Emerging Novel GII.P16 Noroviruses Associated with Multiple Capsid Genotypes

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Data Sources for Norovirus Typing and Outbreak Information
2.2. Norovirus RT-PCR
2.3. Complete Genome Sequencing (NGS)
2.4. Epidemiological Characteristics of GII.P16 Viruses
2.5. Sequence Selection and Data Analysis
3. Results
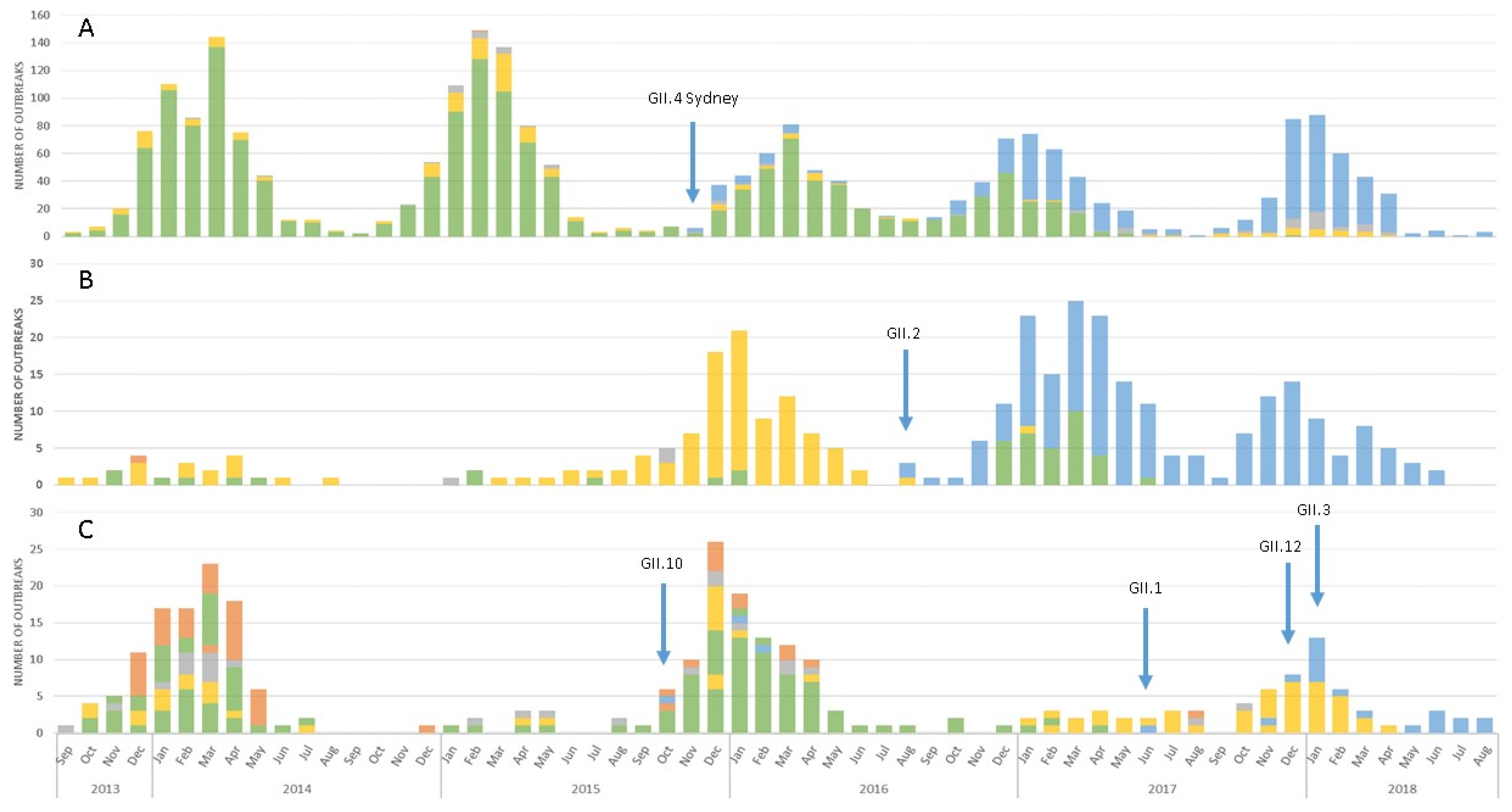
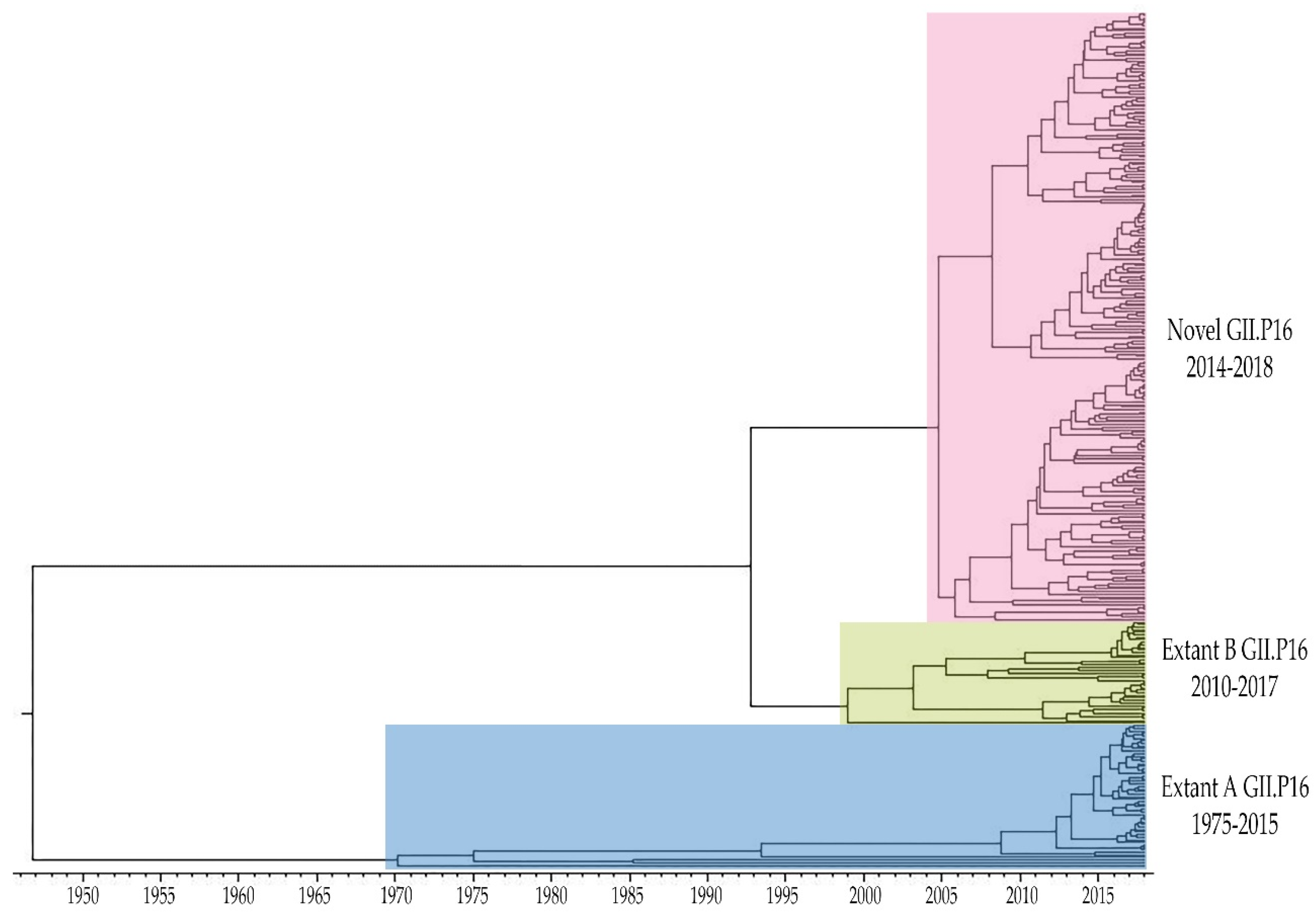
3.1. Emergence of A Novel GII.P16 Polymerase Lineage Detected in Outbreak Samples
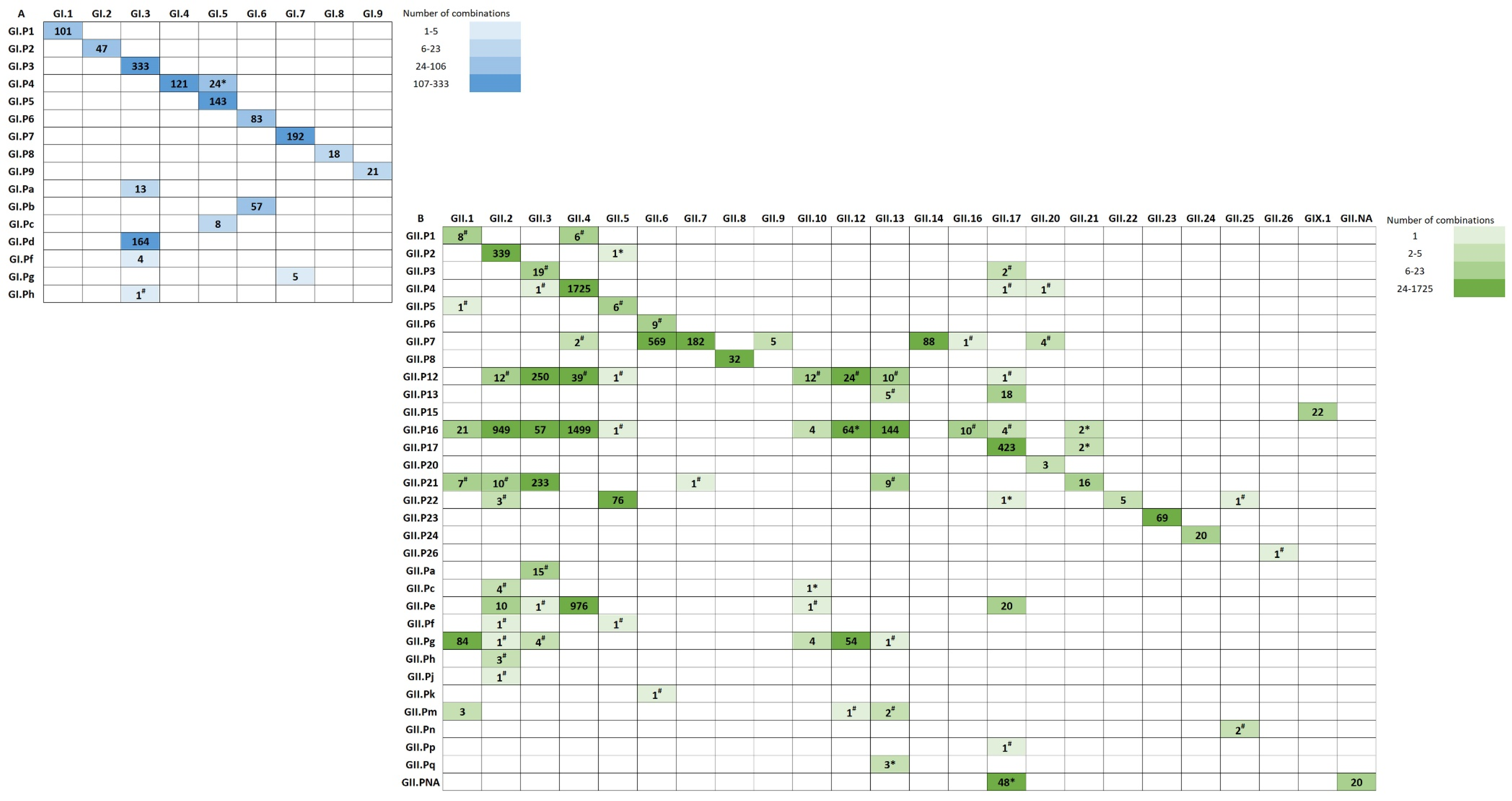
3.2. Polymerase-Capsid Combinations
3.3. Epidemiologic Characteristics and Outcomes for Novel GII.P16 Strains
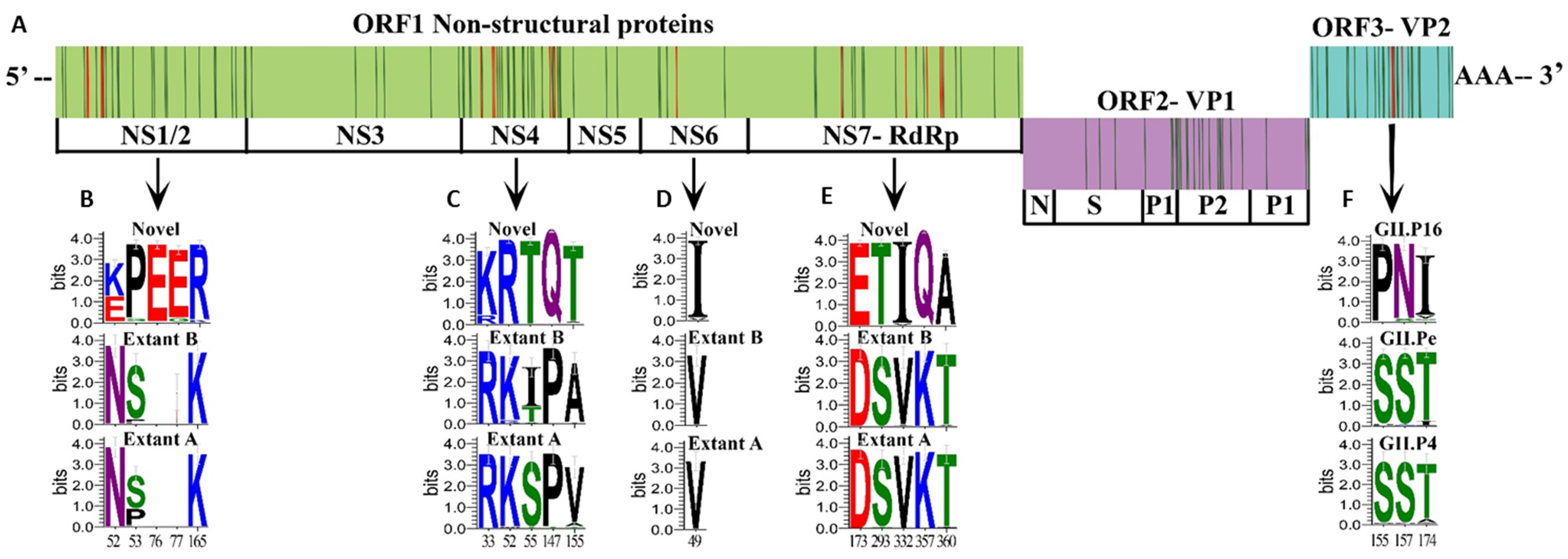
3.4. Unique Markers of Viruses with the Novel GII.P16 Polymerase
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Disclaimer
References
- Ahmed, S.M.; Hall, A.J.; Robinson, A.E.; Verhoef, L.; Premkumar, P.; Parashar, U.D.; Koopmans, M.; Lopman, B.A. Global prevalence of norovirus in cases of gastroenteritis: A systematic review and meta-analysis. Lancet Infect. Dis. 2014, 14, 725–730. [Google Scholar] [CrossRef]
- Kirk, M.D.; Pires, S.M.; Black, R.E.; Caipo, M.; Crump, J.A.; Devleesschauwer, B.; Dopfer, D.; Fazil, A.; Fischer-Walker, C.L.; Hald, T.; et al. World Health Organization Estimates of the Global and Regional Disease Burden of 22 Foodborne Bacterial, Protozoal, and Viral Diseases, 2010: A Data Synthesis. PLoS Med. 2015, 12, e1001921. [Google Scholar]
- Wikswo, M.E.; Kambhampati, A.; Shioda, K.; Walsh, K.A.; Bowen, A.; Hall, A.J. Outbreaks of Acute Gastroenteritis Transmitted by Person-to-Person Contact, Environmental Contamination, and Unknown Modes of Transmission—United States, 2009–2013. Morb. Mortal. Wkly. Rep. Surveill. Summ. 2015, 64, 1–16. [Google Scholar] [CrossRef]
