A Highly Sensitive Method to Detect Avocado Sunblotch Viroid for the Maintenance of Infection-Free Avocado Germplasm Collections


Abstract
1. Introduction
2. Materials and Methods
2.1. Avocado Trees
2.2. Viroid Indexing
2.2.1. Sampling
2.2.2. Tissue Disruption
2.3. RNA Extraction
2.4. Primers and Probes
2.5. Reverse Transcription and Pre-amplification
2.6. TaqMan Real-Time PCR Assays
2.7. Roguing Infected Trees
2.8. Creation of a Backup Germplasm Collection
3. Results
3.1. Assay Design and Testing
3.2. Sensitivity Determination
3.3. Distribution of ASBVd in A Single Tree
3.4. Pooling Samples and Multiplex of ASBVd and GAPDH
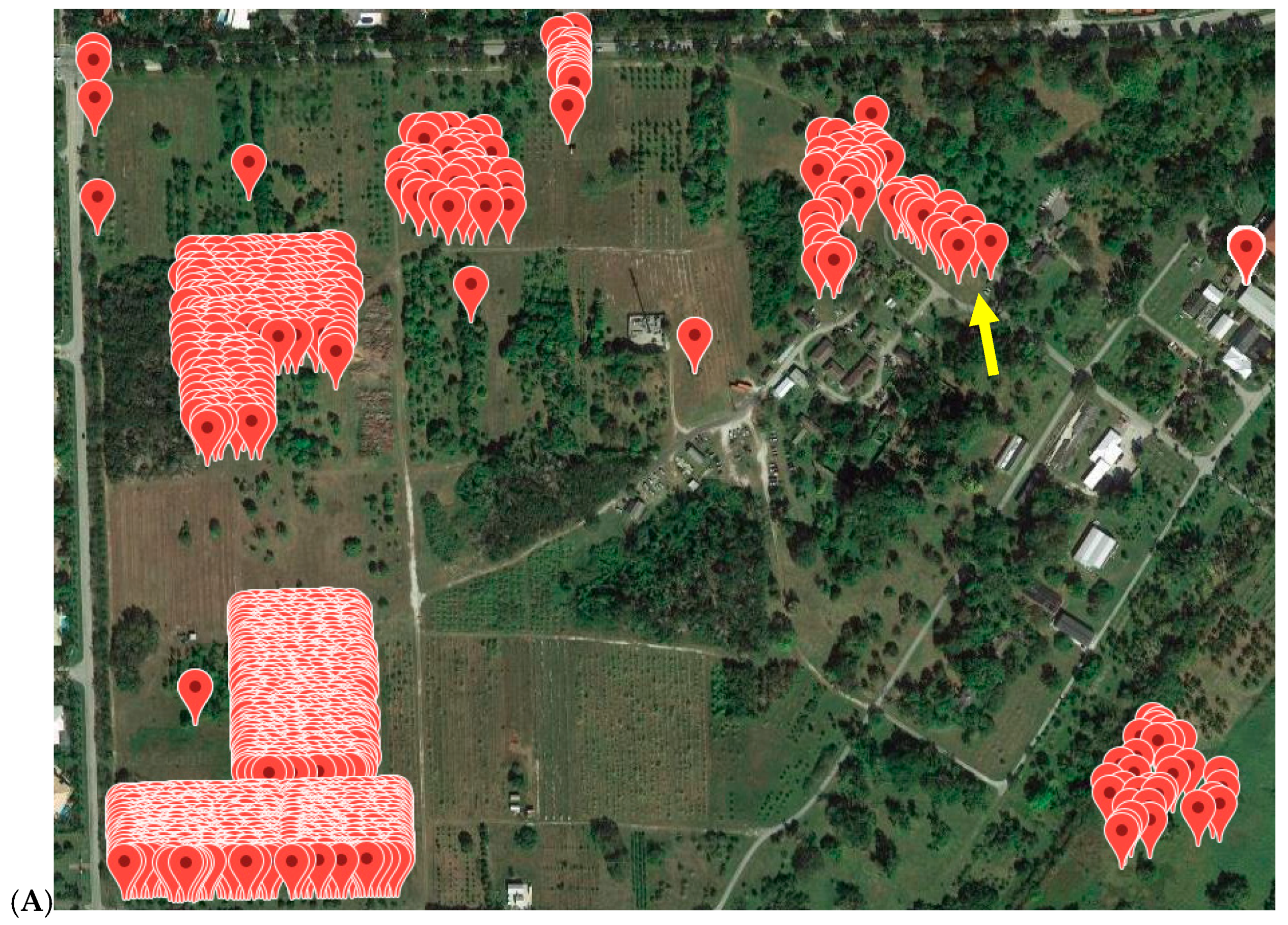
3.5. Application
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Di Serio, F.; Flores, R.; Verhoeven, J.T.J.; Li, S.-F.; Pallás, V.; Randles, J.W.; Sano, T.; Vidalakis, G.; Owens, R.A. Current status of viroid taxonomy. Arch. Virol. 2014, 159, 3467–3478. [Google Scholar] [CrossRef] [PubMed]
- Saucedo-Carabez, J.R.; Téliz-Ortiz, D.; Ochoa-Ascencio, S.; Ochoa-Martínez, D.; Vallejo-Pérez, M.R.; Beltrán-Peña, H. Effect of Avocado sunblotch viroid (ASBVd) on avocado yield in Michoacan, Mexico. Eur. J. Plant Pathol. 2014, 13, 799–805. [Google Scholar] [CrossRef]
- Kuhn, D.N.; Geering, A.D.W.; Dixon, J. Avocado sunblotch viroid. In Viroids and Satellites; Hadidi, A., Flores, R., Randles, J.W., Palukaitis, P., Eds.; Academic Press Ltd-Elsevier Science Ltd.: London, UK, 2017; pp. 297–305. [Google Scholar] [CrossRef]
- Desjardins, P.R.; Drake, R.J.; Sasaki, P.J.; Atkins, E.L.; Bergh, B.O. Pollen transmission of of avocado sunblotch viroid and the fate of the pollen recipient tree. Phytopathology 1984, 74, 845. [Google Scholar]
- Suarez, I.E.; Schnell, R.A.; Kuhn, D.N.; Litz, R.E. Recovery and indexing of avocado plants (persea americana) from embryogenic nucellar cultures of an avocado sunblotch viroid-infected tree. Plant Cell Tissue Organ Cult. 2006, 84, 27–37. [Google Scholar] [CrossRef]
- Suarez, I.E.; Schnell, R.A.; Kuhn, D.N.; Litz, R.E. Micrografting of asbvd-infected avocado (persea americana) plants. Plant Cell Tissue Organ Cult. 2005, 80, 179–185. [Google Scholar] [CrossRef]
- Schnell, R.J.; Tondo, C.L.; Kuhn, D.N.; Winterstein, M.C.; Ayala-Silva, T.; Moore, J.M. Spatial analysis of avocado sunblotch disease in an avocado germplasm collection. J. Phytopathol. 2011, 159, 773–781. [Google Scholar] [CrossRef]
