google Scholar] [CrossRef]
- Toolan, H.W.; Rhode, S.L., 3rd; Gierthy, J.F. Inhibition of 7,12-dimethylbenz(a)anthracene-induced tumors in Syrian hamsters by prior infection with H-1 parvovirus. Cancer Res. 1982, 42, 2552–2555. [Google Scholar]
- Cotmore, S.F.; Agbandje-McKenna, M.; Chiorini, J.A.; Mukha, D.V.; Pintel, D.J.; Qiu, J.; Soderlund-Venermo, M.; Tattersall, P.; Tijssen, P.; Gatherer, D.; et al. The family Parvoviridae. Arch. Virol. 2014, 159, 1239–1247. [Google Scholar] [CrossRef]
- Cotmore, S.F.; Tattersall, P. Parvoviruses: Small Does Not Mean Simple. Annu. Rev. Virol. 2014, 1, 517–537. [Google Scholar] [CrossRef]
- Li, L.; Cotmore, S.F.; Tattersall, P. Parvoviral left-end hairpin ears are essential during infection for establishing a functional intranuclear transcription template and for efficient progeny genome encapsidation. J. Virol. 2013, 87, 10501–10514. [Google Scholar] [CrossRef]
- Nuesch, J.P.; Lacroix, J.; Marchini, A.; Rommelaere, J. Molecular pathways: Rodent parvoviruses—Mechanisms of oncolysis and prospects for clinical cancer treatment. Clin. Cancer Res. 2012, 18, 3516–3523. [Google Scholar] [CrossRef] [PubMed]
- Cotmore, S.F.; Christensen, J.; Nuesch, J.P.; Tattersall, P. The NS1 polypeptide of the murine parvovirus minute virus of mice binds to DNA sequences containing the motif [ACCA]2-3. J. Virol. 1995, 69, 1652–1660. [Google Scholar] [PubMed]
- Nuesch, J.P.; Rommelaere, J. Tumor Suppressing Properties of Rodent Parvovirus NS1 Proteins and Their Derivatives. Adv. Exp. Med. Biol. 2014, 818, 99–124. [Google Scholar]
- Hristov, G.; Kramer, M.; Li, J.; El-Andaloussi, N.; Mora, R.; Daeffler, L.; Zentgraf, H.; Rommelaere, J.; Marchini, A. Through Its Nonstructural Protein NS1, Parvovirus H-1 Induces Apoptosis via Accumulation of Reactive Oxygen Species. J. Virol. 2010, 84, 5909–5922. [Google Scholar] [CrossRef]
- Hashemi, H.; Condurat, A.L.; Stroh-Dege, A.; Weiss, N.; Geiss, C.; Pilet, J.; Cornet Bartolome, C.; Rommelaere, J.; Salome, N.; Dinsart, C. Mutations in the Non-Structural Protein-Coding Sequence of Protoparvovirus H-1PV Enhance the Fitness of the Virus and Show Key Benefits Regarding the Transduction Efficiency of Derived Vectors. Viruses 2018, 10, 150. [Google Scholar] [CrossRef] [PubMed]
- Halder, S.; Nam, H.J.; Govindasamy, L.; Vogel, M.; Dinsart, C.; Salome, N.; McKenna, R.; Agbandje-McKenna, M. Structural characterization of H-1 parvovirus: Comparison of infectious virions to empty capsids. J. Virol. 2013, 87, 5128–5140. [Google Scholar] [CrossRef] [PubMed]
- Harbison, C.E.; Chiorini, J.A.; Parrish, C.R. The parvovirus capsid odyssey: From the cell surface to the nucleus. Trends Microbiol. 2008, 16, 208–214. [Google Scholar] [CrossRef]
- Cotmore, S.F.; D’Abramo, A.M., Jr.; Ticknor, C.M.; Tattersall, P. Controlled conformational transitions in the MVM virion expose the VP1 N-terminus and viral genome without particle disassembly. Virology 1999, 254, 169–181. [Google Scholar] [CrossRef]
- Mani, B.; Baltzer, C.; Valle, N.; Almendral, J.M.; Kempf, C.; Ros, C. Low pH-dependent endosomal processing of the incoming parvovirus minute virus of mice virion leads to externalization of the VP1 N-terminal sequence (N-VP1), N-VP2 cleavage, and uncoating of the full-length genome. J. Virol. 2006, 80, 1015–1024. [Google Scholar] [CrossRef]