- Shah, M.P.; Wikswo, M.E.; Barclay, L.; Kambhampati, A.; Shioda, K.; Parashar, U.D.; Vinje, J.; Hall, A.J. Near Real-Time Surveillance of U.S. Norovirus Outbreaks by the Norovirus Sentinel Testing and Tracking Network—United States, August 2009–July 2015. MMWR Morb. Mortal. Wkly. Rep. 2017, 66, 185–189. [Google Scholar] [CrossRef] [PubMed]
- Hall, A.J.; Lopman, B.A.; Payne, D.C.; Patel, M.M.; Gastanaduy, P.A.; Vinje, J.; Parashar, U.D. Norovirus disease in the United States. Emerg. Infect. Dis. 2013, 19, 1198–1205. [Google Scholar] [CrossRef] [PubMed]
- Green, K. Caliciviridae: The Noroviruses. In Fields’ Virology, 6th ed.; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; Volume 1. [Google Scholar]
- Vinje, J. Advances in laboratory methods for detection and typing of norovirus. J. Clin. Microbiol. 2015, 53, 373–381. [Google Scholar] [CrossRef]
- Lindesmith, L.C.; Donaldson, E.F.; Lobue, A.D.; Cannon, J.L.; Zheng, D.P.; Vinje, J.; Baric, R.S. Mechanisms of GII.4 norovirus persistence in human populations. PLoS Med. 2008, 5, e31. [Google Scholar] [CrossRef]
- Debbink, K.; Donaldson, E.F.; Lindesmith, L.C.; Baric, R.S. Genetic mapping of a highly variable norovirus GII.4 blockade epitope: Potential role in escape from human herd immunity. J. Virol. 2012, 86, 1214–1226. [Google Scholar] [CrossRef]
- Roth, A.N.; Karst, S.M. Norovirus mechanisms of immune antagonism. Curr. Opin. Virol. 2016, 16, 24–30. [Google Scholar] [CrossRef]
- Sosnovtsev, S.V.; Belliot, G.; Chang, K.O.; Onwudiwe, O.; Green, K.Y. Feline calicivirus VP2 is essential for the production of infectious virions. J. Virol. 2005, 79, 4012–4024. [Google Scholar] [CrossRef]