- Olano, C.T.; Schnell, R.J.; Kuhn, D.N. Current status of asbvd infection among avocado accessions in the national germplasm collection. Proc. Fla. State Hort. Soc. 2002, 115, 280–282. [Google Scholar]
- Running, C.; Schnell, R.; Kuhn, D.N. Detection of avocado sunblotch viroid and estimation of infection among accessions in the national germplasm collection for avocado. Proc. Fla. State Hort. Soc. 1996, 109, 235–236. [Google Scholar]
- Ploetz, R.; Peña, J.; Smith, J.; Dreaden, T.; Crane, J.; Schubert, T.; Dixon, W. Laurel wilt, caused by raffaelea lauricola, is confirmed in miami-dade county, center of florida’s commercial avocado production. Plant Dis. 2011, 95, 1589. [Google Scholar] [CrossRef] [PubMed]
- Schnell, R.; Kuhn, D.; Ronning, C.; Harkins, D. Application of rt-pcr for indexing avocado sunblotch viroid. Plant Dis. 1997, 81, 1023–1026. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Da Graça, J.; Mason, T. Detection of avocado sunblotch viroid in flower buds by polyacrylamide gel electrophoresis. J. Phytopathol. 1983, 108, 262–266. [Google Scholar] [CrossRef]
- Semancik, J.; Szychowski, J. Avocado sunblotch disease: A persistent viroid infection in which variants are associated with differential symptoms. J. Gen. Virol. 1994, 75, 1543–1549. [Google Scholar] [CrossRef] [PubMed]
- Morey-León, G.; Ortega-Ramirez, E.; Julca-Chunga, C.; Santos-Chanta, C.; Graterol-Caldera, L.; Mialhe, E. The detection of avocado sunblotch viroid in avocado using a real-time reverse transcriptase polymerase chain reaction. BioTechnologia 2018, 99, 99–107. [Google Scholar] [CrossRef]
- Olano, C.; Borrone, J.; Brown, J.S.; Violi, H.; Ploetz, R.; Schnell, R. Development of mapping populations for avocado. Proc. Fla. State Hort. Soc. 2007, 120, 26–29. [Google Scholar]
- Tondo, C.L.; Schnell, R.J.; Kuhn, D.N. Results of the 2009 asbvd survey of avocado accessions of the national germplasm collection in florida. Proc. Fla. State Hort. Soc. 2010, 123, 5–7. [Google Scholar]
- Geering, A.D.W.; Steele, V.; Kopittke, R. Final report for Horticulture Australia Limited Project AV03009: Development of Avocado sunblotch viroid indexing protocols for the avocado nursery industry; University of Queensland: St Lucia, Australia, 2006. [Google Scholar]