- Zadori, Z.; Szelei, J.; Lacoste, M.C.; Li, Y.; Gariepy, S.; Raymond, P.; Allaire, M.; Nabi, I.R.; Tijssen, P. A viral phospholipase A2 is required for parvovirus infectivity. Dev. Cell 2001, 1, 291–302. [Google Scholar] [CrossRef]
- Allaume, X.; El-Andaloussi, N.; Leuchs, B.; Bonifati, S.; Kulkarni, A.; Marttila, T.; Kaufmann, J.K.; Nettelbeck, D.M.; Kleinschmidt, J.; Rommelaere, J.; et al. Retargeting of rat parvovirus H-1PV to cancer cells through genetic engineering of the viral capsid. J. Virol. 2012, 86, 3452–3465. [Google Scholar] [CrossRef] [PubMed]
- Ros, C.; Bayat, N.; Wolfisberg, R.; Almendral, J.M. Protoparvovirus Cell Entry. Viruses 2017, 9, 313. [Google Scholar] [CrossRef] [PubMed]
- Cotmore, S.F.; Tattersall, P. Parvoviral host range and cell entry mechanisms. Adv. Virus Res. 2007, 70, 183–232. [Google Scholar] [PubMed]
- Angelova, A.L.; Geletneky, K.; Nuesch, J.P.; Rommelaere, J. Tumor Selectivity of Oncolytic Parvoviruses: From in vitro and Animal Models to Cancer Patients. Front. Bioeng. Biotechnol. 2015, 3, 55. [Google Scholar] [CrossRef] [PubMed]
- Deleu, L.; Pujol, A.; Faisst, S.; Rommelaere, J. Activation of promoter P4 of the autonomous parvovirus minute virus of mice at early S phase is required for productive infection. J. Virol. 1999, 73, 3877–3885. [Google Scholar] [PubMed]
- Bashir, T.; Rommelaere, J.; Cziepluch, C. In vivo accumulation of cyclin A and cellular replication factors in autonomous parvovirus minute virus of mice-associated replication bodies. J. Virol. 2001, 75, 4394–4398. [Google Scholar] [CrossRef]
- Porwal, M.; Cohen, S.; Snoussi, K.; Popa-Wagner, R.; Anderson, F.; Dugot-Senant, N.; Wodrich, H.; Dinsart, C.; Kleinschmidt, J.A.; Pante, N.; et al. Parvoviruses cause nuclear envelope breakdown by activating key enzymes of mitosis. PLoS Pathog. 2013, 9, e1003671. [Google Scholar] [CrossRef] [PubMed]
- Bar, S.; Rommelaere, J.; Nuesch, J.P. PKCeta/Rdx-driven phosphorylation of PDK1: A novel mechanism promoting cancer cell survival and permissiveness for parvovirus-induced lysis. PLoS Pathog. 2015, 11, e1004703. [Google Scholar] [CrossRef]
- Fuks, F.; Deleu, L.; Dinsart, C.; Rommelaere, J.; Faisst, S. ras oncogene-dependent activation of the P4 promoter of minute virus of mice through a proximal P4 element interacting with the Ets family of transcription factors. J. Virol. 1996, 70, 1331–1339. [Google Scholar]
- Majumder, K.; Etingov, I.; Pintel, D.J. Protoparvovirus Interactions with the Cellular DNA Damage Response. Viruses 2017, 9, 323. [Google Scholar] [CrossRef]
- Riolobos, L.; Valle, N.; Hernando, E.; Maroto, B.; Kann, M.; Almendral, J.M. Viral oncolysis that targets Raf-1 signaling control of nuclear transport. J. Virol. 2010, 84, 2090–2099. [Google Scholar] [CrossRef] [PubMed]
- Bar, S.; Daeffler, L.; Rommelaere, J.; Nuesch, J.P. Vesicular egress of non-enveloped lytic parvoviruses depends on gelsolin functioning. PLoS Pathog. 2008, 4, e1000126. [Google Scholar] [CrossRef] [PubMed]
- Nuesch, J.P.; Bar, S.; Lachmann, S.; Rommelaere, J. Ezrin-radixin-moesin family proteins are involved in parvovirus replication and spreading. J. Virol. 2009, 83, 5854–5863. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Eichwald, V.; Daeffler, L.; Klein, M.; Rommelaere, J.; Salome, N. The NS2 proteins of parvovirus minute virus of mice are required for efficient nuclear egress of progeny virions in mouse cells. J. Virol. 2002, 76, 10307–10319. [Google Scholar] [CrossRef] [PubMed]