- Subba-Reddy, C.V.; Goodfellow, I.; Kao, C.C. VPg-primed RNA synthesis of norovirus RNA-dependent RNA polymerases by using a novel cell-based assay. J. Virol. 2011, 85, 13027–13037. [Google Scholar] [CrossRef] [PubMed]
- Vongpunsawad, S.; Venkataram Prasad, B.V.; Estes, M.K. Norwalk Virus Minor Capsid Protein VP2 Associates within the VP1 Shell Domain. J. Virol. 2013, 87, 4818–4825. [Google Scholar] [CrossRef] [PubMed]
- Conley, M.J.; McElwee, M.; Azmi, L.; Gabrielsen, M.; Byron, O.; Goodfellow, I.G.; Bhella, D. Calicivirus VP2 forms a portal-like assembly following receptor engagement. Nature 2019, 565, 377–381. [Google Scholar] [CrossRef] [PubMed]
- Ludwig-Begall, L.F.; Mauroy, A.; Thiry, E. Norovirus recombinants: Recurrent in the field, recalcitrant in the lab—A scoping review of recombination and recombinant types of noroviruses. J. Gen. Virol. 2018, 99, 970–988. [Google Scholar] [CrossRef] [PubMed]
- Bull, R.A.; Hansman, G.S.; Clancy, L.E.; Tanaka, M.M.; Rawlinson, W.D.; White, P.A. Norovirus recombination in ORF1/ORF2 overlap. Emerg. Infect. Dis. 2005, 11, 1079–1085. [Google Scholar] [CrossRef] [PubMed]
- Bull, R.A.; Tanaka, M.M.; White, P.A. Norovirus recombination. J. Gen. Virol. 2007, 88, 3347–3359. [Google Scholar] [CrossRef] [PubMed]
- Eden, J.S.; Tanaka, M.M.; Boni, M.F.; Rawlinson, W.D.; White, P.A. Recombination within the pandemic norovirus GII.4 lineage. J. Virol. 2013, 87, 6270–6282. [Google Scholar] [CrossRef]
- Waters, A.; Coughlan, S.; Hall, W.W. Characterisation of a novel recombination event in the norovirus polymerase gene. Virology 2007, 363, 11–14. [Google Scholar] [CrossRef][Green Version]