- Randles, J.W.; Hanold, D.; Harding, R.M.; Skrzeckzkowski, L.; Whattam, M. Viroids in Australasia. In Viroids; Hadidi, A., Flores, R., Randles, J.W., Semancik, J.S., Eds.; CSIRO Publishing: Collingwood, Australia, 2003. [Google Scholar]
- Ainsworth, C. Isolation of RNA from floral tissue ofrumex acetosa (sorrel). Plant Mol. Biol. Report. 1994, 12, 198–203. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Population | Original Source 1 | Current Location 1 | Number Individuals Sampled | Number Individuals with Inconsistent Results from Previous Assay |
|---|---|---|---|---|
| Backup germplasm collection | SHRS ARS | FDWSRU ARS Ft. Detrick, Maryland | 131 | 10 |
| Avocado germplasm collection | SHRS ARS | SHRS ARS Miami, Florida | 383 | 50 |
| Avocado mapping population 2 | SHRS ARS | SHRS ARS Miami, Florida | 50 | NPA 3 |
| Backup germplasm collection | SHRS ARS | PBARC ARS Hilo, Hawaii | 102 | NPA |
| Primer Name | Primer Color in Figure 1 | Primer Sequence (5′→ 3′) | Position in ASBVd Genome | Reaction | Reference |
|---|---|---|---|---|---|
| GAPTM-F1 | TGGAGTGGACAGTGGTCATCAG | RT and TaqMan | Geering, ADW [17] | ||
| GAPTM-R1new | CCCATTGGCCAAGGTGATC | RT and TaqMan | This study | ||
| GAPDHTM-probe | [VIC]-CCCTCAACAATGCC-MGBNFQ | TaqMan | Geering, ADW [17] | ||
| SB1-F1 | Red | TGGGAAGAACACTGATGAG | 180–198 | RT and preAmp | Semancik, JS. [13] |
| SB1-R1 | Red | TCTTTCCCTGAAGAGACGA | 179–161 | RT and preAmp | Semancik, JS. [13] |
| ASBTM-F1 | Blue | TTCCGACTCTGAGTTTCGACTT | 66–87 | TaqMan | Geering, ADW [17] |
| AVFL1 | Green | CAAGAGATTGAAGACGAGTGAACTA | 179–155 | TaqMan | Randles et al. [18] |
| ASBTM-probe | Hatched | [6FAM]TTCCGACTCTGAGTTTCGACTT-MGBNFQ | 89–107 | TaqMan | Geering, ADW [17] |
| ASB-F1 | Purple | GTGAGAGAAGGAGGAGT | 88–104 | Schnell et al. [11] | |
| ASB-R1 | Purple | AAGTCGAAACTCAGAGTCGG | 87–68 | Schnell et al. [11] | |
| AVFL2 | Green | ATCACTTCGTCTCTTCAGGGAAAGA | 130–154 | Randles et al. [18] | |
| ASBTM-R1 | Blue | GTTCTTCCCATCTTTCCCTGA | 189–168 | Geering, ADW [17] |
| Dilution | CT avg | CT SD |
|---|---|---|
| 1:103 | 17.6 | 2.0 |
| 1:104 | 24.3 | 1.9 |
| 1:105 | 25.5 | 1.4 |
| 1:106 | 29.5 | 2.4 |
| 1:107 | 31.1 | 2.4 |
| 1:108 | No amp | 0.0 |
| 1:109 | No amp | 0.0 |
| Sample ID | Avg CT | Avg CT all Locations * | FDWSRU Graft av CT | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Cultivar | Location (SHRS) | Genotype ID | East | North | South | Top North | Top South | West | ||