- Grekova, S.; Aprahamian, M.; Giese, N.; Schmitt, S.; Giese, T.; Falk, C.S.; Daeffler, L.; Cziepluch, C.; Rommelaere, J.; Raykov, Z. Immune cells participate in the oncosuppressive activity of parvovirus H-1PV and are activated as a result of their abortive infection with this agent. Cancer Biol. Ther. 2010, 10, 1280–1289. [Google Scholar] [CrossRef] [PubMed]
- Mattei, L.M.; Cotmore, S.F.; Tattersall, P.; Iwasaki, A. Parvovirus evades interferon-dependent viral control in primary mouse embryonic fibroblasts. Virology 2013, 442, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Schlehofer, J.R.; Rentrop, M.; Mannel, D.N. Parvoviruses are inefficient in inducing interferon-beta, tumor necrosis factor-alpha, or interleukin-6 in mammalian cells. Med. Microbiol. Immunol. 1992, 181, 153–164. [Google Scholar] [CrossRef]
- Angelova, A.; Rommelaere, J. Immune System Stimulation by Oncolytic Rodent Protoparvoviruses. Viruses 2019, 11, 415. [Google Scholar] [CrossRef]
- Angelova, A.L.; Witzens-Harig, M.; Galabov, A.S.; Rommelaere, J. The Oncolytic Virotherapy Era in Cancer Management: Prospects of Applying H-1 Parvovirus to Treat Blood and Solid Cancers. Front. Oncol. 2017, 7, 93. [Google Scholar] [CrossRef]
- Di Piazza, M.; Mader, C.; Geletneky, K.; Herrero y Calle, M.; Weber, E.; Schlehofer, J.; Deleu, L.; Rommelaere, J. Cytosolic Activation of Cathepsins Mediates Parvovirus H-1-Induced Killing of Cisplatin and TRAIL-Resistant Glioma Cells. J. Virol. 2007, 81, 4186–4198. [Google Scholar] [CrossRef]
- Marchini, A.; Li, J.; Schroeder, L.; Rommelaere, J.; Geletneky, K. Cancer therapy with a parvovirus combined with a Bcl-2 inhibitor. U.S. Patent 9,889,169, 13 February 2018. [Google Scholar]
- Angelova, A.L.; Grekova, S.P.; Heller, A.; Kuhlmann, O.; Soyka, E.; Giese, T.; Aprahamian, M.; Bour, G.; Ruffer, S.; Cziepluch, C.; et al. Complementary induction of immunogenic cell death by oncolytic parvovirus H-1PV and gemcitabine in pancreatic cancer. J. Virol. 2014, 88, 5263–5276. [Google Scholar] [CrossRef] [PubMed]
- Moehler, M.; Zeidler, M.; Schede, J.; Rommelaere, J.; Galle, P.R.; Cornelis, J.J.; Heike, M. Oncolytic parvovirus H1 induces release of heat-shock protein HSP72 in susceptible human tumor cells but may not affect primary immune cells. Cancer Gene Ther. 2003, 10, 477–480. [Google Scholar] [CrossRef] [PubMed]
- Moehler, M.H.; Zeidler, M.; Wilsberg, V.; Cornelis, J.J.; Woelfel, T.; Rommelaere, J.; Galle, P.R.; Heike, M. Parvovirus H-1-Induced Tumor Cell Death Enhances Human Immune Response In Vitro via Increased Phagocytosis, Maturation, and Cross-Presentation by Dendritic Cells. Hum. Gene Ther. 2005, 16, 996–1005. [Google Scholar] [CrossRef] [PubMed]
- Geletneky, K.; Kiprianova, I.; Ayache, A.; Koch, R.; Herrero Y Calle, M.; Deleu, L.; Sommer, C.; Thomas, N.; Rommelaere, J.; Schlehofer, J.R. Regression of advanced rat and human gliomas by local or systemic treatment with oncolytic parvovirus H-1 in rat models. Neuro-Oncology 2010, 12, 804–814. [Google Scholar] [CrossRef] [PubMed]
- Kiprianova, I.; Thomas, N.; Ayache, A.; Fischer, M.; Leuchs, B.; Klein, M.; Rommelaere, J.; Schlehofer, J.R. Regression of glioma in rat models by intranasal application of parvovirus h-1. Clin. Cancer Res. 2011, 17, 5333–5342. [Google Scholar] [CrossRef] [PubMed]