- Rohayem, J.; Munch, J.; Rethwilm, A. Evidence of recombination in the norovirus capsid gene. J. Virol. 2005, 79, 4977–4990. [Google Scholar] [CrossRef]
- Bull, R.A.; Eden, J.S.; Rawlinson, W.D.; White, P.A. Rapid evolution of pandemic noroviruses of the GII.4 lineage. PLoS Pathog. 2010, 6, e1000831. [Google Scholar] [CrossRef]
- Ruis, C.; Roy, S.; Brown, J.R.; Allen, D.J.; Goldstein, R.A.; Breuer, J. The emerging GII.P16-GII.4 Sydney 2012 norovirus lineage is circulating worldwide, arose by late-2014 and contains polymerase changes that may increase virus transmission. PLoS ONE 2017, 12, e0179572. [Google Scholar] [CrossRef] [PubMed]
- Tohma, K.; Lepore, C.J.; Ford-Siltz, L.A.; Parra, G.I. Phylogenetic Analyses Suggest that Factors Other Than the Capsid Protein Play a Role in the Epidemic Potential of GII.2 Norovirus. mSphere 2017, 2, e00187-17. [Google Scholar] [CrossRef] [PubMed]
- Siebenga, J.J.; Vennema, H.; Zheng, D.P.; Vinje, J.; Lee, B.E.; Pang, X.L.; Ho, E.C.; Lim, W.; Choudekar, A.; Broor, S.; et al. Norovirus illness is a global problem: Emergence and spread of norovirus GII.4 variants, 2001-2007. J. Infect. Dis. 2009, 200, 802–812. [Google Scholar] [CrossRef] [PubMed]
- Cannon, J.L.; Barclay, L.; Collins, N.R.; Wikswo, M.E.; Castro, C.J.; Magana, L.C.; Gregoricus, N.; Marine, R.L.; Chhabra, P.; Vinje, J. Genetic and Epidemiologic Trends of Norovirus Outbreaks in the United States from 2013 to 2016 Demonstrated Emergence of Novel GII.4 Recombinant Viruses. J. Clin. Microbiol. 2017, 55, 2208–2221. [Google Scholar] [CrossRef] [PubMed]
- CDC. CaliciNet Data. Available online: https://www.cdc.gov/norovirus/reporting/calicinet/data.html (accessed on 28 March 2019).