| AYCOCK RED NO. 19 | W3-1-08-01 | PC | 22.3 | 9.3 | 7.3 | 8.4 | 8.2 | 8.0 | 10.6 | NG |
| CELLON’S HAWAII SDLG | W3-1-02-01 | CFN01 | 26.9 | 25.0 | 39.4 | 33.8 | 35.6 | No amp | 33.6 | NG |
| SHARWIL | WA2-12-37 | CFN19 | 26.9 | 25.0 | 22.9 | 23.9 | 25.2 | 21.9 | 24.3 | NG |
| JOSE ANTONIO | WA2-20-32.2 | CFN22 | No amp | No amp | No amp | 17.9 | 24.0 | No amp | 34.3 | NG |
| R06-T05 | WB4-02-13 | FtD3 | No amp | No amp | 18.0 | No amp | No amp | 11.5 | 32.2 | 18.3 |
| LA PISCINA | WB3-18-08 | FtD30 | 22.3 | 21.8 | 23.9 | No amp | 24.8 | 34.0 | 28.0 | 18.9 |
| DARIAN | WB3-19-11 | FtD36 | 20.0 | 22.3 | No amp | 28.2 | 17.3 | No amp | 28.3 | 11.0 |
| SEMIL 43 | WA2-13-41 | FtD96 | 22.2 | 20.5 | 22.2 | 16.4 | 17.3 | 12.7 | 18.6 | 29.1 |
| P. NUBIGENA | WB3-10-03 | FtD97 | 16.2 | 23.3 | 24.8 | No amp | 22.1 | 20.7 | 24.7 | 8.2 |
| PIC 9615 | WB3-13-10 | FtD105 | 19.7 | No amp | No amp | No amp | 13.4 | No amp | 32.8 | 16.2 |
| PINKERTON | WB4-09-01 | FtD107 | No amp | No amp | 23.2 | No amp | 22.2 | No amp | 34.9 | 6.1 |
| DADE SDLG | WB4-04-17 | FtD110 | No amp | 18.5 | 38.3 | No amp | No amp | No amp | 36.8 | 7.8 |
| HONALINDO 1 | WB3-10-07 | FtD117 | 19.2 | 21.6 | No amp | 9.5 | 9.0 | No amp | 23.5 | 8.8 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kuhn, D.N.; Freeman, B.; Geering, A.; Chambers, A.H. A Highly Sensitive Method to Detect Avocado Sunblotch Viroid for the Maintenance of Infection-Free Avocado Germplasm Collections. Viruses 2019, 11, 512. https://doi.org/10.3390/v11060512
Kuhn DN, Freeman B, Geering A, Chambers AH. A Highly Sensitive Method to Detect Avocado Sunblotch Viroid for the Maintenance of Infection-Free Avocado Germplasm Collections. Viruses. 2019; 11(6):512. https://doi.org/10.3390/v11060512
Chicago/Turabian StyleKuhn, David N., Barbie Freeman, Andrew Geering, and Alan H. Chambers. 2019. "A Highly Sensitive Method to Detect Avocado Sunblotch Viroid for the Maintenance of Infection-Free Avocado Germplasm Collections" Viruses 11, no. 6: 512. https://doi.org/10.3390/v11060512
APA StyleKuhn, D. N., Freeman, B., Geering, A., & Chambers, A. H. (2019). A Highly Sensitive Method to Detect Avocado Sunblotch Viroid for the Maintenance of Infection-Free Avocado Germplasm Collections. Viruses, 11(6), 512. https://doi.org/10.3390/v11060512