- Geletneky, K.; Leoni, A.L.; Pohlmeyer-Esch, G.; Loebhard, S.; Leuchs, B.; Hoefer, C.; Jochims, K.; Dahm, M.; Huber, B.; Rommelaere, J.; et al. Bioavailability, biodistribution, and CNS toxicity of clinical-grade parvovirus H1 after intravenous and intracerebral injection in rats. Comp. Med. 2015, 65, 36–45. [Google Scholar]
- Geletneky, K.; Leoni, A.L.; Pohlmeyer-Esch, G.; Loebhard, S.; Baetz, A.; Leuchs, B.; Roscher, M.; Hoefer, C.; Jochims, K.; Dahm, M.; et al. Pathology, organ distribution, and immune response after single and repeated intravenous injection of rats with clinical-grade parvovirus H1. Comp. Med. 2015, 65, 23–35. [Google Scholar]
- Geletneky, K.; Nuesch, J.P.; Angelova, A.; Kiprianova, I.; Rommelaere, J. Double-faceted mechanism of parvoviral oncosuppression. Curr. Opin. Virol. 2015, 13, 17–24. [Google Scholar] [CrossRef]
- Bhat, R.; Dempe, S.; Dinsart, C.; Rommelaere, J. Enhancement of NK cell antitumor responses using an oncolytic parvovirus. Int. J. Cancer 2011, 128, 908–919. [Google Scholar] [CrossRef]
- Angelova, A.L.; Aprahamian, M.; Grekova, S.P.; Hajri, A.; Leuchs, B.; Giese, N.A.; Dinsart, C.; Herrmann, A.; Balboni, G.; Rommelaere, J.; et al. Improvement of gemcitabine-based therapy of pancreatic carcinoma by means of oncolytic parvovirus H-1PV. Clin. Cancer Res. 2009, 15, 511–519. [Google Scholar] [CrossRef]
- Dempe, S.; Stroh-Dege, A.Y.; Schwarz, E.; Rommelaere, J.; Dinsart, C. SMAD4: A predictive marker of PDAC cell permissiveness for oncolytic infection with parvovirus H-1PV. Int. J. Cancer 2010, 126, 2914–2927. [Google Scholar] [CrossRef] [PubMed]
- Toolan, H.W.; Saunders, E.L.; Southam, C.M.; Moore, A.E.; Levin, A.G. H-1 virus viremia in the human. Proc. Soc. Exp. Biol. Med. 1965, 119, 711–715. [Google Scholar] [CrossRef]
- Johnson, D.R.; O’Neill, B.P. Glioblastoma survival in the United States before and during the temozolomide era. J. Neurooncol. 2012, 107, 359–364. [Google Scholar] [CrossRef]
- Malvezzi, M.; Bertuccio, P.; Levi, F.; La Vecchia, C.; Negri, E. European cancer mortality predictions for the year 2014. Ann. Oncol. 2014, 25, 1650–1656. [Google Scholar] [CrossRef] [PubMed]
- Hajda, J.; Lehmann, M.; Krebs, O.; Kieser, M.; Geletneky, K.; Jager, D.; Dahm, M.; Huber, B.; Schoning, T.; Sedlaczek, O.; et al. A non-controlled, single arm, open label, phase II study of intravenous and intratumoral administration of ParvOryx in patients with metastatic, inoperable pancreatic cancer: ParvOryx02 protocol. BMC Cancer 2017, 17, 576. [Google Scholar] [CrossRef] [PubMed]
- Ottolino-Perry, K.; Diallo, J.S.; Lichty, B.D.; Bell, J.C.; McCart, J.A. Intelligent design: Combination therapy with oncolytic viruses. Mol. Ther. 2010, 18, 251–263. [Google Scholar] [CrossRef] [PubMed]
- Wennier, S.T.; Liu, J.; McFadden, G. Bugs and drugs: Oncolytic virotherapy in combination with chemotherapy. Curr. Pharm. Biotechnol. 2012, 13, 1817–1833. [Google Scholar] [CrossRef] [PubMed]
- Geletneky, K.; Hartkopf, A.D.; Krempien, R.; Rommelaere, J.; Schlehofer, J.R. Improved killing of human high-grade glioma cells by combining ionizing radiation with oncolytic parvovirus H-1 infection. J. Biomed. Biotechnol. 2010, 2010, 350748. [Google Scholar] [CrossRef] [PubMed]
- Wennerberg, E.; Vanpouille-Box, C.; Bornstein, S.; Yamazaki, T.; Demaria, S.; Galluzzi, L. Immune recognition of irradiated cancer cells. Immunol. Rev. 2017, 280, 220–230. [Google Scholar] [CrossRef] [PubMed]