- Vega, E.; Barclay, L.; Gregoricus, N.; Williams, K.; Lee, D.; Vinje, J. Novel surveillance network for norovirus gastroenteritis outbreaks, United States. Emerg. Infect. Dis. 2011, 17, 1389–1395. [Google Scholar] [CrossRef] [PubMed]
- Nelson, M.I.; Mahfuz, M.; Chhabra, P.; Haque, R.; Seidman, J.C.; Hossain, I.; McGrath, M.; Ahmed, A.M.S.; Knobler, S.; Vinje, J.; et al. Genetic Diversity of Noroviruses Circulating in a Pediatric Cohort in Bangladesh. J. Infect. Dis. 2018, 218, 1937–1942. [Google Scholar] [CrossRef] [PubMed]
- Chhabra, P.; Rouhani, S.; Browne, H.; Yori, P.P.; Salas, M.S.; Olortegui, M.P.; Kosek, M.N.; Vinjé, J. Birth cohort study demonstrates multiple norovirus natural infections, homotypic and heterotypic protection and risk of re-infections in children from a highly endemic resource constrained setting. 2019; Unpublished work. [Google Scholar]
- Diez-Valcarce, M.; Lopez, M.R.; Lopez, B.; Morales, O.; Sagastume, M.; Cadena, L.; Kaydos-Daniels, S.; Jarquin, C.; McCracken, J.P.; Bryan, J.P.; et al. Prevalence and genetic diversity of viral gastroenteritis viruses in children younger than 5 years of age in Guatemala, 2014–2015. J. Clin. Virol. 2019, 114, 6–11. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, F.; Vielot, N.A.; Reyes, Y.; Toval, C.; Paniagua, M.; McCarter Bowman, N.; Vinjé, J.; Bucardo, F.; Becker-Dreps, S. Natural History of Sapovirus Infection in a Nicaraguan Birth Cohort: The Sapovirus-Associated Gastroenteritis (Sage) Study; American Society of Tropical Medicine and Hygiene: New Orleans, LA, USA, 2018. [Google Scholar]
- Hall, A.J.; Wikswo, M.E.; Manikonda, K.; Roberts, V.A.; Yoder, J.S.; Gould, L.H. Acute gastroenteritis surveillance through the National Outbreak Reporting System, United States. Emerg. Infect. Dis. 2013, 19, 1305–1309. [Google Scholar] [CrossRef] [PubMed]
- Montmayeur, A.M.; Ng, T.F.; Schmidt, A.; Zhao, K.; Magana, L.; Iber, J.; Castro, C.J.; Chen, Q.; Henderson, E.; Ramos, E.; et al. High-Throughput Next-Generation Sequencing of Polioviruses. J. Clin. Microbiol. 2017, 55, 606–615. [Google Scholar] [CrossRef] [PubMed]
- Ng, T.F.; Chen, L.F.; Zhou, Y.; Shapiro, B.; Stiller, M.; Heintzman, P.D.; Varsani, A.; Kondov, N.O.; Wong, W.; Deng, X.; et al. Preservation of viral genomes in 700-y-old caribou feces from a subarctic ice patch. Proc. Natl. Acad. Sci. USA 2014, 111, 16842–16847. [Google Scholar] [CrossRef] [PubMed]
- Ng, T.F.; Marine, R.; Wang, C.; Simmonds, P.; Kapusinszky, B.; Bodhidatta, L.; Oderinde, B.S.; Wommack, K.E.; Delwart, E. High variety of known and new RNA and DNA viruses of diverse origins in untreated sewage. J. Virol. 2012, 86, 12161–12175. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Greninger, A.L.; Isa, P.; Phan, T.G.; Martinez, M.A.; de la Luz Sanchez, M.; Contreras, J.F.; Santos-Preciado, J.I.; Parsonnet, J.; Miller, S.; et al. Discovery of a novel polyomavirus in acute diarrheal samples from children. PLoS ONE 2012, 7, e49449. [Google Scholar] [CrossRef] [PubMed]