- Burris, H.A., 3rd; Moore, M.J.; Andersen, J.; Green, M.R.; Rothenberg, M.L.; Modiano, M.R.; Cripps, M.C.; Portenoy, R.K.; Storniolo, A.M.; Tarassoff, P.; et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: A randomized trial. J. Clin. Oncol. 1997, 15, 2403–2413. [Google Scholar] [CrossRef] [PubMed]
- Minucci, S.; Pelicci, P.G. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat. Rev. Cancer 2006, 6, 38–51. [Google Scholar] [CrossRef]
- Li, J.; Bonifati, S.; Hristov, G.; Marttila, T.; Valmary-Degano, S.; Stanzel, S.; Schnolzer, M.; Mougin, C.; Aprahamian, M.; Grekova, S.P.; et al. Synergistic combination of valproic acid and oncolytic parvovirus H-1PV as a potential therapy against cervical and pancreatic carcinomas. EMBO Mol. Med. 2013, 5, 1537–1555. [Google Scholar] [CrossRef] [PubMed]
- Andersen, M.H.; Svane, I.M.; Kvistborg, P.; Nielsen, O.J.; Balslev, E.; Reker, S.; Becker, J.C.; Straten, P.T. Immunogenicity of Bcl-2 in patients with cancer. Blood 2005, 105, 728–734. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Twumasi-Boateng, K.; Pettigrew, J.L.; Kwok, Y.Y.E.; Bell, J.C.; Nelson, B.H. Oncolytic viruses as engineering platforms for combination immunotherapy. Nat. Rev. Cancer 2018, 18, 419–432. [Google Scholar] [CrossRef] [PubMed]
- Moehler, M.; Goepfert, K.; Heinrich, B.; Breitbach, C.J.; Delic, M.; Galle, P.R.; Rommelaere, J. Oncolytic Virotherapy as Emerging Immunotherapeutic Modality: Potential of Parvovirus H-1. Front. Oncol. 2014, 4, 92. [Google Scholar] [CrossRef] [PubMed]
- Geletneky, K.; Bartsch, A.; Weiss, C.; Bernhard, H.; Marchini, A.; Rommelaere, J. ATIM-40. High rate of objective anti.tumor response in 9 patients with glioblastoma after viro-immunotherapy with oncolytic parvovirus H-1 in combination with bavacicumab and PD-1 checkpoint blockade. Neuro-Oncology 2018, 20 (Suppl. 6), vi10. [Google Scholar] [CrossRef][Green Version]
- Faisst, S.; Faisst, S.R.; Dupressoir, T.; Plaza, S.; Pujol, A.; Jauniaux, J.C.; Rhode, S.L.; Rommelaere, J. Isolation of a fully infectious variant of parvovirus H-1 supplanting the standard strain in human cells. J. Virol. 1995, 69, 4538–4543. [Google Scholar]
- Weiss, N.; Stroh-Dege, A.; Rommelaere, J.; Dinsart, C.; Salome, N. An in-frame deletion in the NS protein-coding sequence of parvovirus H-1PV efficiently stimulates export and infectivity of progeny virions. J. Virol. 2012, 86, 7554–7564. [Google Scholar] [CrossRef]
- Nüesch, J.; Thomas, N.; Plotzky, C.; Jean, R. Modified Rodent Parvovirus Capable of Propagating and Spreading through Human Gliomas. Patent No. EP2384761B1, 4 September 2013. [Google Scholar]
- El-Andaloussi, N.; Endele, M.; Leuchs, B.; Bonifati, S.; Kleinschmidt, J.; Rommelaere, J.; Marchini, A. Novel adenovirus-based helper system to support production of recombinant parvovirus. Cancer Gene Ther. 2011, 18, 240–249. [Google Scholar] [CrossRef]
- Raykov, Z.; Grekova, S.; Leuchs, B.; Aprahamian, M.; Rommelaere, J. Arming parvoviruses with CpG motifs to improve their oncosuppressive capacity. Int. J. Cancer 2008, 122, 2880–2884. [Google Scholar] [CrossRef]
- Grekova, S.P.; Aprahamian, M.; Giese, N.A.; Bour, G.; Giese, T.; Grewenig, A.; Leuchs, B.; Hörlein, R.; Heller, A.; Angelova, A.L.; et al. Genomic CpG Enrichment of Oncolytic Parvoviruses as a Potent Anticancer Vaccination Strategy for the Treatment of Pancreatic Adenocarcinoma. J. Vaccines Vaccin 2014, 5. [Google Scholar] [CrossRef]