- Greninger, A.L.; Chen, E.C.; Sittler, T.; Scheinerman, A.; Roubinian, N.; Yu, G.; Kim, E.; Pillai, D.R.; Guyard, C.; Mazzulli, T.; et al. A metagenomic analysis of pandemic influenza A (2009 H1N1) infection in patients from North America. PLoS ONE 2010, 5, e13381. [Google Scholar] [CrossRef] [PubMed]
- Nurk, S.; Bankevich, A.; Antipov, D.; Gurevich, A.A.; Korobeynikov, A.; Lapidus, A.; Prjibelski, A.D.; Pyshkin, A.; Sirotkin, A.; Sirotkin, Y.; et al. Assembling single-cell genomes and mini-metagenomes from chimeric MDA products. J. Comput. Biol. 2013, 20, 714–737. [Google Scholar] [CrossRef] [PubMed]
- Burke, R.M.; Shah, M.P.; Wikswo, M.E.; Barclay, L.; Kambhampati, A.; Marsh, Z.; Cannon, J.L.; Parashar, U.D.; Vinje, J.; Hall, A.J. The Norovirus Epidemiologic Triad: Predictors of Severe Outcomes in US Norovirus Outbreaks, 2009–2016. J. Infect. Dis. 2019. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Rozewicki, J.; Yamada, K.D. MAFFT online service: Multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. 2017. [Google Scholar] [CrossRef] [PubMed]
- Kuraku, S.; Zmasek, C.M.; Nishimura, O.; Katoh, K. aLeaves facilitates on-demand exploration of metazoan gene family trees on MAFFT sequence alignment server with enhanced interactivity. Nucleic Acids Res. 2013, 41, W22–W28. [Google Scholar] [CrossRef]
- Drummond, A.J.; Rambaut, A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 2007, 7, 214. [Google Scholar] [CrossRef]
- Bouckaert, R.; Heled, J.; Kuhnert, D.; Vaughan, T.; Wu, C.H.; Xie, D.; Suchard, M.A.; Rambaut, A.; Drummond, A.J. BEAST 2: A software platform for Bayesian evolutionary analysis. PLoS Comput. Biol. 2014, 10, e1003537. [Google Scholar] [CrossRef]
- Lindesmith, L.C.; Brewer-Jensen, P.D.; Mallory, M.L.; Debbink, K.; Swann, E.W.; Vinje, J.; Baric, R.S. Antigenic Characterization of a Novel Recombinant GII.P16-GII.4 Sydney Norovirus Strain with Minor Sequence Variation Leading to Antibody Escape. J. Infect. Dis. 2018, 217, 1145–1152. [Google Scholar] [CrossRef]
- Parra, G.I.; Squires, R.B.; Karangwa, C.K.; Johnson, J.A.; Lepore, C.J.; Sosnovtsev, S.V.; Green, K.Y. Static and Evolving Norovirus Genotypes: Implications for Epidemiology and Immunity. PLoS Pathog. 2017, 13, e1006136. [Google Scholar] [CrossRef] [PubMed]
- Niendorf, S.; Jacobsen, S.; Faber, M.; Eis-Hubinger, A.M.; Hofmann, J.; Zimmermann, O.; Hohne, M.; Bock, C.T. Steep rise in norovirus cases and emergence of a new recombinant strain GII.P16-GII.2, Germany, winter 2016. Euro Surveill. 2017, 22, 30447. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.T.; Kuo, T.Y.; Wu, C.Y.; Liao, W.T.; Hall, A.J.; Wu, F.T. Recombinant GII.P16-GII.2 Norovirus, Taiwan, 2016. Emerg. Infect. Dis. 2017, 23, 1180–1183. [Google Scholar] [CrossRef] [PubMed]
- Kwok, K.; Niendorf, S.; Lee, N.; Hung, T.N.; Chan, L.Y.; Jacobsen, S.; Nelson, E.A.S.; Leung, T.F.; Lai, R.W.M.; Chan, P.K.S.; et al. Increased Detection of Emergent Recombinant Norovirus GII.P16-GII.2 Strains in Young Adults, Hong Kong, China, 2016–2017. Emerg. Infect. Dis. 2017, 23, 1852–1855. [Google Scholar] [CrossRef] [PubMed]
- Gastanaduy, P.A.; Hall, A.J.; Curns, A.T.; Parashar, U.D.; Lopman, B.A. Burden of norovirus gastroenteritis in the ambulatory setting—United States, 2001–2009. J. Infect. Dis. 2013, 207, 1058–1065. [Google Scholar] [CrossRef] [PubMed]
- Rha, B.; Burrer, S.; Park, S.; Trivedi, T.; Parashar, U.D.; Lopman, B.A. Emergency department visit data for rapid detection and monitoring of norovirus activity, United States. Emerg. Infect. Dis. 2013, 19, 1214–1221. [Google Scholar] [CrossRef] [PubMed]
- Ozaki, K.; Matsushima, Y.; Nagasawa, K.; Motoya, T.; Ryo, A.; Kuroda, M.; Katayama, K.; Kimura, H. Molecular Evolutionary Analyses of the RNA-Dependent RNA Polymerase Region in Norovirus Genogroup II. Front. Microbiol. 2018, 9, 3070. [Google Scholar] [CrossRef] [PubMed]
- McCune, B.T.; Tang, W.; Lu, J.; Eaglesham, J.B.; Thorne, L.; Mayer, A.E.; Condiff, E.; Nice, T.J.; Goodfellow, I.; Krezel, A.M.; et al. Noroviruses Co-opt the Function of Host Proteins VAPA and VAPB for Replication via a Phenylalanine-Phenylalanine-Acidic-Tract-Motif Mimic in Nonstructural Viral Protein NS1/2. mBio 2017, 8, e00668-17. [Google Scholar] [CrossRef] [PubMed]
- Ettayebi, K.; Hardy, M.E. Norwalk virus nonstructural protein p48 forms a complex with the SNARE regulator VAP-A and prevents cell surface expression of vesicular stomatitis virus G protein. J. Virol. 2003, 77, 11790–11797. [Google Scholar] [CrossRef] [PubMed]
- Sharp, T.M.; Crawford, S.E.; Ajami, N.J.; Neill, F.H.; Atmar, R.L.; Katayama, K.; Utama, B.; Estes, M.K. Secretory pathway antagonism by calicivirus homologues of Norwalk virus nonstructural protein p22 is restricted to noroviruses. Virol. J. 2012, 9, 181. [Google Scholar] [CrossRef]
- Matsui, S.M.; Kim, J.P.; Greenberg, H.B.; Su, W.; Sun, Q.; Johnson, P.C.; DuPont, H.L.; Oshiro, L.S.; Reyes, G.R. The isolation and characterization of a Norwalk virus-specific cDNA. J. Clin. Investig. 1991, 87, 1456–1461. [Google Scholar] [CrossRef] [PubMed]
- Peersen, O.B. Picornaviral polymerase structure, function, and fidelity modulation. Virus Res. 2017, 234, 4–20. [Google Scholar] [CrossRef] [PubMed]
- Ettayebi, K.; Crawford, S.E.; Murakami, K.; Broughman, J.R.; Karandikar, U.; Tenge, V.R.; Neill, F.H.; Blutt, S.E.; Zeng, X.L.; Qu, L.; et al. Replication of human noroviruses in stem cell-derived human enteroids. Science 2016, 353, 1387–1393. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.K.; Watanabe, M.; Zhu, S.; Graves, C.L.; Keyes, L.R.; Grau, K.R.; Gonzalez-Hernandez, M.B.; Iovine, N.M.; Wobus, C.E.; Vinje, J.; et al. Enteric bacteria promote human and mouse norovirus infection of B cells. Science 2014, 346, 755–759. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
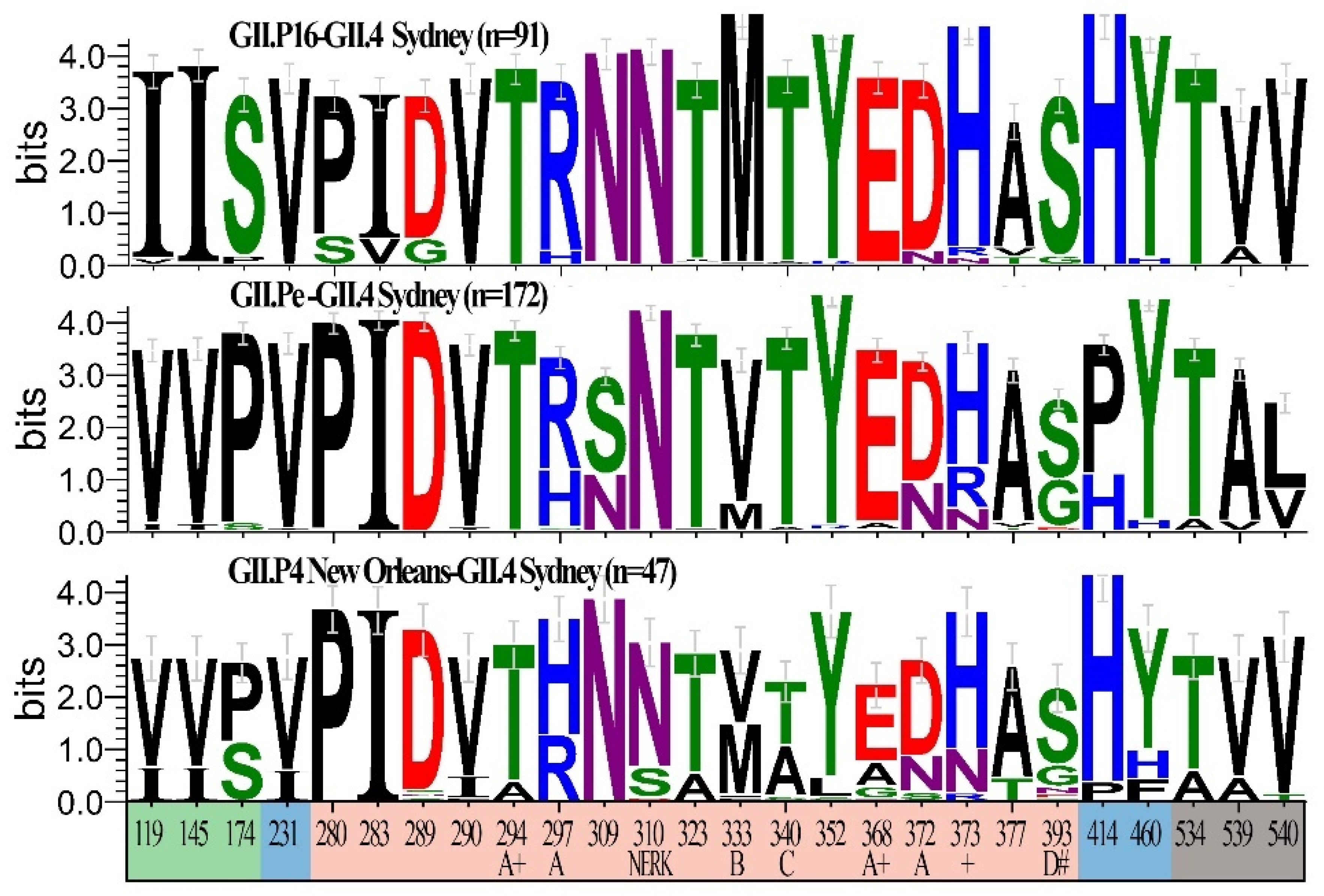{kind=link}
| Transmission | Other Polymerases Total (%) | Novel GII.P16 Total (%) | OR (99% CI) | Total No. |
| Foodborne | 74 (54.4) | 62 (45.6) | 1.00 | 136 |
| Person-to-person | 184 (48.8) | 193 (51.2) | 1.25 (0.75–2.11) | 377 |
| Other | 47 (55.3) | 38 (44.7) | 0.97 (0.47–1.98) | 85 |