- Illarionova, A.; Rommelaere, J.; Leuchs, B.; Marchini, A. Modified Parvovirus Useful for Gene Silencing. Patent No. EP2620503, 31 July 2013. [Google Scholar]
- Cornelis, J.J.; Salome, N.; Dinsart, C.; Rommelaere, J. Vectors based on autonomous parvoviruses: Novel tools to treat cancer? J. Gene Med. 2004, 6 (Suppl. 1), S193–S202. [Google Scholar] [CrossRef] [PubMed]
- Zitzmann, S.; Ehemann, V.; Schwab, M. Arginine-glycine-aspartic acid (RGD)-peptide binds to both tumor and tumor-endothelial cells in vivo. Cancer Res. 2002, 62, 5139–5143. [Google Scholar]
- El-Andaloussi, N.; Bonifati, S.; Kaufmann, J.K.; Mailly, L.; Daeffler, L.; Deryckere, F.; Nettelbeck, D.M.; Rommelaere, J.; Marchini, A. Generation of an adenovirus-parvovirus chimera with enhanced oncolytic potential. J. Virol. 2012, 86, 10418–10431. [Google Scholar] [CrossRef] [PubMed]
- Cornelis, J.J.; Lang, S.I.; Stroh-Dege, A.Y.; Balboni, G.; Dinsart, C.; Rommelaere, J. Cancer gene therapy through autonomous parvovirus–mediated gene transfer. Curr. Gene Ther. 2004, 4, 249–261. [Google Scholar] [CrossRef]
- Olijslagers, S.; Dege, A.Y.; Dinsart, C.; Voorhoeve, M.; Rommelaere, J.; Noteborn, M.H.; Cornelis, J.J. Potentiation of a recombinant oncolytic parvovirus by expression of Apoptin. Cancer Gene Ther. 2001, 8, 958–965. [Google Scholar] [CrossRef]
- Dempe, S.; Lavie, M.; Struyf, S.; Bhat, R.; Verbeke, H.; Paschek, S.; Berghmans, N.; Geibig, R.; Rommelaere, J.; Van Damme, J.; et al. Antitumoral activity of parvovirus-mediated IL-2 and MCP-3/CCL7 delivery into human pancreatic cancer: Implication of leucocyte recruitment. Cancer Immunol. Immunother. 2012, 61, 2113–2123. [Google Scholar] [CrossRef]
- Lavie, M.; Struyf, S.; Stroh-Dege, A.; Rommelaere, J.; Van Damme, J.; Dinsart, C. Capacity of wild-type and chemokine-armed parvovirus H-1PV for inhibiting neo-angiogenesis. Virology 2013, 447, 221–232. [Google Scholar] [CrossRef][Green Version]
- Dinsart, C.; Pervolaraki, K.; Stroh-Dege, A.; Lavie, M.; Ronsse, I.; Rommelaere, J.; Van Damme, J.; Van Raemdonck, K.; Struyf, S. Recombinant Parvoviruses Armed to Deliver CXCL4L1 and CXCL10 Are Impaired in Their Antiangiogenic and Antitumoral Effects in a Kaposi Sarcoma Tumor Model Due To the Chemokines’ Interference with the Virus Cycle. Hum. Gene Ther. 2017, 28, 295–306. [Google Scholar] [CrossRef]
- El-Andaloussi, N.; Leuchs, B.; Bonifati, S.; Rommelaere, J.; Marchini, A. Efficient recombinant parvovirus production with the help of adenovirus-derived systems. J. Vis. Exp. 2012, 62, e3518. [Google Scholar] [CrossRef] [PubMed]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bretscher, C.; Marchini, A. H-1 Parvovirus as a Cancer-Killing Agent: Past, Present, and Future. Viruses 2019, 11, 562. https://doi.org/10.3390/v11060562
Bretscher C, Marchini A. H-1 Parvovirus as a Cancer-Killing Agent: Past, Present, and Future. Viruses. 2019; 11(6):562. https://doi.org/10.3390/v11060562
Chicago/Turabian StyleBretscher, Clemens, and Antonio Marchini. 2019. "H-1 Parvovirus as a Cancer-Killing Agent: Past, Present, and Future" Viruses 11, no. 6: 562. https://doi.org/10.3390/v11060562
APA StyleBretscher, C., & Marchini, A. (2019). H-1 Parvovirus as a Cancer-Killing Agent: Past, Present, and Future. Viruses, 11(6), 562. https://doi.org/10.3390/v11060562