| Setting | Other Polymerases Total (%) | Novel GII.P16 Total (%) | OR (99% CI) | Total No. |
| Non-Healthcare | 154 (56.6) | 118 (43.4) | 1.00 | 272 |
| Healthcare | 144 (45.4) | 173 (54.6) | 1.57 (1.02–2.41) ** | 317 |
| Characteristics of Outbreaks | Other Polymerases Total (%) | Novel GII.P16 Total (%) | OR (99% CI) | Total No. | |
|---|---|---|---|---|---|
| Gender | <50% Female | 88 (55.0) | 72 (45.0) | 1.00 | 160 |
| ≥50% Female | 187 (54.5) | 156 (45.5) | 1.02 (0.62–1.68) | 343 | |
| Age, y | <50% 75 or older | 181 (58.6) | 128 (41.4) | 1.00 | 309 |
| ≥50% 75 or older | 68 (52.3) | 62 (47.7) | 1.29 (0.75–2.22) | 130 | |
| Symptoms | |||||
| Abdominal cramps | <50% cases | 153 (54.1) | 130 (45.9) | 1.00 | 283 |
| ≥50% cases | 63 (55.8) | 50 (44.2) | 0.93 (0.52–1.66) | 113 | |
| Fever | <50% cases | 211 (52.2) | 193 (47.8) | 1.00 | 404 |
| ≥50% cases | 24 (66.7) | 12 (33.3) | 0.55 (0.20–1.39) | 36 | |
| Vomiting | <50% cases | 130 (51.8) | 121 (48.2) | 1.00 | 251 |
| ≥50% cases | 155 (56.6) | 119 (43.4) | 0.83 (0.52–1.30) | 274 | |
| Diarrhea | <50% cases | 87 (43.4) | 76 (46.6) | 1.00 | 163 |
| ≥50% cases | 195 (53.6) | 169 (46.4) | 0.99 (0.61–1.62) | 364 | |
| Magnitude | 0–23 cases | 165 (55.2) | 134 (44.8) | 1.00 | 299 |
| 24–313 cases | 140 (46.8) | 159 (53.2) | 1.40 (0.92–2.14) | 299 | |
| Outcome | Other Polymerases | Novel GII.P16 | RR (99% CI) | ||||
|---|---|---|---|---|---|---|---|
| Case Counts | Total No. | Rate per 10,000 Cases | Case Counts | Total No. | Rate per 10,000 Cases | ||
| Outpatient Visit | 322 | 5086 | 633.11 | 282 | 4922 | 572.94 | 0.90 (0.73–1.11) |
| Emergency Department Visit | 194 | 5428 | 357.41 | 156 | 6260 | 249.20 | 0.69 (0.52–0.91) ** |
| Hospitalized | 155 | 7848 | 197.50 | 179 | 8415 | 212.72 | 1.08 (0.81–1.43) |
| Death | 17 | 8447 | 20.13 | 18 | 8718 | 20.65 | 1.03 (0.42–2.51) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barclay, L.; Cannon, J.L.; Wikswo, M.E.; Phillips, A.R.; Browne, H.; Montmayeur, A.M.; Tatusov, R.L.; Burke, R.M.; Hall, A.J.; Vinjé, J. Emerging Novel GII.P16 Noroviruses Associated with Multiple Capsid Genotypes. Viruses 2019, 11, 535. https://doi.org/10.3390/v11060535
Barclay L, Cannon JL, Wikswo ME, Phillips AR, Browne H, Montmayeur AM, Tatusov RL, Burke RM, Hall AJ, Vinjé J. Emerging Novel GII.P16 Noroviruses Associated with Multiple Capsid Genotypes. Viruses. 2019; 11(6):535. https://doi.org/10.3390/v11060535
Chicago/Turabian StyleBarclay, Leslie, Jennifer L. Cannon, Mary E. Wikswo, Annie R. Phillips, Hannah Browne, Anna M. Montmayeur, Roman L. Tatusov, Rachel M. Burke, Aron J. Hall, and Jan Vinjé. 2019. "Emerging Novel GII.P16 Noroviruses Associated with Multiple Capsid Genotypes" Viruses 11, no. 6: 535. https://doi.org/10.3390/v11060535
APA StyleBarclay, L., Cannon, J. L., Wikswo, M. E., Phillips, A. R., Browne, H., Montmayeur, A. M., Tatusov, R. L., Burke, R. M., Hall, A. J., & Vinjé, J. (2019). Emerging Novel GII.P16 Noroviruses Associated with Multiple Capsid Genotypes. Viruses, 11(6), 535. https://doi.org/10.3390/v11060535